

SUMMARY
The signaling of prostaglandin D2 (PGD2) through a G protein-coupled receptor (GPCR), CRTH2, is a major pathway in type 2 inflammation. Compelling evidence suggests the therapeutic benefits of blocking CRTH2 signaling in many inflammatory disorders. Currently, a number of CRTH2 antagonists are under clinical investigation and one compound, fevipiprant, has advanced to phase 3 clinical trials for asthma. Here, we present the crystal structures of human CRTH2 with two antagonists, fevipiprant and CAY10471. The structures, together with docking and ligand binding data, reveal a semioccluded pocket covered by a well-structured amino terminus and different binding modes of chemically diverse CRTH2 antagonists. Structural analysis suggests a ligand entry port and a binding process that is facilitated by opposite charge attraction for PGD2, which differs significantly from the binding pose and binding environment of lysophospholipids and endocannabinoids, revealing a new mechanism for lipid recognition by GPCRs.
Graphical Abstract
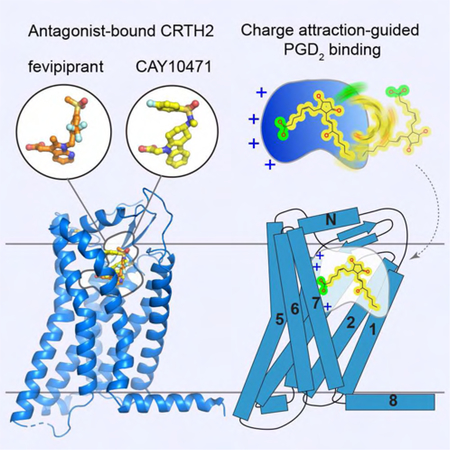
In Brief
Wang et al. reported crystal structures of antagonist-bound human CRTH2 as a new asthma drug target. Chemically diverse antagonists occupy a similar semioccluded pocket with distinct binding modes. Structural analysis suggests a potential ligand entry port and an opposite charge attraction-facilitated binding process for the endogenous CRTH2 ligand prostaglandin D2.
INTRODUCTION
Eicosanoid lipid prostaglandin D2 (PGD2) is the major prostaglandin produced by activated mast cells (Lewis and Austen, 1981). The physiological function of PGD2 is mainly mediated by two G protein-coupled receptors (GPCRs), PGD2 receptor 1 and 2 (DP1 and DP2), which share modest sequence similarity and couple to different G proteins (Monneret et al., 2001; Nagata et al., 1999). DP2 is more commonly called the chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2). While DP1 is closely related to other prostaglandin receptors, CRTH2 is more akin to a group of leukocyte non-chemokine chemoattractant GPCRs, which also includes the receptors for anaphylatoxin C3a and C5a, formylpeptides, leukotrienes and some other eicosanoids (Fredriksson et al., 2003; Nagata and Hirai, 2003; Serhan, 2014) (Figure S1A). These non-chemokine chemoattractant receptors share a relatively high sequence similarity and the same preference for Gi protein, but they recognize diverse ligands, including lipids, peptides and large proteins. Despite much evidence linking this group of receptors to a number of inflammatory diseases, no drugs that specifically target this group of GPCRs are currently commercially available.
CRTH2 is highly expressed in type 2 helper T cells (Th2), innate lymphoid cells (ILCs), eosinophils and basophils (Cosmi et al., 2000; Hirai et al., 2001; Mjosberg et al., 2011; Nagata et al., 1999). PGD2-CRTH2 signaling is a major pathway in type 2 inflammation, leading to the activation of immune cells and the production of type 2 cytokines (Monneret et al., 2001; Xue et al., 2005). Thus, CRTH2 has emerged as a promising new target in treating type 2 inflammation-driven diseases, such as asthma and allergic rhinitis, which has spurred intensive research efforts in developing CRTH2 antagonists for clinical investigation (Kupczyk and Kuna, 2017; Pettipher et al., 2007; Pettipher and Whittaker, 2012; Schuligoi et al., 2010). The first nonlipid CRTH2 antagonist, ramatroban, was discovered by serendipity (Hirai et al., 2002; Sugimoto et al., 2003). Ramatroban was initially developed as a thromboxane receptor antagonist drug used in Japan for treating allergic diseases; it was then proven to also be a CRTH2 antagonist. Modification of ramatroban led to the discovery of the first potent and selective CRTH2 antagonist, CAY10471 (also named TM30089), which exhibits insurmountable action, in contrast to the reversible action of ramatroban in some assays (Mathiesen et al., 2006; Ulven and Kostenis, 2005). Such early studies have inspired a number of companies to develop numerous CRTH2 antagonists with diverse chemical scaffolds and pharmacological properties in the past decade (Kupczyk and Kuna, 2017; Pettipher and Whittaker, 2012; Santus and Radovanovic, 2016). Several of these antagonists have been tested in asthma patients, but the results were mixed (Barnes et al., 2012; Busse et al., 2013; Erpenbeck et al., 2016; Kuna et al., 2016; Miller et al., 2017; Pettipher et al., 2014). It has been suggested that a subpopulation of asthmatic patients whose airway inflammation is largely driven by Th2-type inflammation would benefit most from CRTH2 antagonists (Kupczyk and Kuna, 2017). Recently, a potent CRTH2 antagonist, fevipiprant, showed promising clinical efficacy in patients with uncontrolled asthma in a few clinical trials (White et al., 2018). Thus, CRTH2 antagonists hold the promise of being a new class of asthma drugs, and the development of new CRTH2 antagonists remains highly competitive, as evidenced by the continuing clinical investigation initiated by many companies with their own compounds (Kupczyk and Kuna, 2017; Pettipher and Whittaker, 2012).
Similar to PGD2, nearly all of the CRTH2 antagonists are carboxylic acid derivatives with a carboxylate moiety, which is believed to be a critical pharmacophore that interacts with the receptor (Pettipher and Whittaker, 2012) (Figure 1A). To understand the molecular mechanisms for the action of CRTH2 ligands, we solved the crystal structures of human CRTH2 bound to two antagonists, fevipiprant and CAY10471. The structures, together with the results from computational docking studies and ligand binding assays, reveal conserved and divergent structural features for the binding of diverse CRTH2 antagonists, which occupy a semioccluded ligand-binding pocket covered by a well-structured N-terminal region with a novel conformation. Interesting characteristics of the ligand binding pocket, including a widely open end as the potential ligand entry port and a gradually increased positive charge distribution, allow us to propose a novel mechanism for the binding of PGD2. Structural comparison analysis suggests a distinct binding pose of PGD2 compared to the lysophospholipids and endocannabinoids.
Figure 1.

CRTH2 ligands and overall structures. (A) Chemical structures of PGD2, fevipiprant, CAY10471 and ramatroban and the conserved carboxylate group. (B) Competition radioactive ligand binding assays with HEK-293T cell membranes expressing wtCRTH2 and CRTH2-mT4L. For each experiment, 2 nM [3H] PGD2 was used and various concentrations of PGD2 (top), CAY10471 (middle) and fevipirant (bottom) were added as competing ligands. Data points are presented as the mean values +/− SEM, n=3. (C) Overall structures of fevipiprant-bound and CAY10471-bound CRTH2 are colored in blue and slate, respectively. Fevipiprant and CAY10471 are shown as orange and yellow spheres.
