

Abstract
Summary
Rapid advances in high-throughput sequencing technologies have enabled more efficient acquisition of massive amount of multi-omics data. However, interpretation of the underlying relationships across multi-omics networks has not been fully succeeded, partly due to the lack of effective methods in visualization. To aid interpretation of the results from such multi-omics data, we here present Grimon (Graphical interface to visualize multi-omics networks), an R package that visualizes high-dimensional multi-layered data sets in three-dimensional parallel coordinates. Grimon enables users to intuitively and interactively explore their analyzed data, helping their understanding of multiple inter-layer connections embedded in high-dimensional complex data.
Availability and implementation
Grimon is freely available at https://github.com/mkanai/grimon as an R package with example omics data sets.
Supplementary information
Supplementary data are available at bioinformatics online.
1 Introduction
Elucidation of hidden relationships across multi-omics networks is a key step to understand human biology as a whole system, which enables us to disentangle contributions and interactions of each omic layer (Boyle et al., 2017; Kanai et al., 2018; Okada et al., 2016). Although recent rapid developments in next-generation sequencing technologies facilitates us to acquire massive amount of data from multiple biological perspectives, methods to analyze and interpret such multi-layered high-dimensional data are still under active development.
In particular, an efficient data visualization technique plays an important role to interpret high-dimensional data. Previous studies applied typical dimensionality reduction techniques, such as principal component analysis (PCA) or t-Distributed Stochastic Neighbor Embedding (t-SNE), to represent complex data in two-dimensional space (Lake et al., 2018). However, these methods are only applicable to a single high-dimensional data, and are difficult to illustrate inter-layer relationships across multiple heterogeneous data sets. Parallel coordinates are another option to visualize high-dimensional multivariate data sets (Okada et al., 2015), but one dimension is often not sufficient for representing complex data within a single layer. As a result, we often see multiple two-dimensional plots only highlighting a single aspect of data, and have no clues about complex interplay embedded within multiple data.
To address these issues, we here propose Grimon (Graphical interface to visualize multi-omics networks), which enables us to smoothly explore relationships across multi-layered high-dimensional data visualized in 3D. Grimon is implemented as an R package using rgl (R wrapper for OpenGL), allowing users easily visualize their analyzed data within the same R environment and understand multiple inter-layer connections embedded in high-dimensional complex data.
2 Materials and methods
We expanded two-dimensional parallel coordinates into three-dimensional space to illustrate complex relationships across multi-layered high-dimensional data sets. To efficiently visualize inter-layer connections, we optimized an edge layout by rotating each layer to minimize sum of angles or lengths of edges when represented in three-dimensional space (see Supplementary Note for details). To scale our edge optimization efficiently to larger data points, we employed simulated annealing which stochastically approximates global optimization even in a large search space.
To highlight the main features of Grimon, we used real omics data, including metagenome and multi-omics (phenome, genome and transcriptome), as an example to illustrate how efficiently Grimon could visualize multi-layered relationships. For metagenome data, we acquired taxonomic composition of the fecal microbiota from early rheumatoid arthritis (RA) patients (n = 17) and healthy controls (n = 14) (Maeda et al., 2016). We then applied PCA at different taxonomy levels to represent each taxonomy in two-dimensional space. For multi-omics data, the deep RNA-sequencing of mRNA and miRNA of lymphoblastoid cell lines (LCL) were retrieved from the Geuvadis Project (Lappalainen et al., 2013) in addition to the whole-genome sequencing of the same samples from the 1000 Genomes Project (The 1000 Genomes Project Consortium, 2015). We removed genes and samples with low expression, normalized genotype and expression, and then applied PCA at each omic layer. Additional examples, including transcriptome data (Fairfax et al., 2014) and multiple molecular phenotype data (Li et al., 2016), are also detailed in Supplementary Note.
3 Results
Schematic overview of Grimon is illustrated in Figure 1, which recapitulates its capability to efficiently visualize multi-layered high-dimensional data. Even when there are more than ten clusters, users can intuitively and interactively explore their correspondence across multiple layers in a three-dimensional space. Taking advantage of an R package, Grimon could easily illustrate any analyzed data within the same R environment.
Fig. 1.
