

Abstract
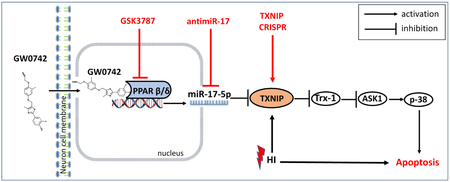
Activation of peroxisome proliferator-activated receptor beta/delta (PPAR-β/δ), a nuclear receptor acting as a transcription factor, was shown to be protective in various models of neurological diseases. However, there is no information about the role of PPAR-β/δ as well as its molecular mechanisms in neonatal hypoxia-ischemia (HI). In the present study, we hypothesized that PPAR-β/δ agonist GW0742 can activate miR-17-5p, consequently inhibiting TXNIP and ASK1/p38 pathway leading to attenuation of apoptosis. Ten-day-old rat pups were subjected to right common carotid artery ligation followed by 2.5 h hypoxia. GW0742 was administered intranasally 1 and 24 h post HI. PPAR-β/δ receptor antagonist GSK3787 was administered intranasally 1 h before and 24 h after HI, antimir-17-5p and TXNIP CRISPR activation plasmid were administered intracerebroventricularly 24 and 48 h before HI, respectively. Brain infarct area measurement, neurological function tests, western blot, reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR), Fluoro-Jade C and immunofluorescence staining were conducted. GW0742 reduced brain infarct area, brain atrophy, apoptosis, and improved neurological function at 72 hours(h) and 4 weeks post HI. Furthermore, GW0742 treatment increased PPAR-β/δ nuclear expression and miR-17-5p level and reduced TXNIP in ipsilateral hemisphere after HI, resulting in inhibition of ASK1/p38 pathway and attenuation of apoptosis. Inhibition of PPAR-β/δ receptor and miR-17-5p and activation of TXNIP reversed the protective effects. For the first time, we provide evidence that intranasal administration of PPAR-β/δ agonist GW0742 attenuated neuronal apoptosis at least in part via PPARyg-β/δ/miR-17/TXNIP pathway. GW0742 could represent a therapeutic target for treatment of neonatal hypoxic ischemic encephalopathy (HIE).
Keywords: PPAR-β/δ, GW0742, HI, apoptosis, miR-17, TXNIP
Graphical Abstract
1. Introduction
Despite major advances in monitoring technology, increasing knowledge of fetal pathologies and widespread use of hypothermia, hypoxic-ischemic encephalopathy (HIE) remains one of the leading causes of neonatal mortality and permanent neurological disability worldwide. Most severe cases of neonatal HIE are unresolved1 and it accounts for 23% of all neonatal deaths globally.2 The incidence of HIE is as high as 26 per 1000 live births in underdeveloped countries, yet it also affects 1 to 8 per 1000 live newborns in developed countries.3 Thus far no pharmacological treatment has consistently improved the long-term survival rate of infants following perinatal HIE,4 which makes search for new therapies necessary.
Peroxisome proliferator–activated receptor β/δ (PPAR-β/δ) is a ligand activated nuclear receptor, that belongs to the nuclear hormone receptor family. There are three members of the PPAR subfamily: α, β/δ, and -γ. They regulate gene expression and multiple biological processes by acting as transcription factors, binding to specific DNA sequence elements within the promoter region of target genes.5 Despite being the predominant subtype in the CNS,5,6 the information about the role of PPAR-β/δ in ischemic stroke is sparse and biology of PPAR-β/δ in the brain is still much less understood than that of PPAR-α and -γ,7 nonetheless neuroprotective benefits of PPAR-β/δ agonists have been observed in models of multiple sclerosis, Alzheimer’s and Parkinson’s disease,8-10 suggesting that PPAR-β/δ may have protective effects against the progression of CNS disorders through promoting cell survival.11,12
Cell death due to neuronal apoptosis plays a major role in brain injury associated with HIE.13 Increasing evidence has shown that PPAR-β/δ is able to promote survival of neurons in vitro under stress conditions due to inhibition of apoptosis.11,14 There is also evidence for PPAR-β/δ beneficial role from animal models, where GW0742, specific PPAR-β/δ agonist, exerted beneficial effects in renal and gut I/R injury.15,16 Interestingly, pretreatment with GW0742 significantly reduced brain infarct volumes of rats submitted to MCAO by reducing apoptosis.17 However, the mechanisms of PPAR-β/δ–mediated protection during ischemic stroke remains unclear. Thioredoxin interacting protein (TXNIP) activation is a key event leading to apoptosis in neurons.18 TXNIP can inhibit antioxidative function of thioredoxin (Trx), resulting in activation of ASK1/p38 MAPK apoptotic pathway.19 It was previously shown, that TXNIP expression can be repressed by PPAR-α20 and PPAR-γ,18 but the mechanism of this regulation is unknown. It is also unknown whether PPAR-β/δ can regulate TXNIP. Moreover, recent evidence indicates that the expression of miRNA’s can be under the transcriptional control of PPAR’s.21,22,23 Concomitantly, miR-17 was shown to bind to and down-regulate TXNIP in rat pancreatic β-cells,24 while miR-17 downregulation stabilized TXNIP and removed thioredoxin inhibition on ASK1 leading to apoptosis in mouse neural stem cells.25
Based on aforementioned evidence, we hypothesized that intranasally administered GW0742 can activate miR-17-5p resulting in TXNIP downregulation, restoration of thioredoxin-1(Trx-1) and inhibition of ASK1/p38 pathway, leading to attenuation of neuronal apoptosis and improvement of neurobehavioral outcomes after HI.
2. Methods
2.1. Animals
All protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University. All studies were conducted in accordance with the United States Public Health Service's Policy on Humane Care and Use of Laboratory Animals. Sprague Dawley rat mothers, with litters of 12 pups were purchased from Envigo (Livermore, CA). Ten-day old rat pups (n=217) were used for this study. The model used was the modified Rice-Vannucci neonatal hypoxia-ischemia (HI) model.26 Briefly, rat pups were anesthetized with 3% isoflurane and maintained throughout the surgery with 2%. Common carotid artery (CCA) was isolated and double ligated with a 5.0 silk surgical suture and cut between ligation sites. For the sham treatment, CCA was exposed but without ligation and the hypoxic treatment. After the surgical procedure, the rats were allowed to recover for 1 h on a heated blanket. Thereafter, they were placed in an airtight jar partially submerged in a 37°C water bath. A gas mixture of 8% oxygen and 92% nitrogen was delivered into the jars for 2 h and 30 min. Thereafter, animals were returned to their mothers.