RESULTS
Crystallization of CRTH2 and overall structures
To crystallize human CRTH2, a construct of human CRTH2 was generated by inserting an engineered T4 lysozyme (mT4L) (Thorsen et al., 2014) with an additional N-terminal 8-amino acid linker into the intracellular loop 3 (ICL3) for crystallization (Figure S1B). The 8-amino acid linker greatly improved crystal quality, which was achieved unintentionally. To further facilitate crystallogenesis, the flexible C-terminal region from R340 to S395 was removed before crystallization, and the potential glycosylation site N25 was mutated to alanine. No other mutations were introduced. Ligand competition binding assays showed that the sequence modifications did not significantly affect the ligand-binding properties of CRTH2 (Figure 1B). Using this construct, we solved the crystal structures of human CRTH2 in complex with two antagonists, fevipiprant and CAY10471, at 2.80 Å and 2.74 Å resolution, respectively (Figure 1C, Table 1).
Table 1.
Data collection and refinement statistics.
| CRTH2-fevipiprant | CRTH2-CAY10471 | |
|---|---|---|
| PDB ID | 6D26 | 6D27 |
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 50.457, 61.710, 266.517 | 52.175, 62.636, 272.215 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50 − 2.8 (2.85 − 2.8) | 50 − 2.7 (2.75 − 2.7) |
| aRmerge (%) | 18.3 (77.4) | 16.6 (62.8) |
| I/σ(I) | 8.9 (1.2) | 11.6 (1.3) |
| bCC1/2 | (0.71) | (0.87) |
| Completeness (%) | 91.9 (76.5) | 92.6 (74.1) |
| Redundancy | 4.9 (3.9) | 5.5 (3.6) |
| Refinement | ||
| Resolution (Å) | 50 − 2.8 (2.95 − 2.8) | 50 − 2.74 (2.86 − 2.74) |
| No. reflections | 19525 (2293) | 25865 (1211) |
| cRcryst / Rfree(%) | 25.0 (35.9) / 28.0 (44.4) | 24.2 (33.8) / 28.0 (42.3) |
| No. atoms | ||
| Protein | 3498 | 3447 |
| Ligand | 149 | 163 |
| Water | 3 | 8 |
| B factors (Å2) | ||
| Protein | 84.7 | 94.7 |
| Ligand | 93.3 | 107.2 |
| Water | 55.4 | 70.0 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.002 | 0.002 |
| Bond angles (°) | 0.6 | 0.6 |
| Ramachandran plot | ||
| Favored (%) | 96.4 | 96.1 |
| Allowed (%) | 3.6 | 3.9 |
| Outline (%) | 0 | 0 |
| Molprobity score | 1.47 | 1.24 |
| All atom crash score | 4.53 | 2.07 |
Values in parentheses are for highest-resolution shell.
Rmerge = ∑|Ii − Im|/∑Ii, where Ii is the intensity of the measured reflection and Im is the mean intensity of all symmetry related reflections.
CC1/2 is the correlation coefficient of the half datasets.
Rcryst = Σ||Fobs| − |Fcalc||/Σ|Fobs|, where Fobs and Fcalc are observed and calculated structure factors.
Rfree = ΣT||Fobs| − |Fcalc||/ΣT|Fobs|, where T is a test data set of about 5% of the total reflections randomly chosen and set aside prior to refinement.
The high-quality electron density maps allowed us to model all residues of CRTH2 from A5 to L327, except for G237, which was replaced by mT4L, in the structure of CRTH2 with fevipiprant. The structure of CRTH2 with CAY10471 is nearly identical to the structure with fevipiprant, except for a few loop residues near the ligand binding pocket and the disordered S22-A25 region at the N-terminus. The overall structure of CRTH2, especially the second extracellular loop (ECL2) with a conserved β-hairpin structure, is similar to the structures of two other non-chemokine chemoattractant GPCRs that are close phylogenetic neighbors, namely, the LTB4 receptor (BLT1) and the C5a receptor (C5aR) (Hori et al., 2018; Liu et al., 2018; Robertson et al., 2018) (Figure S2A). Interestingly, even though C5aR recognizes peptide ligands, which are chemically distinct from the lipid mediators recognized by CRTH2, if the structures of C5aR and CRTH2 are superimposed on each other, the antagonists of CRTH2 nearly overlap with part of the peptide antagonist PMX53 for C5aR (Figure S2B). However, unlike BLT1 and C5aR, CRTH2 contains a well-folded N-terminal structure with a central alpha helix (N-helix) connected to the transmembrane helix 1 (TM1) by a long loop (N-loop) (Figures 2A and S3A). The N-helix and N-loop pack tightly against the second extracellular loop (ECL2), forming a lid between ECL1 and ECL3 to largely cover the ligand binding pocket (Figure 2A). Besides the disulfide bond between C182 in ECL2 and C1043.52 (Ballesteros-Weinstein numbering) in TM3 that is conserved in most rhodopsin-like GPCRs, an additional disulfide bond was found between C11 and C1995.31, linking the N-terminal helix to TM5 and thereby fixing the position of the N-terminal region (Figure 2A). The N-terminal region of TM1 in CRTH2 tilts toward TM2 compared to those in BLT1 and C5aR and thereby creates a gap between TM1 and TM7 as the only open end of the ligand-binding pocket (Figures 2B and S2C). Since the extracellular ligand access is restricted to the pocket by the lid domain, this gap in the lateral side of CRTH2 is likely to be the ligand entry port.
Figure 2.

N-terminal region and ligand entry port in CRTH2. (A) Well-folded N-terminal region with an N-helix and N-loop in the structure of CRTH2 bound to fevipiprant (green). The disulfide bond connecting the N-terminus and TM5 is indicated with an arrow. Fevipiprant is shown as orange spheres. (B) Structural comparison of N-terminal region and TM1 in CRTH2 (blue), BLT1 (cyan) and C5aR (grey). In both A and B, the open end of the ligand binding pocket in CRTH2 as the potential ligand entry port is marked with a red dashed circle, which is occupied by the extracellular regions of TM1 in BLT1 and C5aR.
The C-terminal region in CRTH2, D310-L327, immediately after TM7 forms an unusually long helical structure, namely, helix 8. Helix 8 in CRTH2 exhibits an interesting amphipathic nature characterized by four leucine residues and one valine residue lining the membrane-facing side, suggesting a strong membrane association, and positively charged residues lining the cytoplasmic side (Figure S3B). A previous study showed that the C-terminal tail of CRTH2 negatively regulated receptor signaling and that a truncation of the C-terminus after R317 could enhance Gi signaling (Schroder et al., 2009), indicating an important role of the long helix 8 in CRTH2 signaling. In addition, the entire cytoplasmic surface of CRTH2 is highly positively charged with a number of sulfate ions modeled in the region (Figure S3C). Although speculative, such characteristics may suggest a potential regulation of receptor signaling by negatively charged phospholipids (Huynh et al., 2009).
Structural basis for the binding of CAY10471 and fevipiprant
The high-quality electron density maps allowed unambiguous modeling of fevipiprant and CAY10471 as two slow-dissociating CRTH2 antagonists in the structures (Mathiesen et al., 2006; Royer et al., 2007; Sykes et al., 2016) (Figure S4A and B). Fevipiprant and CAY10471 bind to a semioccluded ligand-binding pocket with a widely open end surrounded by the N-helix, the N-loop and the extracellular parts of TM1 and TM7, and an occluded distal end surrounded by TMs 3, 5 and 6 (Figure 3A, Figure S4A and B). A majority of residues in the ligand binding pocket are aromatic residues, with a few highly charged residues clustered at the occluded distal end (Figure 3B). The side chains of two charged residues, R1704.64 and K2105.42, and the side chains of two tyrosine residues, Y184 in ECL2 and Y2626.51, point toward the carboxylate head groups of the two antagonists to form a strong polar interaction network, creating a highly charged environment to hold the carboxylate group at the distal end of the ligand binding pocket. Four phenylalanine residues, F872.60, F1113.32, F1123.33 and F2947.43, form the bottom of the ligand binding pocket. These residues, together with F902.63, H1073.28, Y183, Y184, Y2626.51 and L2867.35, engage in extensive aromatic and hydrophobic interactions with the central aromatic groups in the antagonists: the methyl azaindole group in fevipirant and the tetrahydrocarbazole group in CAY10471. Both central aromatic groups also engage in cation-π interactions with the side chain of R1704.64.
