

Abstract
Motivation
The rapid accumulation of both sequence and phenotype data generated by high-throughput methods has increased the need to store and analyze data on distributed storage and computing systems. Efficient data management across these heterogeneous systems requires a workflow management system to simplify the task of analysis through automation and make large-scale bioinformatics analyses accessible and reproducible.
Results
We developed SciApps, a web-based platform for reproducible bioinformatics workflows. The platform is designed to automate the execution of modular Agave apps and support execution of workflows on local clusters or in a cloud. Two workflows, one for association and one for annotation, are provided as exemplar scientific use cases.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Key challenges in big data analysis include, but are not limited to, access to sufficient computing resources, massive data transfer, standardized analysis workflows and reproducibility. To address these challenges, we built a workflow platform by using infrastructure components of the CyVerse project (Goff et al., 2011). The CyVerse project, funded by the National Science Foundation of United States, aims to design, deploy, and expand a national Cyber-Infrastructure (CI) for life science researchers and to train scientists in its use. Two major components of the CyVerse CI, the Data Store built on top of iRODS (rule-oriented data system) (Moore and Rajsekar, 2010) and the Agave platform (Dooley et al., 2012), were used in building the SciApps platform.
Specifically, on the computing side, we registered a local CSHL cluster as an execution system in the Agave platform. The Agave platform or API was designed to be an open-source, platform-as-a-service solution for hybrid cloud computing. It provides a full suite of services to handle the complete cycle of analysis job management, including defining the interface between the scientific application (or app) and the execution system on which the app is executed through an app JSON file, job submission and monitoring, data transfer, archiving of analysis results, and many other aspects. Through the Agave API, SciApps applications that execute on the CSHL cluster as defined by the app JSON are available automatically in the Discovery Environment of CyVerse (or DE). Simply updating the execution system in the app JSON file switches from the CSHL cluster to a cloud or vice versa. SciApps workflows can be constructed with apps that execute on a local cluster or a cloud like XSEDE (Towns et al., 2014). For both cases, to avoid transferring massive amount of data across different sites, the workflow engine keeps all intermediate results close to the cluster and copies them to the final location/site only after the entire workflow is completed. On the data side, SciApps supports retrieval of data from the CyVerse Data Store. The CyVerse Data Store provides researchers with reliable access to secure cloud-based storage. CyVerse users get 100 GB storage space at registration, and this limit can be increased upon request.
2 Architecture
SciApps consists of a web interface for executing apps and building workflows, a workflow engine for execution of workflow, a storage server, a computing cluster, and a web server for various visualization services (Supplementary Fig. S1). It is designed for leveraging cloud-based storage and computing systems through Agave Science API. SciApps’s local storage and computing systems are set up as a remote CyVerse system for handling the analysis of large-scale datasets locally and more efficiently (Wang et al., 2015a).
2.1 Web interface
The SciApps web interface has four areas (Fig. 1). Apps in the left panel are searchable by names and categorized according to the EDAM ontology (Ison et al., 2013). The app search function is interactive: when any letters are typed into the search box, categories with a matched app or apps will be expanded with matched app names. Clicking on an app will bring up the app form in the main panel, along with a short description of the app below the form. The app form is loaded in the main panel with default (or previously used) inputs and parameters (if reloaded from the history panel), and once submitted, the analysis/job history is displayed in the history panel, and can be selected for building a workflow. The history panel only displays outputs predefined in the app JSON file, as shown for Step 5 (Fig. 1). If the user aims to build a workflow, these predefined outputs are the only ones that should be used as inputs for subsequent analysis tasks. More details on how to use SciApps interface to build and run workflows are provided in the SciApps platform guide (https://www.sciapps.org/page/help).
Fig. 1.