2.2. Drug Administration
GW0742 (25, 100 and 400 μg/kg, Tocris, USA) or vehicle (1% DMSO diluted in corn oil) were administered intranasally27 at 1 and 24 h after HI. 2 μl of GW0742 per drop was given every 2 min in alternating nares. PPAR-β/δ antagonist GSK3787 (300 μg/kg, Abcam, USA) or vehicle (1% DMSO diluted in corn oil) were administered intranasally at 1 h before HI and at 24 h post HI. 0.5 nmol of LNA miR-17-5p inhibitor (antimiR, rno-miR-17-5p miRCURY LNA miRNA Power Inhibitor, Exiqon) or control (miRCURY LNA miRNA Power Inhibitor Control, Exiqon) were administered via intracerebroventricular injection28 at 1.5 mm posterior, 1.5 mm lateral to the bregma and 2.5 mm deep on the ipsilateral hemisphere at 24 h before HI. A total of 2 μl of antimiR per pup was injected slowly in 5 min. The needle was left in place for an additional 10 min and then slowly withdrawn over 5 min to prevent backflow. Intranasal route of administration was also tested - 200 pmol of miR-17-5p-LNA inhibitor or same dose of LNA scramble control in 10 μl saline were delivered into each naris at 24 h before HI. 1 μg of TXNIP CRISPR activation plasmid (Santa Cruz, USA) or control CRISPR Activation Plasmid (Santa Cruz, USA) were administered via intracerebroventricular injection to the ipsilateral hemisphere at 48 h pre HI. 2μl drug per pup was injected slowly in 5 min.
2.3. Infarct Area Measurements
Animals were anesthetized and euthanized at 72 h post HI. Brains were removed, sectioned into 2 mm slices and immersed in 2% solution of 2,3,5 triphenyltetrazolium chloride (TTC) (Sigma Aldrich, USA). The infarct area was traced and analyzed by Image J software (NIH).
2.4. Western Blot
Western Blot was performed as described previously.29 Animals were euthanized at 72 h post HI and transcardially perfused with ice cold PBS solution (pH 7.4). Brains were removed, instantly divided into ipsilateral and contralateral cerebrums and snap frozen in liquid nitrogen. Whole-cell lysates were obtained by homogenizing the tissue in RIPA lysis buffer (Santa Cruz Biotechnology, USA) and further centrifuged at 14,000 g at 4°C for 20 min. The supernatant was collected and aliquoted which was later used for measuring protein concentration by using a detergent compatible assay (Bio-Rad, Dc protein assay). Nuclear fractions were extracted by using Nuclear Extraction Kit (Abcam). Equal amounts of protein (40μg) were loaded on a 8%~12% SDS-PAGE gel and electrophoresed. Then they were transferred to a nitrocellulose membrane, which was blocked with 5% non-fat blocking grade milk (Bio-Rad, Hercules, USA) and incubated with primary antibodies for PPAR-β/δ (two different antibodies: recognizing 50 kDa, 1:500 Santa Cruz Biotechnology and recognizing 50 and 39 kDa, 1:1000, Abcam), TXNIP (1:500, Santa Cruz Biotechnology), Trx-1 (1:750, Abcam), p-ASK1 (1:500, Santa Cruz Biotechnology), p-p38 (1:500, Santa Cruz Biotechnology), Bcl-2 (1:500, Abcam), Bax (1:1000, NOVUS Biologicals), cleaved caspase-3 (1:1000, Cell Signaling Technology), actin (1:2000, Santa Cruz Biotechnology), lamin B1 (1:500, Santa Cruz Biotechnology), GAPDH (1:1000, Santa Cruz Biotechnology). The following day, nitrocellulose membranes were incubated with secondary antibodies (1:2000, Santa Cruz Biotechnology, USA) for 2h at room temperature. Immunoblots were then probed via ECL Plus chemiluminescence reagent kit (American Bioscience, Arlington Heights, IL) and analyzed using Image J (NIH, USA).
2.5. Histological Analysis
Pups were anesthetized and perfused with 0.1 M PBS followed by 4% formaldehyde solution (PFA) at 72 h post HI. The brains were removed and post-fixed (4% PFA, 4°C, 24 h), then transferred into a 30% sucrose solution. The brains were then sectioned at 10 mm thickness with a cryostat (Leica LM3050S) for Fluoro-Jade C30, immunofluorescence staining, and 30 mm for Nissl’s staining.
2.6. Evaluation of Brain Tissue Loss
The percentage of brain tissue loss = (contralateral hemisphere – ipsilateral hemisphere)/contralateral hemisphere *100%.
2.7. Neurobehavioral Tests
Neurobehavioral tests were performed in a blinded setup at either 72 h or 4 weeks post HI. To evaluate short-term neurological function, negative geotaxis test was performed 72 h after HI. To evaluate long-term neurological function, water maze, rotarod, and foot-fault were performed at 4 weeks post HI.31
1. Negative geotaxis: pups were placed head downward onto an inclined board (40°), the time taken for the pups to rotate their bodies to head upward position was recorded. The maximum testing time was 60s.
2. Foot-fault test: rats were placed on a horizontal grid floor (square size 20 cm-40 cm with a mesh size of 4 cm2) elevated 1m above ground for 1 min. Foot-fault was defined when the animal inaccurately placed a for- or hindlimb and fell through one of the openings in the grid. The number of foot-faults for each animal was recorded.
3. Rotarod test: assessed motor impairment using an accelerating rotarod (Columbus Instruments Rotamex, USA). A total of two rotarod trials were performed and the average duration (in seconds) was recorded and analyzed.
4. Morris water maze test was set up by submerging a platform in a pool of water and testing the rats’ ability to find the hidden platform using visual cues around the room. Rats were tested in a five-day test in both cued and hidden tests with all trials lasting no more than 60s. In the cued test if the rats had not discovered the platform in 60s, they were manually guided to the platform. A video recording system traced all of the animals’ activities and the swim paths were measured for quantification of distance, latency, and swimming speed by the Video Tracking System SMART-2000 (San Diego Instruments Inc., USA).
2.8. RT-qPCR for miR-17-5p and TXNIP mRNA Quantification
Total RNA was extracted using the Qiazol reagent (Qiagen) and 2 μg of RNA was subjected to reverse transcription with miScript II RT kit (Qiagen), following the manufacturer's instructions. MiR-17-5p levels were determined by using miScript SYBR Green PCR kit with miScript Primer Assay kit (Qiagen) according to manufacturer's instructions. Primers included miScript Universal Primer, miR-17-5p miScript Primer Assay (Rn_miR-17-5p_1; Cat#MS00013118; Qiagen) and SNORD61 miScript Primer Assay (Hs_SNORD61_11; Cat#MS00033705; Qiagen). Two ng of template cDNA were used for miR-17-5p quantification in a final volume of 25 μl containing specific primers and QuantiTect SYBR Green PCR master mix following manufacturer's instructions. PCR was done in duplicate and threshold cycle numbers were averaged for each sample. Successful amplification of products was evidenced by the amplification curve, the dissociation curve, and visualization of the qPCR products on agarose gels. SNORD61 was used for normalization.28 A relative fold change in expression was determined using the comparative cycle threshold method (2 −ΔΔCT). The TXNIP mRNA abundance was determined with RT-qPCR using miScript SYBR Green PCR kit (Qiagen). Primers included: TXNIP, Forward: AGTTACCCGAGTCAAAGCCG; Reverse: TCTCGTTCTCACCTGTAGGC. Actin: Forward: CTTCCTTCCTGGAATC; Reverse: GGCATAGAGGTCTTTACGGATG. Each PCR reaction mixture consisted of 20 ng of template cDNA, specific primers and QuantiTect SYBR Green PCR Master Mix for a total of 25 μl
2.9. Statistical Analysis
Statistical analysis was performed with Prism 6.0 software. Data were presented as mean ± SD. Difference between groups was evaluated by Student's t test (unpaired, 2-tailed) or one-way ANOVA followed by Dunnett’s or Bonferroni-Šídák post hoc tests. Data were considered significant when p<0.05.