Figure 3.

Binding of CAY10471 and fevipiprant. (A) Ligand binding pocket with the open end and the distal end and binding poses of both ligands. (B) Residues involved in the binding of fevipiprant and CAY10471. Hydrogen bonds are shown as black dashed lines. Disulfide bonds are shown as yellow sticks. (C) Different conformations of W2837.32 and L20 in the structures of CRTH2 with two ligands. The disordered region between L20 and A25 in CRTH2-CAY10471 is shown as a slate dashed line.
Despite the conserved features, the tail groups of the two antagonists show distinct binding modes and engage in different additional interactions with the receptor (Figures 3A and B). For fevipiprant, the methylsulfonyl phenyl group extends toward ECL2, with the substituted trifluoromethyl group facing a cleft between TM1 and TM7. Additional aromatic interactions form between the phenyl group and aromatic residues F902.63, H95, Y183 and W2837.32, and hydrogen bonds form between one oxygen atom of the methylsulfonyl moiety and the main chain amine group of C182. Such a binding mode of fevipiprant, together with the details of the binding pocket, well explains the results of the structure and function relationship (SAR) studies for developing fevipiprant (Sykes et al., 2016). For CAY10471, the sulfonyl group also extends toward ECL2, while the fluorophenyl group, as the tail group, extends toward TM7, resulting in a swing of the indole ring of W2837.32 compared to this residue in the structure with fevipiprant. Such a conformation of W2837.32 further causes the movement of L20 at the N-terminus, potentially leading to a disordered region of S22-S24 in the structure of CRTH2 with CAY10471 (Figure 3C). The fluorophenyl group of CAY10471 participates in the aromatic interactions with residues Y183, W2837.32 and P2877.36. Modifications of the central tetracarcazole group of CAY10471, such as substitution with an unsaturated carcazole group with a flat plane and ring opening, would change the position of the sulfonyl fluorophenyl tail group relative to the surrounding aromatic residues and cause steric clash, thus resulting in lower affinities (Pettipher and Whittaker, 2012).
A new binding mode of ramatroban revealed by docking
CAY10471 shares a high structural similarity with its parent compound ramatroban (Figure 1A). The major difference is that, instead of an acetate group, ramatroban has a longer propionate group attached to the central tetracarbazole group. Previous studies have shown that CAY10471 is an insurmountable antagonist with slow dissociation, while ramatroban is a highly reversible antagonist (Mathiesen et al., 2006). In addition, ramatroban is less effective in stabilizing the receptor compared to fevipiprant and CAY10471 in our thermostability assays (Figure 4A), indicating that ramatroban potentially engages in different interactions with the receptor. We simulated the binding of ramatroban to CRTH2 by computational docking. We used the structure of CRTH2 with CAY10471 as template because of the high structural similarity between CAY10471 and ramatroban. We validated our docking methods by reproducing the same binding pose of CAY10471 as that in the crystal structure (Figure S4C). Interestingly, the top-ranked docking poses of ramatroban differ significantly from those of CAY10471 in the crystal structure (Figure 4B). The sulfonyl fluorophenyl tail group of ramatroban occupies a region similar to that occupied by the same moiety in CAY10471. However, compared to CAY10471, the tetracarbazole and propionate moieties of ramatroban, although still buried in the aromatic pocket, adopt a flipped conformation to accommodate the longer propionate head group. This binding mode positions the carboxylate group in ramatroban away from the spatially constrained Y184-K2105.42-Y2626.51 cluster, thereby disrupting the polar interaction network associated with CAY10471. Additionally, the tetracarbazole group of ramatroban resides in an unfavorable polar environment, further compromising ramatroban binding (Figures 4B and S4C). Taken together, our findings well explain the weaker binding of ramatroban compared to that of CAY10471. More importantly, they suggest that even small changes in chemical structures during drug design may lead to significant changes in the binding affinities of CRTH2 antagonists.
Figure 4.

Ramatroban docking results. (A) Thermostability of unliganded CRTH2 and CRTH2 bound to ramatroban, fevipiprant and CAY10471. Apparent melting temperatures, Tms, were calculated and are shown in the brackets. Data points are presented as the mean values +/− SEM, n=2. (B) Binding pose of ramatroban, shown as dark purple sticks, from docking. CAY10471 is shown as thin yellow sticks for comparison. The carboxylate group in ramatroban is circled. See also Figure S4C.
Insights into PGD2 binding
The conservation of the carboxylate group in PGD2 and most CRTH2 antagonists suggests that the carboxylate in PGD2 occupies a similar site with a highly polar environment formed by residues R1704.64, Y183, Y184, K2105.42, Y2626.51 and E2696.58 (Figure 5A). Consistently, previous mutagenesis studies have demonstrated the important roles of K2105.42 and E2696.58 in PGD2 binding (Hata et al., 2005). The rest of the hydrocarbon chain of PGD2 with a central cyclopentyl ring likely occupies the hydrophobic space largely constituted by the aromatic residues (Figure 5A), which can potentially form π-π interactions with the two carbon-carbon double bonds in PGD2 to further stabilize the ligand. The hydrophobic environment of the ligand binding pocket is shielded from the extracellular aqueous milieu by the lid domain formed by the N-terminus and ECL2. The C11A mutation, which presumably disrupts the disulfide bond linking the N-terminal region to TM5 to destabilize the N-terminal region, could significantly compromise PGD2 binding in our assays, suggesting the important role of the N-terminal region in PGD2 binding (Figure 5B).
Figure 5.

PGD2 binding. (A) Potential binding pocket for PGD2 as shown by the transparent grey cavity. The polar residues that may be involved in the interactions with the carboxylate group in PGD2 are labeled in purple. (B) Cell surface expression levels of wild-type CRTH2 (wtCRTH2) and three mutants (left) and their specific saturation binding of 3H-PGD2 (right). The cell surface expression of each construct in HEK-293T cells was assessed by measuring the binding of fluorescent anti-FLAG antibodies to the FLAG epitope displayed at the N-terminus of the receptor through flow cytometry. The specific saturation binding assays were performed using cell membranes. The W283A mutant is shown as a positive control, in which the binding of PGD2 was not significantly altered. Data points are presented as the mean values +/− SEM, n=3. (C) Succinate (pink) and propylene glycol (brown) molecules modeled in the structures of fevipiprant-bound CRTH2 (blue) and CAY10471-bound CRTH2 (slate). The hydrogen bonds are shown as black dashed lines. (D) Charge distribution of the ligand-binding pocket of CRTH2 (open-book view). Fevipiprant is shown as orange sticks. (E) Cartoon diagram of the proposed PGD2 binding process. The positive charge potential is indicated as “+”. The lipid bilayer is shown as the pink background. The carboxylate group in PGD2 is colored in green, while the rest hydrophobic moiety of PGD2 is colored in yellow.