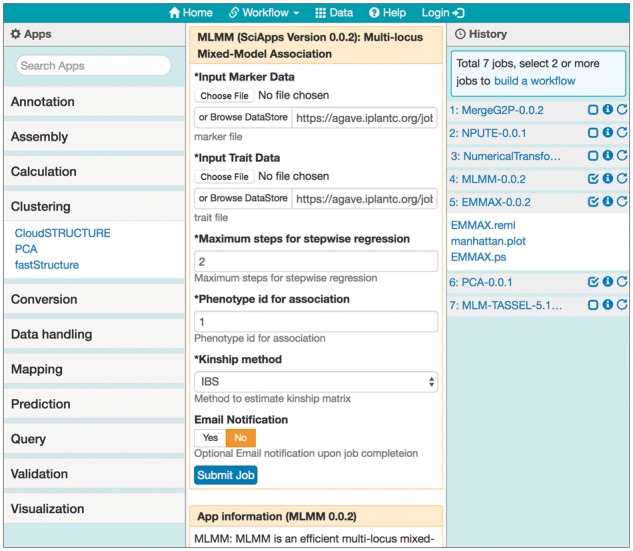
SciApps web interface. The interface, in which users perform data analyses and build workflows, has four areas: the navigation bar, containing workflow functionalities (building, loading, public and private workflows), a link to example data, help, and login for CyVerse authentication; the app panel (left column) for categorized apps (with the Clustering category clicked and expanded); the main panel (middle column) for app form(s) or workflow builder form; and the history panel (right column) for job name followed by three icons: checkbox for building a workflow from executed jobs, job history (i), and job re-launch. Here, a seven-step association workflow is loaded in the history panel, the app form of the fourth step is re-loaded in the main panel, and the results from Step 5 are clicked and expanded in the history panel. As an example, Steps 4–6 are checked (for building a new workflow)
2.2 Authentication
For access to multiple cloud-based systems, SciApps adopts the CyVerse Central Authentication Service (CAS). When a user logs in, they are directed to the CyVerse portal, where they enter their username and password; upon successful authentication, they are redirected back to SciApps. The CyVerse username is captured by SciApps for two uses: First, it directs SciApps to user’s sci_data folder in CyVerse Data Store, as shown in Supplementary Figure S2 for user lwang; Second, once an analysis job is submitted, the job is shared with the user through the Agave API so that, if needed, user can check the detailed job information through Agave’s web interface (https://togo.agaveapi.co/). Sharing is required since SciApps uses a designated CyVerse user account (a ‘superuser’) to execute apps and workflows. The superuser also gains full access to each user’s sci_data folder once the folder is automatically created, immediately after the user enables the SciApps service in the CyVerse user portal (https://user.cyverse.org). In other words, SciApps adopts a platform-centric approach by designating a superuser for managing the entire analysis cycle.
2.3 Workflow
To create a workflow, each analysis job is submitted, recorded and accessed through the web portal. Part or all of a series of recorded jobs can be saved as reproducible, sharable workflows for future execution with the original or modified inputs and parameters. When a user submits an analysis task, the job is recorded in the history panel, and a workflow is built by selecting two or more jobs using the check boxes (Fig. 1). The input/output relationships among individual tasks are built by tracing the origin of intermediate output, which is available in the job history metadata of the Agave API. Once built, a graphic workflow diagram is shown for verification (e.g. see Supplementary Fig. S3). After visual inspection, users can save the workflow from the diagram, and then download the workflow JSON file from ‘My workflows’ page.
The workflow diagram is interactive. Users can mouse over both data and app nodes to check related metadata and full names (long names are truncated in the diagram), and click for more metadata (input nodes), opening or downloading results (other data nodes), and full documentations (app nodes). For a running workflow, the diagram provides real-time job status updates by automatically updating the color of the app node (Supplementary Fig. S4). Alternatively, the status of individual jobs can also be obtained by clicking on the info icon. As shown in both Supplementary Figures S3 and S4, SciApps workflows are implemented as directed acyclic graphs. The execution of a step is only dependent on the availability of its required input(s), making it possible to exploit parallelism. For example, in Supplementary Figure S4, two jobs are running at the same time (app nodes in blue) because their required inputs are available simultaneously.
For one User (A) to share an analysis with another User (B), User A can build a workflow from the analysis jobs, download a lightweight workflow JSON from SciApps, and send it to User B. User B can then load the workflow JSON on SciApps to check all inputs, intermediate data, parameters, final results and related metadata. Through SciApps’ platform-centric approach, User B can also run the same workflow with modified inputs and parameters without any permission issues because jobs are submitted by the superuser, who has gained access to all of the workflow components either through sharing (apps, systems, sci_data folder) or being the owner (analysis jobs). If User B just wants to re-run a subset of the analysis, a new workflow can be built by selecting a subset of analysis jobs (Fig. 1). Alternatively, User B can also add more steps to the workflow by launching new jobs with any outputs from the workflow. For example, it is straightforward to build a three-pass workflow from a two-pass annotation workflow. In other words, any SciApps workflows can serve as a template for building new workflows.