3. Results
3.1. Time Course Expression Levels of Endogenous Proteins after HI
Fig. 1A shows representative western blot bands of endogenous expression levels of PPAR-β/δ, TXNIP, Trx-1, p-ASK1 and p-p38. The endogenous expression of PPAR-β/δ and Trx-1 were significantly downregulated at 72 h post HI when compared with sham group (p<0.05, Fig. 1B and D). In contrast, the expression of TXNIP, p-ASK1 and p-p38 was increased at 72 h post HI (p > 0.05, Fig. 1C, E and F).
FIGURE 1:

Temporal expression of endogenous PPAR-β/δ, TXNIP, thioredoxin-1 (Trx-1), p-ASK1 and p-p38 in the ipsilateral brain hemisphere after hypoxia-ischemia (HI). (A) Representative pictures of western blot data. (B) Western blot data showed that endogenous PPAR-β/δ expression levels significantly decreased at 72 hours (h) post HI. (C) TXNIP was increased at 3, 6, 24 and 72 h post HI. (D) Trx-1 level decreased at 0 and 72 h after HI. (E) p-ASK1 was increased at 12 and 72 h post HI. (F) p-p38 was increased from 0 to 72 h after HI. *p < 0.05 vs. sham. n= 4 for each group.
3.2. Intranasal administration of GW0742 reduced infarct area, improved shortterm neurological function and attenuated neuronal death in the hippocampus at 72 h post HI
Treatment with low dose of GW0742 (25μg/kg) showed to significantly reduce infarct area in the ipsilateral hemisphere compared to vehicle-treated group (17.4%± 6.44 c.f. 31.6% ± 4.64, p<0.05, Fig. 2A and B). No significant reduction in infarct area was observed with medium and high treatment doses (100 and 400 μg/kg). Short term neurological function was evaluated using negative geotaxis test. Vehicle-treated pups spent more time rotating to upward head position compared to the sham group at 72 h after HI (45.5 s ±9.9 c.f. 10.0 s±3.4, p<0.01, Fig. 2C). Low dose of GW0742 significantly improved short term neurological function compared with the vehicle group (27.2 s ± 12.1 c.f. 45.5 s ± 9.9, p < 0.05, Fig. 2C). No abnormal behavior was observed due to GW0742 administration. Thus, 25 μg/kg of GW0742 was selected as the optimal dose in following experiments. Since HI injury results in neuronal degeneration and apoptosis, to test whether GW0742 treatment can attenuate those adverse effects, we used Fluoro-Jade C staining. In Figure 2D, we found that there was a strong green contrast FJC positive staining in CA1 and CA2 region of ipsilateral hippocampus in vehicle animals when compared to sham. GW0742 treatment reduced FJC positive degenerated neurons in ipsilateral hippocampus compared to vehicle.
FIGURE 2:

Effect of intranasal administration of GW0742 on brain infarct area (A–B), short-term neurological function (C) and hippocampal apoptosis at 72 hours post hypoxia-ischemia (HI) (D). (A–B) TTC staining showed that low (25μg/kg) dose of GW0742 treatment significantly reduced infarct area when compared with vehicle. (C) Negative geotaxis showed that low dose (25 μg/kg) of GW0742 significantly improved neurological function compared with vehicle animals. n = 6/group. (D) Fluoro-Jade C staining showed massive positively stained neurons undergoing apoptosis in CA1 and CA2 area of the hippocampus in vehicle group compared to sham. GW0742 treatment reduced positive staining neurons (n = 2/group). *p < 0.01 vs. sham; #p < 0.05 vs. Hl+vehicle. Scale bar = 100 μm.
3.3. GW0742 Upregulated Nuclear PPAR-β/δ and Induced Nuclear Translocation
PPAR-β/δ in neurons from sham animals was expressed both in cytoplasm and nucleus, whereas after HI insult PPAR-β/δ was expressed mainly in the cytoplasm (Fig.3A). After intranasal treatment with GW0742 we detected PPAR-β/δ mainly in the nucleus of stained neurons (Fig.3A). Expression pattern found in immunofluorescence studies was confirmed by western blot. Expression of PPAR-β/δ in the nuclear fraction after HI was very low (Fig.3C), while it was largely increased after the treatment. We detected 2 bands of PPAR-β/δ - at 50 and 39 kDa (Fig.3C). Both 50 and 39 kDa forms were largely increased in the nuclear fraction after GW0742 treatment (p<0.05, Fig.3D). Whole cell extract analysis showed, that 39 kDa transcript was decreased at 72 h after the treatment, while 50 kDa was not significantly different (fig.3E and F). PPAR-β/δ inhibition halted nuclear translocation - GSK3787 applied together with GW0742 resulted in attenuation of PPAR-β/δ induction in the nuclear fraction (p<0.05, Fig. 3F). Nuclear/total PPAR-β/δ ratio in HI+GW0742 group was higher for 39 kDa isoform (Fig. 3G).
FIGURE 3:

Immunofluorescence staining and nuclear localization of PPAR-β/δ in the brain at 72 hours post hypoxia-ischemia (HI). (A) Immunofluorescence staining showed nuclear PPAR-β/δ (green) expression was decreased on neurons (red) in vehicle treated animals compared with sham and increased after GW0742 treatment. Blue was for nucleus (DAPI). Arrows indicate colocalization of PPAR-β/δ and NeuN on neurons that are showed in higher magnification in lower panels. Nuclei are outlined in black. (B) The schematic diagram shows the location of immunofluorescence staining (small black box). (C-D) Western blot data showed that PPAR-β/δ expression in the nuclear fraction was significantly decreased at 72 h post HI and it was increased after GW0742 treatment. (E-F) Western blot results showing PPAR-β/δ expression in nuclear and whole cell fraction. Inhibition of PPAR-β/δ receptor with GSK3787 prevented nuclear translocation of PPAR-β/δ. S, sham; V, HI+vehicle; GW, HI+GW0742; GSK, HI+GW0742+GSK3787. (G) Nuclear to whole cell PPAR-β/δ expression ratio showed that 39 kDa isoform was overrepresented in nuclear fraction compared to 50 kDa form. Scale bar: upper panels = 100 μm, lower panels= 10 μm; *p < 0.05 vs. sham; #p < 0.05 vs. HI+vehicle, &p <0.05 vs. HI+GW0742. n=6 for whole cell (total) extracts groups, n = 3 for nuclear extract groups.