The well-structured N-terminal region results in a gap between the N-loop and TM7 as the only open end of the ligand binding pocket, which could serve as a ligand entry port for lipid agonists and antagonists (Figure 2). Three positively charged residues, H95, R175 and R179 from ECL1 and ECL2, which do not directly interact with the antagonists, project their side chains into this entry port (Figure 5C). Interestingly, strong electron density was observed around R175 and R179 in our structures. We modeled a succinate or a propylene glycol molecule from the crystallization conditions in the two structures to fit the electron density (Figures 5C and S5). Both compounds contain polar groups that are the same as or similar to the carboxylate group in PGD2, forming salt bridges or hydrogen bonds with R175 and R179. These observations suggest that the carboxylate group of PGD2 may form similar interactions with those two residues at the ligand entry port during the early stage of ligand recognition. Moreover, the entire ligand binding pocket of CRTH2 exhibits a gradually increased positive charge distribution from the entry port to the distal end (Figure 5D). We propose that such a feature plays an important role in guiding PGD2 to access the pocket by attracting its carboxylate group to reach the distal end. Collectively, as shown in Figure 5E, the process of PGD2 binding suggested by our results includes the anchoring of the carboxylate group of PGD2 to the ligand entry port and the following access to the ligand-binding pocket facilitated by the positive charge gradient. The nonuniform charge distribution may also help PGD2 to change its orientation in the lipid bilayer to enter the ligand-binding pocket. Considering the negative charge property of the carboxylate group, the change in the orientation of PGD2 likely does not occur spontaneously in the lipid bilayer. Supporting this mechanism for PGD2 recognition, previous studies have shown that mutations of R179 could reduce the affinity of PGD2 by 5- to 10-fold (Hata et al., 2005), and we also showed that mutating the positively charged residue R1704.64 at the distal end of the ligand binding pocket nearly abolished PGD2 binding (Figure 5B).
Structural comparison with other lipid GPCRs
BLT1, as the receptor for the lipid LTB4, is closely related to CRTH2, and their endogenous ligands LTB4 and PGD2 are both eicosanoids with a common precursor (Smith, 1989). To the best of our knowledge, BLT1 and CRTH2 are the only two eicosanoid GPCRs with solved structures. Although their structures share a high similarity (Figure S2A), compared to CRTH2, the N-terminal region of BLT1 is completely disordered in the structure of guinea pig BLT1 bound to an atypical antagonist BIIL260 (Hori et al., 2018), leaving the ligand binding pocket open to the extracellular milieu. This is likely an inherent feature of BLT1 since its short N-terminal sequence does not favor a structured motif and no cysteine residue is present at the N-terminus to form an additional disulfide bond. The ligand binding port between TM1 and TM7 in CRTH2 is also absent in BLT1 because of a difference TM1 conformation (Figure 2B). Such a large structural divergence of the ligand binding pockets in BLT1 and CRTH2 suggests that those two receptors adopt different mechanisms for the lipid recognition, even though their ligands are both eicosanoids with a high chemical similarity (Figure S2A).
The feature of a structured N-terminal region as a lid domain covering the ligand-binding pocket has also been observed in a few other GPCRs for diffusible lipid ligands, such as sphingosine-1-phosphate (S1P1) (Hanson et al., 2012), lysophosphatidic acid (LPA1) (Chrencik et al., 2015) and endocannabinoids (CB1) (Hua et al., 2017; Hua et al., 2016; Shao et al., 2016), and has been proposed to be a conserved feature for many lipid GPCRs. However, CRTH2 differs significantly from these lipid GPCRs in the extracellular region. First, the overall conformation of the N-terminal region is largely different in CRTH2 than in other lipid GPCRs. The N-termini of S1P1 and LPA1 form a helical structure on top of the extracellular surface that packs against ECL1 and ECL2, while the N-terminus of CB1 forms a loop structure followed by a very short helix, which is buried in the helical bundle and packs against ECL1, ECL2 and TM7 (Figure 6A). In CRTH2, the N-helix is nearly parallel to the β-hairpin of ECL2, forming the lid domain together with the long N-loop. Second, the ECL2s in the S1P1, LPA1 and CB1 receptors lack a β-hairpin motif and project toward the inside of the helical bundle (Figure 6A). As a result, the conserved extracellular disulfide bond linking ECL2 to TM3 in almost all class A GPCRs, including CRTH2, is missing in those receptors. Third, compared to those in S1P1, LPA1 and CB1, the long N-loop in CRTH2 results in a wider ligand entry port on the side of the helical bundle (Figure S6). This may provide enough space for the orientation change of PGD2 at the ligand entry port to prepare the ligand for entering the ligand binding pocket (Figure 5E).
Figure 6.

Structural comparison of CRTH2 to lipid GPCRs S1P1, LPA1 and CB1. (A) Extracellular regions in the structures of CRTH2 (blue), S1P1 (magenta, PDB ID 3V2W), LPA1 (pink, PDB ID 4Z35), CB1 with an inverse agonist (green, PDB ID 5U09) and CB1 with an agonist (cyan, PDB ID 5XRA). Fevipiprant (orange) and ligands in other GPCRs (light blue) are shown as spheres. For S1P1 and LPA1, only antagonist-bound structures are available. See also Figure S6A. (B) Charge distribution of the ligand-binding pockets pointed by arrows of these GPCRs shown in A. The potential ligand access ports are marked with black dashed circles. (C) Cartoon diagrams of the ligand-binding modes of four lipid GPCRs. From the left to the right: CRTH2 with prostanglandin D2 (PGD2), S1P1 with sphingosine-1-phosphate (S1P), LPA1 with lysophosphatidic acid (LPA) and CB1 with N-arachidonoylethanolamine (AEA). The lipid bilayer is shown as the pink background. For all ligands, the polar head groups are colored in green, while the rest hydrophobic moieties are colored in yellow.
In addition to the large structural divergence of the extracellular regions, the ligand binding pocket in CRTH2 also exhibits different characteristics compared to that of S1P1, LPA1 and CB1. The most striking difference is the polar environment of the distal end of the ligand binding pocket in CRTH2, whereas the corresponding regions in S1P1, LPA1 and CB1 are largely hydrophobic (Figure S6). Consequently, the carboxylate head group of PGD2 is buried deeply inside the distal end of the pocket, while the hydrocarbon chain possibly extends toward the ligand entry port. In contrast, the fatty-acyl chains of the endogenous ligands for S1P1, LPA1 and CB1 are buried deep inside the binding pocket, while the polar head groups are close to the extracellular surface (Chrencik et al., 2015; Hanson et al., 2012; Hua et al., 2017). Additionally, the electrostatic charge distributions of the ligand binding pockets in these lipid GPCRs are largely different (Figure 6B). In the structures of antagonist-bound S1P1 and LPA1 and agonist-bound CB1, the ligand access port is positively charged, while the rest of the binding pockets are highly negatively charged, contrasting with the highly positively charged ligand binding pocket in CRTH2. Such a charge distribution may help to position the phosphate head groups of lysophospholipids or the hydroxyl groups of endocannabinoids at the ligand access port through both electrostatic attraction and repulsion to ensure that their acyl chains are buried in the binding pocket (Figure 6C). Therefore, it is likely that for those receptors the acyl chains of the lysophospholipids and the endocannabinoids go into the ligand-binding pockets without a large orientation change of the lipid molecules, which is different from the proposed multistep process for the recognition of PGD2 by CRTH2 (Figure 5E).