3 Use cases
Two example use cases, one for association and one for annotation analysis, demonstrate the major features of SciApps.
3.1 Annotation
The workflow presented in this use case study implements a three-pass iterative annotation pipeline with two apps, MAKER (Holt and Yandell, 2011), a portable genome annotation pipeline with an integrated suite of gene prediction tools, and SNAP (Korf, 2004), a Semi-HMM-based Nucleic Acid Parser. In this workflow, MAKER annotation results for novel genomes without much prior knowledge of gene models are used to estimate HMM parameters with SNAP. The estimated parameters are then supplied to MAKER for re-annotation. The pipeline iterates MAKER and SNAP several times for a three-pass annotation (Supplementary Fig. S3) to improve the final gene models. The MAKER app has two outputs, annotation result in GFF format (my.all.gff.gz), which is also the input of SNAP for HMM parameter estimation, and maker_output.jbrowse, which points to a JBrowse (Skinner et al., 2009) view of the annotation result and evidences (Supplementary Fig. S5). The user can visualize annotation results from each pass on JBrowse and compare them to determine whether annotation results have been improved by additional passes.
3.2 Association
A genome-wide association study is an examination of a genome-wide set of genetic variants in a population of individuals aimed at determining whether any variant is associated with a trait. Supplementary Figure S4 shows an association workflow with individual components previously built in the DE (Wang et al., 2015b). From left to right, trait data and marker data are intersected by accession ID (or sample ID) with the MergeG2P app; missing markers in merged marker data (m_marker.txt) are imputed with NPUTE, an app based on the nearest-neighbor algorithm (Roberts et al., 2007); and then the imputed marker file (imputed.txt) is used by the PCA app to estimate population structure or converted for use in association studies with three mixed-model analysis methods, EMMAX (Zhou and Stephens, 2012), MLM-TASSEL (Zhang et al., 2010), and MLMM (Segura et al., 2012). For direct visualization of the association results from each mixed model, a web-based interactive Manhattan plot (Supplementary Fig. S6) was built using the Shiny framework (Chang et al., 2015). List of nearby genes can be retrieved from Gramene (Ware et al., 2002) by clicking on the plot.
4 Implementation
The back end of SciApps was built using Perl and the MySQL database engine, and the front-end was built with React, an open-source JavaScript library. The workflow engine uses the database to track job status and perform the submission of a subsequent job once its inputs are ready. Most components of the SciApps web interface are rendered from JSON data, including the app category and app list in the left panel, app form in the main panel, history in the right panel, and the workflow forms and diagram. The schemas of all JSON data are custom-designed for fast rendering, with the exception of the app JSON schema, which is adopted from the Agave API. In addition to defining inputs and parameters for rendering the app form, the app JSON specifies the system where the app will be executed. This makes it possible for SciApps to leverage the Agave API for job management on both local and cloud-based systems. To add a new app, storage, or execution system, users can follow the CyVerse tutorial (https://github.com/cyverse/cyverse-sdk).
The diagram is built with mermaid (https://knsv.github.io/mermaid/) and modified for interactivity on metadata and real-time job status. The latter is acquired through Agave API’s Webhook notification for jobs, which is also used for automatic updating of the MySQL database and automated execution of a workflow. In regard to genome browsers, both JBrowse and Biodalliance (https://www.biodalliance.org/) are supported for visualizing alignments, variants, and genome annotation results. h5ai (https://larsjung.de/h5ai/) is used to display the directory of example data and folders containing complete lists of jobs.
5 Discussion
SciApps provides a web-based workflow platform, accessible to all CyVerse users (currently ∼50 000), for automating the execution of modular Agave apps. Such automation is currently not supported by the DE. SciApps workflows can be used to process large amount of data either remotely on a cloud or locally. Local processing has the benefit of reducing cross-country data transfers. For the 12 jobs used in the Association or Annotation workflows, a comparison of the total time required when launching from SciApps or DE shows that SciApps always outperforms DE by avoiding such transfers (Supplementary Fig. S7).