3.4. GW0742 Reduced Brain Atrophy at 4 Weeks Post HI
Intranasal administration of GW0742 treatment significantly attenuated brain atrophy in the ipsilateral hemisphere, as demonstrated by reduction of brain weight loss (p< 0.05) and tissue loss (p < 0.05). HI insult resulted in severe brain atrophy of the ipsilateral hemispheres at 4 weeks post-HI, marked by a weight loss in ipsilateral hemisphere (p<0.01, Fig.4A). It was remarkably attenuated after GW0742 treatment at 4 weeks post-insult (18.6% ± 8.2 c.f. 31.5% ± 10.1, p<0.05, Fig.4B). Nissl-stained coronal brain sections at 4 weeks post-injury showed brain atrophy in vehicle group at 4 weeks post HI, as demonstrated by lesion, neuronal loss, and tissue breakdown (Fig. 4C). Morphology quantification studies of brain volume loss confirmed the statistical significance between sham and vehicle+HI group (p< 0.01). GW0742 treatment significantly reduced the brain tissue loss (16.7% ± 9.9 c.f. 33.7% ± 18.1, P < 0.05, Fig. 4D). HI resulted in hippocampal degeneration in ipsilateral hemisphere, while GW0742 attenuated these changes (Fig.4E).
FIGURE 4:

Effects of GW0742 treatment on brain atrophy and hippocampal morphology at 4 weeks post hypoxia-ischemia (HI). (A-B) Significant loss of right-to-left hemisphere (ipsilateral/contralateral) weight ratio is evident in vehicle rats, and which was significantly improved by GW0742 treatment at 4 weeks post HI. (C-D) Representative pictures of Nissl's stained brain slices showed tissue loss in ipsilateral hemisphere. GW0742 treatment significantly reduced the percent of tissue loss when compared with vehicle. (E) Representative images of hippocampus in ipsilateral hemisphere. HI resulted in degeneration of ipsilateral hippocampus, while GW0742 attenuated hippocampal damage. *p < 0.01 vs. sham; #p < 0.05 vs. HI+vehicle. n = 9 for sham and HI+ GW0742, n=8 for HI+vehicle group.
3.5. GW0742 Improved Long-term Neurological Function at 4 Weeks Post HI
To investigate the effects of GW0742 treatment on the long-term neurological impairments induced by HI, neurological functions were assessed by foot-fault, rotarod and water maze at 4 weeks post HI. In the foot fault test, we noted that vehicle controls displayed significantly more total foot-faults (19.9 ± 5.1 c.f. 10.2 ± 3.4, p<0.05), more foot slips on left-contralateral side compared to the sham group, in both forelimbs (19.7 ± 10.2 c.f. 5.4 ± 2.9, p < 0.05) and hindlimbs (32.4 ± 9.4 c.f. 18.0 ± 7.5, p < 0.05) and that the performance was significantly better in GW0742-treated group for total foot-faults and contralateral foot-faults compared to the vehicle controls (Total 15.0 ± 3.5 c.f. 19.9 ± 5.1, p < 0.05; Contralateral 17.4 ± 3.5 c.f. ± 8.8, p<0.05, Fig. 5A). Moreover, GW0742 treatment significantly increased the falling latency compared to vehicle controls in rotarod test (5 RPM accelerating: 42.5± 5.8 c.f. 35.3 ± 6.8, p<0.05, Fig. 5B). In the water maze test, vehicle-treated animals spent more time finding the platform, which meant they had cognitive impairment in memorizing the platform location compared to sham animals. (p<0.05, Fig. 5C). However, GW0742-treated animals showed to significantly improve memory and learning abilities as it took them less time to get to the platform (p<0.05, Fig. 5C). Meanwhile, there was no significant difference in velocity amongst the three groups, indicating that it is the spatial memory loss, not the slower velocity, that led to the longer escape latency (Fig. 5D).
FIGURE 5:

Effects of GW0742 treatment on long-term neurological function at 4 weeks post hypoxia-ischemia (HI). (A-B) GW0742 treatment significantly improved motor function as shown by foot-fault test and rotarod. (C) GW0742 treatment group showed significant improvement in spatial memory loss shown as decrease in escape latency when compared with vehicle. (D) Swim speed had no statistical differences among three groups, meaning that observed differences in escape latency were due to spatial memory loss, and not the speed differences. *p < 0.05 vs. sham; #p < 0.05 vs. HI+vehicle. n = 9 for sham and HI+GW0742, n=8 for HI+vehicle group.
3.6. In vivo Inhibition of PPAR-β/δ and miR-17-5p and Activation of TXNIP Abolished GW0742 Neuroprotective Effects at 72 h post HI
To evaluate whether pathway interventions can affect apoptosis and increase brain infarction we inhibited PPAR-β/δ and miR-17-5p, and activated TXNIP. Our TTC staining data (Fig. 6A) showed that GSK3787, miR-17-5p inhibitor and TXNIP CRISPR activation significantly reversed the protective effects of GW0742, seen as the significant increase in percent of infarcted area (p<0.05, Fig. 6B). Negative geotaxis test showed that animals treated with GW0742 and either GSK3787, miR-17-5p inhibitor or TXNIP CRISPR activation plasmid, had significantly impaired neurological function compared with corresponding controls (p < 0.05, Fig. 6C).
FIGURE 6:

Effects of GSK3787, TXNIP CRISPR activation plasmid and antimir-17-5p with GW0742 on infarct volume (A–B) and neurological function (C) at 72 hours post hypoxia-ischemia (HI). (A-B) The infarct volume was significantly increased in all 3 treatment groups with interventions when compared with respective controls treated with GW0742. (C) Negative geotaxis showed that animal groups treated with GW0742 and either GSK3787, TXNIP CRISPR activation plasmid or antimir-17-5p had significantly impaired neurological function compared with respective controls treated with GW0742. *p<0.05 vs. sham; #p < 0.05 vs. HI+vehicle; &p <0.05 vs. HI+GW0742+DMSO in corn oil; @p < 0.05 vs. HI+GW0742+control CRISPR; $p < 0.05 vs. HI+GW0742+control LNA. n = 6 for each group.