DISCUSSION
GPCRs recognize a broad range of molecules with a vast chemical diversity through different mechanisms. Our understanding of the recognition of lipid mediators by GPCRs primarily comes from the structural studies of receptors for lysophospholipids and endocannabinoids including S1P1, LPA1, LPA6 and CB1, which have revealed two different types of extracellular ligand recognition domains (Taniguchi et al., 2017). In S1P1, LPA1 and CB1, the N-terminal region folds on top of the ligand binding pocket and the ECL2 projects toward the inside of the 7-TM bundle to interact with the ligands, while in LPA6, the ligand binding pocket is open to the extracellular environment, with the ECL2 extending away from the 7-TM bundle, similar to BLT1. Our structures of CRTH2 reveal a new conformation of the extracellular region that, to the best of our knowledge, has not been observed in other GPCR structures. In the structures, the well-folded N-terminal region packs tightly against the ECL2, resulting in a widely open end of the ligand binding pocket as the ligand entry port. The structural analysis allows us to propose a novel mechanism for the binding of the lipid molecule PGD2 to CRTH2, in which the carboxylate group of PGD2 first binds to the ligand entry port through interactions with positively charged residues and then extends deeply into the ligand-binding pocket following the positive charge gradient, while the rest of the hydrocarbon chain is stabilized by many aromatic residues in the ligand binding pocket (Figure 5E). Our studies thus offer new insights into how GPCRs recognize chemically diverse endogenous lipid mediators. Additionally, despite the structural divergence of the extracellular domains in CRTH2, S1P1, LPA1 and CB1, these receptors share a similar feature characterized by a gap between the N-terminal segments of TM1 and TM7 (Figure S6), which also extends to the photoreceptor rhodopsin (Palczewski et al., 2000; Park et al., 2008). Such a feature may be highly conserved in a majority of lipid-activated GPCRs, providing a common structural basis for the uptake and release of lipophilic ligands.
CRTH2 belongs to a group of non-chemokine chemoattractant GPCRs that are phylogenetically close to each other but recognize very diverse ligands from lipids to peptides to large proteins (Figures S1A and S2A). The structures of CRTH2 reported here, together with the previously reported structures of BLT1 and C5aR, show a large structural divergence of the extracellular region in those receptors, likely accounting for the recognition of diverse ligands by those GPCRs. On the other hand, the structures also reveal a conserved structural feature in these receptors. One residue in TM6, Y6.51, which is conserved as a Y or F in other non-chemokine chemoattractant GPCRs, directly interacts with the ligands of all three receptors (Figure S7). This residue sits on top of a structural motif F6.44XXCW6.48XP6.50 that is highly conserved in rhodopsin-like GPCRs and interacts with W6.48, which has been suggested to function as a toggle switch in the activation of some GPCRs (Smit et al., 2007). We propose that for the group of non-chemokine chemoattractant GPCRs, the three conserved residues, F6.44, W6.48 and Y/F6.51, line up in TM6 to constitute a critical structural motif that mediates the propagation of signal from the extracellular ligand binding pocket to the cytoplasmic region that interacts with intracellular signaling molecules in receptor activation.
The two receptors, CRTH2 and BLT1, apparently adopt different mechanisms for lipid recognition, with distinct ligand binding pockets, even though the endogenous ligands for BLT1 and CRTH2, LTB4 and PGD2 respectively, are both eicosanoid lipid mediators with a high chemical similarity (Figure S2A). Some other members of this group of GPCRs including FPR2/ALX, ChemR23 (CMKLR1) and GPR32 recognize a special group of eicosanoid lipids called specialized pro-resolving lipid mediators (SPMs). SPMs can promote the resolution of inflammation, in contrast to the primary pro-inflammatory function of most eicosanoid lipids, including LTB4 and PGD2. Whether the recognition of SPMs by their receptors is similar to the lipid recognition by BLT1 or by CRTH2 needs further investigation. This is important considering the increasing research interests in developing new pro-resolving mediators as a novel therapy for treating inflammatory diseases (Dalli and Serhan, 2018). In addition, one member of this group, FPR2/ALX, can sense both formyl peptides and SPMs. The molecular mechanism for such promiscuous ligand recognition remains elusive.
Our structures also provide new insights into CRTH2 drug development. The ligand binding pocket revealed by our structures comprises many aromatic residues and a few polar residues at the distal end. Correspondingly, most CRTH2 antagonists share a similar structural feature characterized by an acetate polar group attached to a central aromatic group to fit the ligand binding pocket (Pettipher and Whittaker, 2012). The different binding poses of the tail groups of CAY10471 and fevipiprant associated with the different conformations of W2837.32 and the N-loop indicate that the open end of the ligand binding pocket, which we propose to be the ligand entry port, exhibits certain structural flexibility. Additional structures of CRTH2 with other antagonists that have distinct tail groups are needed to further investigate the conformational diversity of residues in this region, as it may significantly affect the results of structure-based virtual screening for developing novel CRTH2 antagonists. Furthermore, the unexpected small molecules modeled in this region suggest that the ligand entry port may offer an additional site for designing new synthetic CRTH2 antagonists, which compared to CAY10471 and fevipiprant would engage in additional interactions with the receptor to achieve stronger binding and a longer duration of action.
Collectively, our structures offer novel structural insights into the action of diverse CRTH2 antagonists, which will facilitate CRTH2 drug development for a number of inflammatory diseases, including asthma. They also reveal interesting features of the ligand binding pocket and suggest a novel mechanism for the binding of the endogenous lipid molecule PGD2, thus shedding light on the structural and mechanistic diversity of GPCRs for the recognition of lipid mediators.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Cheng Zhang (chengzh@pitt.edu). DNA constructs and other research reagents generated by the authors will be distributed upon request to other research investigators under a Material Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Spodoptera frugiperda Sf9 cells were purchased from Expression Systems. Cells were cultured in ESF 921 medium (Expression Systems) at 27°C and infected with baculovirus at a density of 4 × 106 cells per ml for large-scale protein expression. HEK-293T cells were initially obtained form the American Type Culture Collection (ATCC). Cell were cultured in the DMEM medium with 4.5 g/L glucose, L-glutamine & sodium pyruvate plus 10% FBS at 37°C with 5% CO2. 300 μg/ml geneticin and neomycin were used to select the clones for constructing stable cell lines. Plasmocin (Fisher Scientific) was used to prevent mycoplasma contamination.