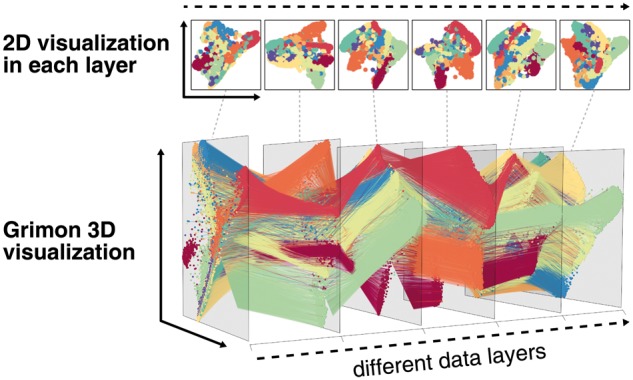
Schematic overview of Grimon visualization for high-dimensional multi-layered data. Grimon depicts cluster relationships across multiple layers by transforming multiple two-dimensional visualizations into three-dimensional space. Here, we used a simulated high-dimensional data with 10 clusters based on the Modified National Institute of Standards and Technology (MNIST) dataset, as a publicly available example. Each plane corresponds to a two-dimensional visualization of t-SNE result for each layer. Each edge connects the same sample in different layers. Edge color corresponds to each cluster. The horizontal axis represents multiple data layers
Here, we show practical examples of Grimon’s visualization. First example is metagenome data from early RA patients and healthy controls. Early RA patients with intestinal microbiota dominated by Prevotella copri are known to develop severe arthritis (Maeda et al., 2016). Although typical PCA visualization could also recapitulate a cluster of early RA patients with Prevotella copri at a specific single taxonomy level (e.g., L5: genus in Supplementary Fig. S1A), their relationships between different taxonomy levels are not observable. Furthermore, choice of the taxonomy level for visualization could be arbitrary. On the other hand, Grimon could illustrate that while the RA patients and controls were localized together in the upper taxonomy levels (L2: class and L3: order), the cluster of RA patients with Prevotella copri actually isolated at approximately L4 (family), and the clusters became consolidated at further taxonomy levels (L5: genus and L6: species; Supplementary Fig. S1B). Moreover, Grimon could also visualize relationships of bacteria, which illuminates Grimon’s functionality to visualize heterogeneous data with different number of samples (Supplementary Fig. S2).
Grimon visualization of multi-omics data was highlighted using transcriptome data (mRNA and miRNA) from the Geuvadis Project with phenome (population) and genome data from the 1000 Genomes Project. It is well-known that principal components of genotype forms distinct clusters of ancestral populations, while these of transcriptome spread more diversely relatively independent from populations (Martin et al., 2014). We visually recaptured this observation, but from different perspective, by connecting every individual from population to genotype, and transcriptome (Supplementary Fig. S3). We also applied Grimon to additional multi-omics data of molecular phenotypes (Supplementary Fig. S4), both of which clearly show Grimon’s applicability to any multi-omics data set.
Application of Grimon is not limited to visualize PCA or t-SNE results. Shared differentially expressed genes (DEG) across multiple conditions are another interest when interpreting multiple transcriptome data sets. By contrasting volcano plots among multiple sets of stimulation condition comparisons, Grimon could effectively highlight how DEGs in one comparison pattern are relatively distributed in the other comparison patterns (Supplementary Fig. S5).
4 Conclusions
We developed an intuitive, efficient data visualization method, Grimon, which facilitates interactive exploration and efficient interpretation of multi-layered high-dimensional data. Grimon helps users to smoothly understand complex interplay across multi-omics networks by visualizing data on the intuitive, user-friendly graphical interface.
Supplementary Material
Acknowledgements
We thank Prof. Kiyoshi Takeda, Drs. Shota Nakamura, Daisuke Motooka, Eiryo Kawakami, and Towfique Raj for their kind supports on the study.
Funding
This study was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (15H05911), AMED (16km0405206h0001, 16gm6010001h0001 and 17ek0410041h0001). M.K. was supported by a Nakajima Foundation Fellowship.
Conflict of Interest: none declared.
References
- Boyle E.A. et al. (2017) An expanded view of complex traits: from polygenic to omnigenic. Cell, 169, 1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfax B.P. et al. (2014) Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science, 343, 1246949–1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai M. et al. (2018) Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet., 50, 390–400. [DOI] [PubMed] [Google Scholar]
- Lake B.B. et al. (2018) Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol., 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T. et al. (2013) Transcriptome and genome sequencing uncovers functional variation in humans. Nature, 501, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.I. et al. (2016) RNA splicing is a primary link between genetic variation and disease. Science, 352, 600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y. et al. (2016) Dysbiosis contributes to arthritis development via activation of autoreactive T cells in the intestine. Arthritis Rheumatol., 68, 2646–2661. [DOI] [PubMed] [Google Scholar]
- Martin A.R. et al. (2014) Transcriptome sequencing from diverse human populations reveals differentiated regulatory architecture. PLoS Genet., 10, e1004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y. et al. (2015) Construction of a population-specific HLA imputation reference panel and its application to Graves’ disease risk in Japanese. Nat. Genet., 47, 798–802. [DOI] [PubMed] [Google Scholar]
- Okada Y. et al. (2016) Significant impact of miRNA—target gene networks on genetics of human complex traits. Sci. Rep., 6, 22223.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.