3.7. GW0742 Suppressed Apoptosis, Downregulated TXNIP Levels and Inhibited p-ASK1/p-p38 Axis Signaling at 72 h Post HI
To investigate the mechanism by which GW0742 attenuated apoptosis, animals were divided into the following groups: sham, HI+vehicle, HI+GW0742, HI+ GW0742+GSK3787, HI+GW0742+DMSO+corn oil, HI+GW0742+LNA antimimiR-17-5p, HI+GW0742+control LNA, HI+GW0742+TXNIP CRISPR, HI+GW0742+control CRISPR. Western blot data (Fig.7A) showed that TXNIP, p-ASK1, p-p38 and cleaved caspase-3 expression significantly increased in HI+vehicle group when compared with sham (p<0.05, Fig. 7B, D, E, G) while Trx-1 and Bcl-2/Bax ratio were decreased (p<0.05, Fig.7C and F). GW0742 treatment decreased TXNIP, p-ASK1, p-p38 and cleaved caspase-3 levels and increased Trx-1 and Bcl-2/Bax ratio when compared with HI + vehicle (p<0.05, Fig. 7B, D, E and G). GSK3787 significantly increased TXNIP expression, thereby abolishing the effects of GW0742, leading to decrease of Trx-1, Bcl-2/Bax ratio and increase of p-ASK1, p-p38 and cleaved caspase-3 levels. Inhibition of miR-17-5p was associated with increased TXNIP expression, decreased Trx-1, activation of p-ASK1/p-p38 axis signaling and increased cleaved caspase-3 expression in the ipsilateral hemisphere (p<0.05, Fig.7A–G). Furthermore, direct activation of TXNIP also led to activation of p-ASK1/p-p38 proapoptotic pathway (p < 0.05, Fig. 7A-G).
FIGURE 7:

Effects of GW0742 on apoptosis via the PPAR-β/δ/TXNIP/Trx-1/p-ASK1/p-p38 signaling pathway at 72 hours post hypoxia-ischemia (HI). (A) Representative picture of western blot data showing bands of the expression levels of TXNIP, thioredoxin-1 (Trx-1), p-ASK1, p-p38, Bcl-2, Bax and cleaved caspase-3 either with GW0742 treatment alone or GW0742+DMSO in corn oil, GW0742+GSK3787, GW0742+control CRISPR, GW0742+TXNIP CRISPR, GW0742+control LNA antimir, GW0742+antimir-17-5p groups. (B–G) Western blot data quantification of bands showed that GW0742 significantly increased Trx-1 and Bcl-2/Bax ratio and significantly decreased TXNIP, p-ASK1, p-p38 and caspase-3 cleavage when compared with HI+vehicle. GSK3787, TXNIP CRISPR activation plasmid and antimir-17-5p showed to significantly increase TXNIP (B), p-ASK1 (D), p-p38 (E) and cleavage of caspase-3 (G) expression, but decrease Trx-1 (C), Bcl-2/Bax (F) expression when compared with GW0742+DMSO in corn oil, GW0742+control CRISPR and GW0742+control LNA groups, respectively. *p < 0.05 vs. sham; #p < 0.05 vs. HI+ vehicle; &p < 0.05 vs. HI+GW0742+DMSO in corn oil; @p < 0.05 vs. HI+GW0742+ control CRISPR; $p < 0.05 vs. HI+GW0742+control LNA. n = 6 for each group. n=5 for HI+GW0742+antimir-17-5p.
3.8. GW0742 Treatment Increased miR-17-5p and Decreased TXNIP mRNA Level in Ipsilateral Hemispheres 72 h Post HI
To check whether GW0742 treatment affected miR-17-5p level and to validate miR-17-5p inhibition we performed RT-qPCR. HI resulted in increase of TXNIP mRNA level and GW0742 treatment reduced TXNIP mRNA levels in ipsilateral hemisphere at 72 h after HI (p<0.05, Fig.8A). GW0742 treatment increased miR-17-5p level in ipsilateral hemisphere at 72 h post HI (p<0.05, Fig.8B). Inhibition of miR-17-5p by LNA injected by i.c.v. led to massive decrease of miR-17-5p level (p<0.01, Fig.8B), while intranasal LNA was not as effective, although the reduction of miR-17-5p was still statistically significant (p<0.05, Fig.8C).
FIGURE 8:

Effects of GW0742 on TXNIP mRNA and miR-17-5p levels and effects of miR-17-5p inhibition at 72 hours post hypoxia-ischemia (HI). (A)TXNIP mRNA levels increased in HI+vehicle group, while GW0742 treatment reduced TXNIP mRNA level in ipsilateral hemisphere. n = 3 for each group. (B) GW0742 treatment significantly increased miR-17-5p level in ipsilateral hemisphere. Furthermore RT-qPCR confirmed robust miR-17-5p inhibition after i.c.v. LNA antimir-17-5p administration to GW0742 treated rats. n = 4 for each group. (C) Intranasal administration of LNA antimir-17-5p was not as effective as i.c.v. administration, although it also significantly reduced miR-17-5p level. n = 3 per group. *p < 0.05 vs. sham; #p < 0.05 vs. HI + vehicle; $p < 0.01 vs. HI + GW0742 + control LNA i.c.v., Λp < 0.05 vs. HI + GW0742 + control LNA intranasal.
4. Discussion
The challenge to establish safe and effective neuroprotective therapies for HIE has remained a priority in neonatology for decades, and thus far the only effective treatment is hypothermia. It provides protection, however most severe cases of neonatal HIE are unresolved as only 1 in 6 infants benefit from hypothermia.32 One of the main pathologies encountered after hypoxic-ischemic injury is apoptosis, although involvement of apoptotic mechanisms in the etiology of neonatal HIE is not completely understood.33 Increased caspase-3 activation has been identified in brain sections in children who died after experiencing HIE34 as well as in brains of rats subjected to HI. As apoptosis is also involved in normal brain development, the neonatal brain may be more susceptible to this cell death than the adult35 and therefore interventions at the apoptotic stage of neonatal HIE have potential to be beneficial. The novel finding of our present study was that intranasal administration of GW0742, a potent PPAR-β/δ agonist significantly attenuates HI-induced brain injury by reducing neuronal apoptosis, which was mediated by PPAR-β/δ/miR-17/TXNIP signaling pathway. Treatment with GW0742 improved neurological impairment and was associated with increased expression of nuclear PPAR-β/δ, activation of miR-17-5p and inhibition of TXNIP, p-ASK-1, p-p38 and cleavage of caspase-3, a hallmark of apoptosis. Blockage of PPAR-β/δ receptor and miR-17-5p and activation of TXNIP exacerbated ipsilateral hemisphere infarction rate and was associated with increased Bax/Bcl-2 ratio and cleavage of caspase-3.