METHOD DETAILS
Protein expression and purification
To facilitate crystallization, an engineered CRTH2 construct was designed by inserting a modified T4 lysozyme (eT4L) (Thorsen et al., 2014) into the third intracellular loop (ICL3) between residues R236 an R238, along with a linker (ADLGLQHR) at the N-terminus of eT4L, which was introduced by accident, and introducing a mutation of glycosylation site N25A. A human rhinovirus (3C) protease cleavage site was inserted at residue S339 for C-terminus removal. The construct gene was synthesized by gBlocks (Integrated DNA Technologies) and cloned into a modified pFastBac vector (Invitrogen), which contains a Flag-tag followed by a tobacco etch virus (TEV) protease cleavage site at the N-terminus and an 8 × His-tag at the C-terminus. The engineered protein was expressed in Sf9 cells. Cells were infected by virus at a density of 4 × 106 cells per ml and cultured at 27°C for 48 h with an tagonist CAY10471 (Cayman Chemical) or fevipiprant (MedKoo Biosciences) at a final concentration of 100 nM in the medium. The transfected cells were collected by centrifugation and stored at −80°C. To stabilize the receptor, 1 μM CAY10471 or fevipiprant was added during all purification steps. The frozen Sf9 cells were lysed by stirring in buffer containing 20 mM Tris-HCl, pH7.5, 0.2 μg/ml leupeptin, 100 μg/ml benzamidine and 2mg/ml iodoacetamide. After centrifugation, the pellet was resuspended and solubilized in buffer containing 20mM HEPES, pH7.5, 750mM NaCl, 1% (w/v) n-dodecyl-β-D-maltoside (DDM, Anatrace), 0.2% (w/v) sodium cholate (Sigma), 0.2% (w/v) cholesterol hemisuccinate (CHS, Anatrace), 20% (v/v) glycerol, 0.2 μg/ml leupeptin, 100 μg/ml benzamidine, 500 unit Salt Active Nuclease (Arcticzymes) and 2mg/ml iodoacetamide at 4°C for 2h. The supernatant was is olated by centrifugation at 25,000 g for 30 min and incubated with nickel Sepharose resin (GE healthcare) plus 10 mM imidazole at 4°C overnight. The resin was washed with buffer containing 20 mM HEPES, pH 7.5, 500 mM NaCl, 0.1% (w/v) DDM, 0.02% (w/v) CHS and 30 mM imidazole. The protein was eluted by 400 mM imidazole and directly loaded onto anti-Flag M1 antibody resin (homemade) after adding 2mM CaCl2. The detergent was slowly exchanged to 0.01% (w/v) lauryl maltose neopentyl glycol (MNG, Anatrace) on M1 antibody resin. The receptor was finally eluted with buffer containing 20 mM HEPES, pH 7.5, 100mM NaCl, 0.002% (w/v) MNG, 0.001% (w/v) CHS, 200 μg/ml Flag peptide and 5 mM EDTA. After treatment with TEV protease, 3C protease and PNGase F (NEB) at 4°C overnight, the N-terminal Flag-tag, th e C-terminal His-tag and glycosylation were removed. The monodispersed receptor was collected after size-exclusion chromatography using a Superdex 200 Increase column (GE healthcare). The purified receptor was concentrated to 50–60 mg/ml for crystallization.
Crystallization
The CRTH2 receptor in complex with CAY10471 or fevipiprant was crystallized using the lipidic cubic phase (LCP) method (Caffrey, 2009). The protein sample was mixed with the monoolein/cholesterol lipid (10:1 w/w) at a weight ratio of 1: 1.5 (protein: lipid) using two glass syringes to form clear LCP. Then the LCP mixture was then dispensed onto glass plates in 20 nl drops and overlaid with 700 nl of precipitant solution using a Gryphon robot (Art Robbins). The crystallization condition of CRTH2-CAY10471 was 100 mM MES, pH 6.5, 100 mM ammonium sulfate, 30% (v/v) PEG400 and 2% (v/v) polypropylene glycol P400, while the same condition plus 1 mM succinate salt was used to crystallize CRTH2-fevipiprant. The crystallization plates were placed in a 15°C incuba tor. Crystals appeared in 3 days and grew to full size in 2 weeks, which were then harvested from LCP using micro mounts (MiTeGen) and flash frozen in liquid nitrogen.
Data collection and structure determination
X-ray diffraction data was collected at the Chicago Advanced Photon Source (APS) beam line 23ID-B of GM/CA with a microbeam with a 10 μm diameter. Each crystal was exposed with a 10 μm × 10 μm beam for 0.2 s and 0.2 degree oscillation per frame to collect 20–40 degrees of rotation data. Data from 21 crystals of CRTH2-CAY10471 and 25 crystals of CRTH2-fevipiprant were processed and merged by HKL2000 software (Otwinowski and Minor, 1997).
The initial phase of CRTH2-fevipiprant complex was determined by molecular replacement in Phaser (McCoy et al., 2007) using the C5aR receptor structure (PDB ID 6C1R) and T4L portion of ETBR (PDB ID 5XPR) as search models. The structure model was refined and rebuilt using PHENIX (Adams et al., 2010) and COOT (Emsley and Cowtan, 2004), respectively. The structure of CRTH2-CAY10471 was solved by molecular replacement using the CRTH2-fevipiprant model and refined using the same method. Finally, MolProbity (Chen et al., 2010; Williams et al., 2018) was used to check the quality of the two models. All the structure figures were produced with PyMOL (http://www.pymol.org/). The charge distribution was calculated by using APBS (Baker et al., 2001).
Protein thermostability assay
The wild-type CRTH2 was used to determine the thermostability of the receptor with different ligands. The receptor was expressed and purified in a similar way as for the crystallization trials except that the ligand was finally removed by size exclusion chromatography using buffer without any ligand. The unliganded receptor was incubated with buffer only or with buffers containing 10 μM of the different ligands, ramatroban, fevipiprant and CAY10471, and then labeled with 7-diethylamino-3-(4-maleinidylphenyl)-4-methylcoumarin (CPM) dye (Sigma) at a concentration of 0.1 mg/ml. The CPM fluorescence intensity (excitation 387 nm, emission 463 nm) of each sample at different temperature points (from 30°C to 75°C) was measured by a spectrophotometer (SPECTRAMax Paradigm).
HEK-293T cell surface expression of CRTH2 constructs
All radioactive ligand-binding experiments were performed using the membranes of stably transfected HEK-293T cells. To build the stable cell lines, constructs of the wild-type CRTH2 and CRTH2 mutants C11A, R170A and W283A were cloned into the vector pcDNA3.1+ (Invitrogen) with a FLAG-tag at the N-terminus, and then transfected into HEK-293T cell using FuGENE Transfection Reagent (Promega) for constructing stable cell lines.
To determine the surface expression of each CRTH2 construct, flow cytometry experiments were performed. Cells were suspended in PBS, washed twice, and incubated with 1 μg/ml DyLight 488 (Thermo Fisher)-labeled anti-Flag M1 antibody (homemade) plus 2 mM CaCl2 in the dark for 15 min at room temperature. The cells were then analyzed for the DyLight488 fluorescence on a FACScan flow cytometer (BD CellQuest™ Pro). The protein expression levels were represented by the median fluorescence values of the sorted cells. All of the cells stably expressing wild-type CRTH2 and mutants showed comparable receptor expression levels.
Membrane preparation and radioactive ligand-binding assays
To prepare cell membranes, HEK-239T cells stably expressing different CRTH2 constructs were rinsed and detached with PBS buffer. After centrifugation at 1000 × g for 10 min, the cell pellets were resuspended in buffer containing 20 mM Tris-HCl, pH 7.5, 1 mM EDTA, 0.2 μg/ml leupeptin and 100 μg/ml benzamidine. After homogenization, the samples were centrifuged first at 1000 × g for 10 min, then at 150,000 g for 1 h. The membrane pellet was resuspended in buffer containing 20 mM HEPES, pH7.5, 100 mM NaCl, 1 mM EDTA and homogenized, then frozen in liquid nitrogen and stored at −80°C.