SciApps’ front-end interface is designed with three panels aligned similarly to those of the Galaxy (Giardine et al., 2005) and GenePattern (Reich et al., 2006) platforms. On the back end, SciApps is designed to be flexible in regard to data placement and mixing of local and cloud-based computing resources. In other words, SciApps workflows can work with data located on a local server, a cloud, or a mix of both. Similarly, the computation of each step can happen in either location, depending on the execution system definition in the app JSON. This flexibility is achieved via adoption of the Agave API. Specifically, the Agave job JSON specifies where to retrieve the data and where to archive the results. The Agave app JSON, in addition to describing each step in the Common Workflow Language (CWL) format (Amstutz et al., 2016), specifies where to execute an app and how many processors and how much RAM are needed. Therefore, the SciApps workflow JSON is not interoperable with CWL. However, for single-node jobs, it would be possible to convert SciApps workflows into CWL format by combining the workflow JSON with the Agave app JSON and job JSON, although this would eliminate the flexibility regarding where and how to execute an app and where the data can be placed. Another challenge is that CWL does not yet support the Singularity containers (Kurtzer et al., 2017), which are used by SciApps applications to simplify workflow switching between local and cloud-based computing resources.
Future work will be focused on two goals: first, to continuously optimize SciApps for more efficient processing of local data with local computing resources in order to support data processing and management for project like MaizeCode; and second, to configure and optimize a community version to simplify local installation of SciApps. The community version will pull data directly from the CyVerse Data Store to XSEDE clusters for computing, and will be able to be launched from a laptop or virtual machine. Consequently, it will no longer be necessary to set up a local cluster and storage server, in order to use SciApps.
Supplementary Material
Acknowledgements
We thank our CyVerse colleagues for helping building and testing the platform, and Andrew Olson, Michael Campbell, and Kapeel Chougule for helping building the exemplar workflows.
Funding
This work is supported by the NSF [grant DBI-1265383] (CyVerse), and partially from NSF [grant IOS-1445025] (MaizeCODE).
Conflict of Interest: none declared.
References
- Amstutz P., et al. (2016) Common Workflow Language, v1.0. figshare. https://w3id.org/cwl/v1.0/. [Google Scholar]
- Chang W., et al. (2015) Web Application Framework for R. https://cran.r-project.org/package=shiny. [Google Scholar]
- Dooley R., et al. (2012) Software-as-a-service: the iPlant Foundation API. In: 5th IEEE Workshop on Many-Task Computing on Grids and Supercomputers (MTAGS), Salt Lake City, Utah, USA. [Google Scholar]
- Giardine B., et al. (2005) Galaxy: a platform for interactive large-scale genome analysis. Genome Res. ,15, 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff S.A., et al. (2011) The iPlant collaborative: cyberinfrastructure for plant biology. Front Plant Sci. ,2, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C., Yandell M. (2011) MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics ,12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ison J., et al. (2013) EDAM: an ontology of bioinformatics operations, types of data and identifiers, topics and formats. Bioinformatics ,29, 1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. (2004) Gene finding in novel genomes. BMC Bioinformatics ,5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtzer G.M., et al. (2017) Singularity: scientific containers for mobility of compute. PLoS One, 12, e0177459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R.W., Rajsekar A. (2010) Irods: data sharing technology integrating communities of practice. In: International Geoscience and Remote Sensing, pp. 1984–1987. [Google Scholar]
- Reich M., et al. (2006) GenePattern 2.0. Nat. Genet., 38, 500. [DOI] [PubMed] [Google Scholar]
- Roberts A., et al. (2007) Inferring missing genotypes in large SNP panels using fast nearest-neighbor searches over sliding windows. Bioinformatics ,23, i401–i407. [DOI] [PubMed] [Google Scholar]
- Segura V., et al. (2012) An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. ,44, 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M.E., et al. (2009) JBrowse: a next-generation genome browser. Genome Res. ,19, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towns J., et al. (2014) XSEDE: accelerating Scientific Discovery, Comput .Sci. Eng. ,16, 62–74. [Google Scholar]
- Wang L., et al. (2015a) Architecting a distributed bioinformatics platform with iRODS and iPlant agave API. International Conference on Computational Science and Computational Intelligence (CSCI), Las Vegas, Nevada, USA, pp. 420–423. [Google Scholar]
- Wang L., et al. (2015b) A genome-wide association study platform built on iPlant cyber-infrastructure. Concurr. Comp. Pract. E ,27, 420–432. [Google Scholar]
- Ware D., et al. (2002) Gramene: a resource for comparative grass genomics. Nucleic Acids Res. ,30, 103–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., et al. (2010) Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. ,42, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Stephens M. (2012) Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. ,44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.