At first we checked the time-course expression of pathway proteins after HI. PPAR-β/δ was significantly decreased at 72 h after HI, which may be due to progressive death of cortical neurons, as PPAR-β/δ is highly expressed in those cells in sham but not in vehicle treated rats, which we demonstrated in immunofluorescence experiment. TXNIP was induced after HI, while Trx-1 was decreased and consequently ASK1/p38 apoptotic pathway was activated. Decreased expression of Trx-1 is in agreement with results of Hattori36, who showed that thioredoxin level in the injured core was largely decreased after HI, while it was increased in the surviving neurons in penumbra. Our model is a severe injury model with average total infarct ratio of 30% (60% of ipsilateral hemisphere), therefore Trx-1 expression in our study was significantly decreased in ipsilateral hemispheres at 72 h after HI.
We then tested effectiveness of 3 different doses of GW0742 in HI model. Based on time-course results we decided to treat the pups 1 and 24 h after HI. 25 μg/kg dosage administered intranasally was the most effective, it significantly reduced infarct area of ipsilateral hemisphere at 72 h and improved short-term neurological impairments. Higher doses were not as effective, which may be explained by results from other study, showing that prolonged exposure to high dose of GW0742 was toxic to cerebellar granule neurons.14 Similar protective effects of GW0742 were shown in MCAO and SAH models. In MCAO studies, GW0742 was used as an intraperitoneal pretreatment- injected at 30 min before MCAO it reduced infarction, apoptosis and brain edema in rats,17 while injected to mice 10 min before reperfusion it decreased infarction rate, BBB permeability and proinflammatory cytokines level.37 GW0742 administered intracerebroventricularly at 30 min before SAH enhanced the neurological scoring of rats and protected the BBB.38 Those studies, however, differed from ours, as they utilized aged rats, and used GW0742 as a pretreatment. Our study is first to show protective effect of PPAR-β/δ activation in neonatal rat HI model, which has significance for clinical translation.
In the present study GW0742 protected the neurons, attenuated brain atrophy and improved rat outcomes in neurobehavioral tests accessing neurological function and memory in short as well as long-term after HI. Spatial memory is highly dependent on hippocampus function,39 and FJC showed that GW0742 inhibited neuronal death in CA1 and CA2 regions of the hippocampus at 72 h after HI, while histological staining showed protection of the hippocampus at 4 weeks post HI, which was translated to improved outcome in Morris water maze test. GW0742 improved performance of rats in both short-term and long term tests, as it was shown previously, that degree of atrophy in the ipsilateral hemisphere and hippocampus strongly correlates with short-term sensorimotor deficit and long-term neurofunctional outcome.31
In the previous studies PPAR-β/δ expression was reported in adult mice and human brains5, where it was localized in neurons in numerous brain areas, moreover PPAR-β/δ mRNA was the most abundant in mouse brain among PPAR’s.5 Additionally Challet et al. found PPAR-β/δ immunoreactive neurons in suprachiasmatic nucleus of hamster.40 We found that PPAR-β/δ was highly expressed in neurons of neonatal rats. To our best knowledge our study is the first to show the expression of PPAR-β/δ in neonatal rat brain. In our experiments PPAR-β/δ was expressed both in cytoplasm and nucleus of sham rat neurons, similarly to the expression found in adult mouse brain.5 Whereas after HI insult the expression pattern changed, and PPAR-β/δ was expressed mainly in the cytoplasm. Teng et al. found that PPAR-β/δ expression in adult rat neurons was decreased after SAH injury, but authors didn’t check for cytoplasmic or nuclear localization.38 After intranasal treatment with GW0742, we detected PPAR-β/δ mainly in the nucleus, which suggests that intranasally delivered GW0742 targeted the injured brain, causing PPAR-β/δ activation and translocation, which we later confirmed with western blot. Interestingly we detected 2 bands of PPAR-β/δ - at 50 and ~39 kDa. Database search for PPAR-β/δ transcripts recognized by anti-PPAR-β/δ antibody revealed, that besides canonical 50 kDa transcript, it recognizes ~39 kDa isoform which shares the same immunogen sequence. Both, 50 and 39 kDa forms were largely increased in the nuclear fraction after GW0742 treatment. However, as 39 kDa isoform was overrepresented in the nucleus after the treatment, this may suggest that this form may be responsible for observed protective effect. Transcript coding for this ~40 kDa protein uses an alternate exon in the 3' coding region and 3'UTR. It encodes isoform, which is shorter and has a distinct C-terminus, compared to 50 kDa protein.41 Interestingly, study investigating expression of PPAR isoforms in zebrafish tissues, which identified PPAR-β/δ band at ~50 kDa and a stronger band at ~ kDa, showed that PPAR-β/δ is higher expressed in various juvenile tissues compared to the adults.42 This finding is interesting in context of overrepresentation of this isoform in neonatal rats brain in our study. Authors, however, didn’t check for expression in brain tissue. Expression of truncated ~40-kDa PPAR-β/δ have been also previously found in mouse and gray mullet liver samples,43,44 however number of studies investigating function of this short isoform is very limited. There is evidence supporting its protective role in spinal cord injury (SCI). Studies in adult rat SCI model showed, that this PPAR-β/δ form was decreased after the model injury, which is similar to what we demonstrated in hypoxia-ischemia model. Telmisartan showed to increase level of ~40 kDa PPAR-β/δ expression after spinal cord injury, which was associated with beneficial effects manifested by improved motor function and pain responses.45 Consistent with this finding, increased level of nuclear ~40 kDa PPAR-β/δ in our study was accompanied by improved performance in rotarod test assessing motor function. This protective mechanism in SCI may be mediated through phosphorylation of 5' AMP-activated protein kinase (AMPK).45 The action of telmisartan was inhibited by a blockade of the receptor with GSK0660, which resulted in decrease of PPAR-β/δ expression level. Those results suggest, that the restoration of ~40 kDa PPAR-β/δ expression was responsible for therapeutic potential of telmisartan in rats with SCI. Additionally, blockade of PPAR-β/δ receptor resulted in inhibition of the telmisartan-induced changes of HMGB1 and RAGE expression, suggesting that ~40 kDa PPAR-β/δ may play a role in inhibiting HMGB1/RAGE proinflammatory signaling. Other study showed that decrease of ~40 kDa PPAR-β/δ in spinal cords of streptozotocin (STZ) induced diabetic rats after SCI injury was responsible for exacerbation of SCI injury and higher mortality rate compared to non-diabetic control rats. PPAR-β/δ expression in the spinal cords of STZ-diabetic rats decreased in a time-dependent manner after SCI.46 Thus, authors reasoned, that the activation of this PPAR-β/δ isoform might be helpful to attenuate the injury from SCI in diabetic rats. Indeed, GW0742 significantly increased the survival time of diabetic rats with SCI, but level of ~40 kDa PPAR-β/δ after GW0742 treatment was not measured in this study. On the other hand, transcript coding for ~40 kDa PPAR-β/δ was identified in human placenta and adipose tissues and it was shown in in vitro studies using expression vectors in human cell line, that this isoform may be a negative regulator of full-length PPAR-β/δ.47 To analyze the transactivating ability of short form, the pFABPLUC reporter activity was investigated during overexpression of this form in the absence or presence of the expression vector for full-length PPAR-β/δ and the PPAR-β/δ specific ligand GW501516. Cotransfection of pFABPLuc and the expression vector for full-length PPAR-β/δ in HeLa cells and treatment with agonist increased the trans-activation ability in a dose-dependent manner. Truncated form, on the other hand, had no trans-activation ability in the presence of GW501516 but rather showed tendency to repress the ligand-induced activation of pFABPLuc reporter by full-length PPAR-β/δ. Overall this study suggests, that short PPAR-β/δ isoform may have a role in the control of full-length PPAR-β/δ signaling and function. Further experiments are needed to clarify relationship between PPAR-β/δ proteins, and in vivo studies investigating effect of specific inhibition of particular isoform separately may help to elucidate function of PPAR-β/δ proteins in the context of ischemic brain injury.