For the radioactive ligand-binding assays, ~20 μg of membrane protein was incubated with [3H]PGD2 (Perkin Elmer) and nonradioactive ligands for 2 h at room temperature in 200 μl of binding buffer containing 20 mM HEPES, pH 7.5, 1 mM EDTA, 5 mM MgCl2, 5 mM MnCl2 and 0.1% (w/v) BSA. For the saturation binding, [3H]PGD2 was added in various concentrations from 0 to 20 nM. Total and nonspecific binding was measured in the absence and presence of 10 μM non-radioactive PGD2 (Cayman Chemical), respectively. For the competition binding assays, the cell membranes were incubated with 2 nM [3H]PGD2 and various concentrations of competing ligands (nonradioactive PGD2, CAY10471 or fevipiprant) from 0.01 nM to 10 μM. After incubation, the reaction was terminated by adding 5 ml of cold binding buffer and rapidly filtering through glass giber prefilters (Millipore Sigma). The filters were washed three times with 5 ml cold binding buffer, and the retained receptor-bound [3H]PGD2 was incubated with 5 ml of CytoScint liquid scintillation cocktail (MP Biomedicals) and counted on a Beckman LS6500 scintillation counter.
Molecular docking
We chose the CAY10471-bound CRTH2 structure as the receptor/host for docking ramatroban. Before docking ramatroban, we performed cognate docking of CAY10471 back to this CRTH2 structure to ascertain that the docking protocol could reproduce its crystal geometry. Following validation of the docking protocol, ramatroban (structure obtained from PubChem (Kim et al., 2016)) was docked to this CRTH2 structure. All docking runs were performed using GOLD 5.5 (Jones et al., 1997). Before docking, all crystal waters and ligands were deleted and hydrogen atoms were added to the receptor. The docking solutions were scored on the basis of the ChemScore fitness function (Baxter et al., 1998; Eldridge et al., 1997).
QUANTIFICATION AND STATISTICAL ANALYSES
Protein thermostability assay
Results are represented as the mean ± SEM from 2 independent experiments. Data were analyzed with the Boltzmann sigmoidal equation in GraphPad Prism 7 (GraphPad Software) to calculate the melting temperature (Tm).
Cell surface expression and ligand binding assay
Protein expression levels are represented by the median fluorescence values of the sorted cells. Results are represented as the mean ± SEM from 3 independent measurements.
Ligand binding data are represented as the mean ± SEM from 3 independent experiments. Data were analyzed with the one site competitive and saturation binding methods in GraphPad Prism 7 (GraphPad Software).
DATA AVAILABILITY
The coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes PDB 6D26 and PDB 6D27 for CRTH2-fevipiprant and CRTH2-CAY10471, respectively.
The original data for the protein thermostability assay and the ligand-binding assay have been published on Mendeley: http://dx.doi.org/10.17632/m57xsf7v5n.1.
Supplementary Material
Highlight.
Crystal structures of antagonist-bound human CRTH2 are solved
A well-structured N-terminus covers ligand binding pocket
Conserved and divergent binding features of CRTH2 antagonists are revealed
A multiple-step binding process of prostaglandin D2 is proposed
ACKNOWLEDGMENTS
We thank the staff at the GM/CA @ APS of Argonne National Laboratory at Chicago for their assistance with X-ray diffraction data collection. We thank Dr. James C. Burnett and Dr. Peter Wipf for discussion. We acknowledge the financial support from the University of Pittsburgh and the Maximizing Investigators’ Research Award (MIRA) R35 grant 1R35GM128641 from NIH (C.Z.), the funding support from the Biomedical Research Council, A*STAR (R.N.V.K.D. and H.F.) and the funding support from National Natural Science Foundation of China (31770791 and 315707410) (Z.W.). Z.W. is also supported by the startup funds from Southern University of Science and Technology and the Recruitment Program of Global Youth Experts of China.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA (2001). Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes N, Pavord I, Chuchalin A, Bell J, Hunter M, Lewis T, Parker D, Payton M, Collins LP, Pettipher R, et al. (2012). A randomized, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy 42, 38–48. [DOI] [PubMed] [Google Scholar]
- Baxter CA, Murray CW, Clark DE, Westhead DR, and Eldridge MD (1998). Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins: Structure, Function, and Bioinformatics 33, 367–382. [PubMed] [Google Scholar]
- Busse WW, Wenzel SE, Meltzer EO, Kerwin EM, Liu MC, Zhang N, Chon Y, Budelsky AL, Lin J, and Lin SL (2013). Safety and efficacy of the prostaglandin D2 receptor antagonist AMG 853 in asthmatic patients. J Allergy Clin Immunol 131, 339–345. [DOI] [PubMed] [Google Scholar]
- Caffrey M (2009). Crystallizing membrane proteins for structure determination: use of lipidic mesophases. Annu Rev Biophys 38, 29–51. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrencik JE, Roth CB, Terakado M, Kurata H, Omi R, Kihara Y, Warshaviak D, Nakade S, Asmar-Rovira G, Mileni M, et al. (2015). Crystal Structure of Antagonist Bound Human Lysophosphatidic Acid Receptor 1. Cell 161, 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosmi L, Annunziato F, Galli MIG, Maggi RME, Nagata K, and Romagnani S (2000). CRTH2 is the most reliable marker for the detection of circulating human type 2 Th and type 2 T cytotoxic cells in health and disease. Eur J Immunol 30, 2972–2979. [DOI] [PubMed] [Google Scholar]
- Dalli J, and Serhan CN (2018). Identification and structure elucidation of the proresolving mediators provides novel leads for resolution pharmacology. Br J Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge MD, Murray CW, Auton TR, Paolini GV, and Mee RP (1997). Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. Journal of computer-aided molecular design 11, 425–445. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Erpenbeck VJ, Popov TA, Miller D, Weinstein SF, Spector S, Magnusson B, Osuntokun W, Goldsmith P, Weiss M, and Beier J (2016). The oral CRTh2 antagonist QAW039 (fevipiprant): A phase II study in uncontrolled allergic asthma. Pulm Pharmacol Ther 39, 54–63. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, and Schioth HB (2003). The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63, 1256–1272. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, et al. (2012). Crystal structure of a lipid G protein-coupled receptor. Science 335, 851–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Lybrand TP, and Breyer RM (2005). Identification of determinants of ligand binding affinity and selectivity in the prostaglandin D2 receptor CRTH2. J Biol Chem 280, 32442–32451. [DOI] [PubMed] [Google Scholar]
- Hirai H, Tanaka K, Takano S, Ichimasa M, Nakamura M, and Nagata K (2002). Cutting edge: agonistic effect of indomethacin on a prostaglandin D2 receptor, CRTH2. J Immunol 168, 981–985. [DOI] [PubMed] [Google Scholar]
- Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, et al. (2001). Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med 193, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T, Okuno T, Hirata K, Yamashita K, Kawano Y, Yamamoto M, Hato M, Nakamura M, Shimizu T, Yokomizo T, et al. (2018). Na(+)-mimicking ligands stabilize the inactive state of leukotriene B4 receptor BLT1. Nat Chem Biol 14, 262–269. [DOI] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, Pu M, Korde A, Jiang S, Ho JH, et al. (2017). Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 547, 468–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, Korde A, et al. (2016). Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 167, 750–762 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh J, Thomas WG, Aguilar MI, and Pattenden LK (2009). Role of helix 8 in G protein-coupled receptors based on structure-function studies on the type 1 angiotensin receptor. Mol Cell Endocrinol 302, 118–127. [DOI] [PubMed] [Google Scholar]