GW0742 in our study restored Trx-1 level leading to inhibition of p-ASK1 and p-p38 MAP kinase. It has been established that Trx-1 blocks autophosphorylation of ASK-1,48 and it is known that p-ASK-1/p-p38 pathway promotes apoptosis.49 Consequently, after the treatment with GW0742 we observed increase of Bcl-2/Bax ratio and decrease of caspase-3 cleavage, suggesting that PPAR-β/δ activation led to attenuation of apoptosis. Previous in vitro studies showed that PPAR-β/δ agonists-L-165041 and GW501516, protected SH-SY5Y cells from Thapsigargin, MPP+ and Staurosporin induced cell death11 and mechanism of observed protection involved inhibition of caspase-3 and −7 activity, suggesting that PPAR-β/δ agonists protected cells through their antiapoptotic function. We next wanted to test whether GW0742 exhibits protective properties via the PPAR-β/δ/miR-17/TXNIP signaling pathway. Because of emerging evidence showing that PPAR receptors can regulate micro-RNA’s level, and the previous in vivo experiments showing that PPAR-α and -γ can inhibit TXNIP, we wanted to check if PPAR-β/δ activation can inhibit TXNIP through miR-17. microRNAs are small noncoding RNAs, which fine-tune gene expression, usually by posttranscriptional degradation of target mRNA or translational repression. Recently it was found, that treatment of endothelial cells with a PPAR-β/δ agonist (GW501516) led to an increase of miR-100,50 while PPAR-β/δ activation after ischemia repressed miR-15a.22 miR-17-5p is the most prominent member of the miR-17-92 cluster, and has been identified as the first miRNA with oncogenic potential and with essential role in proliferation and apoptosis. Its role is highlighted by the fact that miR-17 knockout in mice is neonatally lethal.51 miR-17 was shown to have the direct role in inhibition of ROS generation in human microglial cells.52 It has been confirmed that miR-17 is a negative regulator of TXNIP mRNA stability. There are highly conserved seed sequences for miR-17 in the TXNIP 3′-UTR which are found to govern TXNIP mRNA expression at posttranscriptional level.53 miR-17-5p inhibition in our study reversed the protective effect of GW0742, leading to increase of TXNIP, activation of downstream ASK1/p38 pathway and increased caspase-3 cleavage. It was previously shown that PPAR-α20 and -γ18 activation can promote TXNIP inhibition, but the underlying mechanism of this repression was not clear. Here we show, for the first time, that TXNIP- repressing, antiapoptotic effect of PPAR-β/δ can, at least in part, be mediated through miR-17-5p. Supporting our hypothesis is increased miR-17-5p level after PPAR-β/δ activation by GW0742, suggesting that PPAR-β/δ activated transcription of pri-miR-17-5p, while miR-17-5p inhibition abolished protective, antiapoptotic effects of GW0742. We also showed that observed protective effect of GW0742 is dependent on PPAR-β/δ receptor and TXNIP. Both PPAR-β/δ inhibition and TXNIP activation led to aggravation of outcomes and attenuation of antiapoptotic function of GW0742.
TXNIP have been considered as a bridge linking oxidative stress and inflammation and it is a known inducer of NLRP3 inflammasome. Activation of NLRP3 inflammasome results in pro-caspase-1 cleavage and IL-1β secretion thus promoting inflammatory response. Anti-inflammatory properties of PPAR’s are well documented and in vivo data suggests that PPAR-β/δ ligands have activity in a number of disease models that are partly driven by the inflammatory response.54,55,56 As neuroinflammation is a substantial component of HI brain injury,3 we think it is possible that some part of the observed protective effect of GW0742 may be attributed to regulation of cytokines production and attenuation of neuroinflammation, which we will focus on during our future experiments.
In conclusion, the administration of GW0742 after HI reduced infarct area, attenuated neuronal apoptosis and improved neurological outcomes. The neuroprotective effects of GW0742 could be mediated via the PPAR-β/δ/miR-17/TXNIP signaling pathway. Regarding lack of effective pharmacotherapy against HIE, our current evidence supports the idea that GW0742 could be a promising therapeutic candidate for patients, which can salvage the neurons after HIE.
Supplementary Material
Highlights:
PPAR-β/δ nuclear expression is decreased after neonatal brain injury (HI)
GW0742 induces PPAR-β/δ nuclear expression in neurons
GW0742 has a protective effect on brain infarction and neurobehavior after HI
GW0742 protective effect is mediated by PPAR-β/δ, miR-17-5p and TXNIP
PPAR-β/δ activation attenuates apoptosis after HI
Acknowledgments
The study was supported by a grant from National Institutes of Health NS104083 to Dr. John H. Zhang.