- Jones G, Willett P, Glen RC, Leach AR, and Taylor R (1997). Development and validation of a genetic algorithm for flexible docking. Journal of molecular biology 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Kim S, Thiessen PA, Bolton EE, Chen J, Fu G, Gindulyte A, Han L, He J, He S, Shoemaker BA, et al. (2016). PubChem Substance and Compound databases. Nucleic Acids Res 44, D1202–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuna P, Bjermer L, and Tornling G (2016). Two Phase II randomized trials on the CRTh2 antagonist AZD1981 in adults with asthma. Drug Des Devel Ther 10, 2759–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupczyk M, and Kuna P (2017). Targeting the PGD2/CRTH2/DP1 Signaling Pathway in Asthma and Allergic Disease: Current Status and Future Perspectives. Drugs 77, 1281–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RA, and Austen KF (1981). Mediation of local homeostasis and inflammation by leukotrienes and other mast cell-dependent compounds. Nature 293, 103–108. [DOI] [PubMed] [Google Scholar]
- Liu H, Kim HR, Deepak RNVK, Lei W, Chung KY, Fan H, Wei Z, and Zhang C (2018). Orthosteric and allosteric action of the C5a receptor antagonists. Nature Structural & Molecular Biology. [DOI] [PubMed] [Google Scholar]
- Mathiesen JM, Christopoulos A, Ulven T, Royer JF, Campillo M, Heinemann A, Pardo L, and Kostenis E (2006). On the mechanism of interaction of potent surmountable and insurmountable antagonists with the prostaglandin D2 receptor CRTH2. Mol Pharmacol 69, 1441–1453. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D, Wood C, Bateman E, LaForce C, Blatchford J, Hilbert J, Gupta A, and Fowler A (2017). A randomized study of BI 671800, a CRTH2 antagonist, as add-on therapy in poorly controlled asthma. Allergy Asthma Proc 38, 157–164. [DOI] [PubMed] [Google Scholar]
- Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, and Spits H (2011). Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol 12, 1055–1062. [DOI] [PubMed] [Google Scholar]
- Monneret G, Gravel S, Diamond M, Rokach J, and Powell WS (2001). Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood 98, 1942–1948. [DOI] [PubMed] [Google Scholar]
- Nagata K, and Hirai H (2003). The second PGD(2) receptor CRTH2: structure, properties, and functions in leukocytes. Prostaglandins Leukot Essent Fatty Acids 69, 169–177. [DOI] [PubMed] [Google Scholar]
- Nagata K, Tanaka K, Ogawa K, Kemmotsu K, Imai T, Yoshie O, Abe H, Tada K, Nakamura M, Sugamura K, et al. (1999). Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol 162, 1278–1286. [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. (2000). Crystal structure of rhodopsin: A G protein-coupled receptor. Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- Park JH, Scheerer P, Hofmann KP, Choe HW, and Ernst OP (2008). Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187. [DOI] [PubMed] [Google Scholar]
- Pettipher R, Hansel TT, and Armer R (2007). Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat Rev Drug Discov 6, 313–325. [DOI] [PubMed] [Google Scholar]
- Pettipher R, Hunter MG, Perkins CM, Collins LP, Lewis T, Baillet M, Steiner J, Bell J, and Payton MA (2014). Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy 69, 1223–1232. [DOI] [PubMed] [Google Scholar]
- Pettipher R, and Whittaker M (2012). Update on the development of antagonists of chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2). From lead optimization to clinical proof-of-concept in asthma and allergic rhinitis. J Med Chem 55, 2915–2931. [DOI] [PubMed] [Google Scholar]
- Robertson N, Rappas M, Dore AS, Brown J, Bottegoni G, Koglin M, Cansfield J, Jazayeri A, Cooke RM, and Marshall FH (2018). Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 553, 111–114. [DOI] [PubMed] [Google Scholar]
- Royer JF, Schratl P, Lorenz S, Kostenis E, Ulven T, Schuligoi R, Peskar BA, and Heinemann A (2007). A novel antagonist of CRTH2 blocks eosinophil release from bone marrow, chemotaxis and respiratory burst. Allergy 62, 1401–1409. [DOI] [PubMed] [Google Scholar]
- Santus P, and Radovanovic D (2016). Prostaglandin D2 receptor antagonists in early development as potential therapeutic options for asthma. Expert Opin Investig Drugs 25, 1083–1092. [DOI] [PubMed] [Google Scholar]
- Schroder R, Merten N, Mathiesen JM, Martini L, Kruljac-Letunic A, Krop F, Blaukat A, Fang Y, Tran E, Ulven T, et al. (2009). The C-terminal tail of CRTH2 is a key molecular determinant that constrains Galphai and downstream signaling cascade activation. J Biol Chem 284, 1324–1336. [DOI] [PubMed] [Google Scholar]
- Schuligoi R, Sturm E, Luschnig P, Konya V, Philipose S, Sedej M, Waldhoer M, Peskar BA, and Heinemann A (2010). CRTH2 and D-type prostanoid receptor antagonists as novel therapeutic agents for inflammatory diseases. Pharmacology 85, 372–382. [DOI] [PubMed] [Google Scholar]
- Serhan CN (2014). Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Yin J, Chapman K, Grzemska M, Clark L, Wang J, and Rosenbaum DM (2016). High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit MJ, Vischer HF, Bakker RA, Jongejan A, Timmerman H, Pardo L, and Leurs R (2007). Pharmacogenomic and structural analysis of constitutive g protein-coupled receptor activity. Annu Rev Pharmacol Toxicol 47, 53–87. [DOI] [PubMed] [Google Scholar]
- Smith WL (1989). The eicosanoids and their biochemical mechanisms of action. Biochem J 259, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H, Shichijo M, Iino T, Manabe Y, Watanabe A, Shimazaki M, Gantner F, and Bacon KB (2003). An orally bioavailable small molecule antagonist of CRTH2, ramatroban (BAY u3405), inhibits prostaglandin D2-induced eosinophil migration in vitro. J Pharmacol Exp Ther 305, 347–352. [DOI] [PubMed] [Google Scholar]
- Sykes DA, Bradley ME, Riddy DM, Willard E, Reilly J, Miah A, Bauer C, Watson SJ, Sandham DA, Dubois G, et al. (2016). Fevipiprant (QAW039), a Slowly Dissociating CRTh2 Antagonist with the Potential for Improved Clinical Efficacy. Mol Pharmacol 89, 593–605. [DOI] [PubMed] [Google Scholar]
- Taniguchi R, Inoue A, Sayama M, Uwamizu A, Yamashita K, Hirata K, Yoshida M, Tanaka Y, Kato HE, Nakada-Nakura Y, et al. (2017). Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 548, 356–360. [DOI] [PubMed] [Google Scholar]
- Thorsen TS, Matt R, Weis WI, and Kobilka BK (2014). Modified T4 Lysozyme Fusion Proteins Facilitate G Protein-Coupled Receptor Crystallogenesis. Structure 22, 1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulven T, and Kostenis E (2005). Minor structural modifications convert the dual TP/CRTH2 antagonist ramatroban into a highly selective and potent CRTH2 antagonist. J Med Chem 48, 897–900. [DOI] [PubMed] [Google Scholar]
- White C, Wright A, and Brightling C (2018). Fevipiprant in the treatment of asthma. Expert Opin Investig Drugs 27, 199–207. [DOI] [PubMed] [Google Scholar]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L, Gyles SL, Wettey FR, Gazi L, Townsend E, Hunter MG, and Pettipher R (2005). Prostaglandin D2 causes preferential induction of proinflammatory Th2 cytokine production through an action on chemoattractant receptor-like molecule expressed on Th2 cells. J Immunol 175, 6531–6536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates and structure factors have been deposited in the Protein Data Bank under the accession codes PDB 6D26 and PDB 6D27 for CRTH2-fevipiprant and CRTH2-CAY10471, respectively.
The original data for the protein thermostability assay and the ligand-binding assay have been published on Mendeley: http://dx.doi.org/10.17632/m57xsf7v5n.1.