Footnotes
Potential Conflicts of Interest
Nothing to report
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rong Z, Pan R, Xu Y, et al. Hesperidin pretreatment protects hypoxia-ischemic brain injury in neonatal rat. Neuroscience 2013; 255:292–299. [DOI] [PubMed] [Google Scholar]
- 2.Lawn JE, Cousens S, Zupan J. 4 million neonatal deaths: when? where? why? Lancet. 2005;365:891–900. [DOI] [PubMed] [Google Scholar]
- 3.Douglas-Escobar M, Weiss MD. Hypoxic-ischemic encephalopathy: a review for the clinician. JAMA Pediatr. 2015;169(4):397–403. [DOI] [PubMed] [Google Scholar]
- 4.Yildiz EP, Ekici B, Tatli B, Neonatal hypoxic ischemic encephalopathy: an update on disease pathogenesis and treatment. Expert Rev Neurother. 2017; 17(5):449–459. [DOI] [PubMed] [Google Scholar]
- 5.Warden A, Truitt J, Merriman M, Ponomareva et al. Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep. 2016; 10;6:27618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnegg CI, Robbins ME. Neuroprotective Mechanisms of PPARδ: Modulation of Oxidative Stress and Inflammatory Processes. PPAR Res. 2011:373560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aleshin S, Strokin M, Sergeeva M, Reiser G. Peroxisome proliferator-activated receptor (PPAR)β/δ, a possible nexus of PPARα- and PPARγ-dependent molecular pathways in neurodegenerative diseases: Review and novel hypotheses. Neurochem Int. 2013;63(4):322–30. [DOI] [PubMed] [Google Scholar]
- 8.Kanakasabai S, Walline CC, Chakraborty S, Bright JJ. PPARδ deficient mice develop elevated Th1/Th17 responses and prolonged experimental autoimmune encephalomyelitis, Brain Research 2011; vol. 1376, pp. 101–112. [DOI] [PubMed] [Google Scholar]
- 9.Madrigal JL, Kalinin S, Richardson JC, Feinstein DL. Neuroprotective actions of noradrenaline: effects on glutathione synthesis and activation of peroxisome proliferator activated receptor delta, Journal of Neurochemistry 2007; vol. 103, no. 5, pp. 2092–2101. [DOI] [PubMed] [Google Scholar]
- 10.Kalinin S, Richardson JC, Feinstein DL. PPARdelta agonist reduces amyloid burden and brain inflammation in a transgenic mouse model of Alzheimer’s disease, Current Alzheimer Research 2009; vol. 6, no. 5, pp. 431–437. [DOI] [PubMed] [Google Scholar]
- 11.Iwashita A, Muramatsu Y, Yamazaki T. Neuroprotective efficacy of the peroxisome proliferator-activated receptor δ-selective agonists in vitro and in vivo, Journal of Pharmacology and Experimental Therapeutics 2007; vol. 320/3:1087–1096. [DOI] [PubMed] [Google Scholar]
- 12.Yin KJ, Deng Z, Hamblin M, et al. Vascular PPARδ protects against stroke induced brain injury, Arteriosclerosis, Thrombosis, and Vascular Biology 2011; 31/3:574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pulera MR, Adams LM, Liu H et al. Apoptosis in a neonatal rat model of cerebral hypoxia-ischemia. Stroke 1998; 29:2622–2630. [DOI] [PubMed] [Google Scholar]
- 14.Basu-Modak S, Braissant O, Escher P. Effect of the peroxisome proliferator-activated receptor β activator GW0742 in rat cultured cerebellar granule neurons, Journal of Neuroscience Research 2004; 77/2, pp. 240–249. [DOI] [PubMed] [Google Scholar]
- 15.Letavernier E, Perez J, Joye E, et al. Peroxisome proliferator-activated receptor beta/delta exerts a strong protection from ischemic acute renal failure. J Am Soc Nephrol 2005;16:2395–2402. [DOI] [PubMed] [Google Scholar]
- 16.Di Paola R, Esposito E, Mazzon E, et al. GW0742, a selective PPAR-beta/delta agonist, contributes to the resolution of inflammation after gut ischemia/reperfusion injury. J Leukoc Biol 2010;88:291–301. [DOI] [PubMed] [Google Scholar]
- 17.Chao X, Xiong C, Dong W, et al. Activation of peroxisome proliferator-activated receptor β/δ attenuates acute ischemic stroke on middle cerebral ischemia occlusion in rats. J Stroke Cerebrovasc Dis. 2014;23(6):1396–402. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Li R, Wang X, et al. Umbelliferone ameliorates cerebral ischemia–reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome, Neuroscience Letters 2015; (600):182–187 [DOI] [PubMed] [Google Scholar]
- 19.Nishiyama A, Matsui M, Iwata S, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. The Journal of Biological Chemistry 1999; 274 (31): 21645–50 [DOI] [PubMed] [Google Scholar]
- 20.Billiet L, Furman C, Cuaz-Pérolin C, et al. Thioredoxin-1 and its natural inhibitor, vitamin D3 up-regulated protein 1, are differentially regulated by PPAR alpha in human macrophages. J Mol Biol. 2008;384(3):564–76. [DOI] [PubMed] [Google Scholar]
- 21.Portius D, Sobolewski C, Foti M. MicroRNAs-Dependent Regulation of PPARs in Metabolic Diseases and Cancers. PPAR Res. 2017; 2017: 7058424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin KJ, Deng Z, Hamblin M, et al. Peroxisome proliferator-activated receptor delta regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J Neurosci. 2010;30(18):6398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dharap A, Pokrzywa C, Murali S, et al. Mutual induction of transcription factor PPARγ and microRNAs miR-145 and miR-329. J Neurochem. 2015;135(1): 139–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hong K, Xu G, Grayson TB, Shalev A. Cytokines Regulate β-Cell Thioredoxin-interacting Protein (TXNIP) via Distinct Mechanisms and Pathways. J Biol Chem. 2016. ;291 (16):8428–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong D, Fu N, Yang P. MiR-17 Downregulation by High Glucose Stabilizes Thioredoxin-Interacting Protein and Removes Thioredoxin Inhibition on ASK1 Leading to Apoptosis. Toxicol Sci. 2016;150(1):84–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice JE 3rd, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981. ;9:131–41. [DOI] [PubMed] [Google Scholar]
- 27.Lioutas VA, Alfaro-Martinez F, Bedoya F, et al. Intranasal Insulin and Insulin-Like Growth Factor 1 as Neuroprotectants in Acute Ischemic Stroke. Transl Stroke Res. 2015;6:264–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Q, Dasgupta C, Li Y, et al. Inhibition of microRNA-210 provides neuroprotection in hypoxic-ischemic brain injury in neonatal rats. Neurobiol Dis. 2016;89:202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi X, Xu L, Doycheva DM, et al. Sestrin2, as a negative feedback regulator of mTOR, provides neuroprotection by activation AMPK phosphorylation in neonatal hypoxic-ischemic encephalopathy in rat pups. . J Cereb Blood Flow Metab. 2017; 37(4):1447–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmued LC, Stowers CC, Scallet AC, et al. Fluoro-jade C results in ultrahigh resolution and contrast labeling of degenerating neurons. Brain Res 2005; 1035: 24–31. [DOI] [PubMed] [Google Scholar]
- 31.Ten VS, Bradley-Moore M, Gingrich JA, et al. Brain injury and neurofunctional deficit in neonatal mice with hypoxic-ischemic encephalopathy. Behav Brain Res 2003; 145: 209–219 [DOI] [PubMed] [Google Scholar]
- 32.Wachtel EV, Hendricks-Munoz KD. Current management of the infant who presents with neonatal encephalopathy. Curr Probl Pediatr Adolesc Health Care 2011; 41:132–153. [DOI] [PubMed] [Google Scholar]
- 33.Lu Q, Black SM. Neonatal Hypoxic-Ischemic Brain Injury: Apoptotic and Non-Apoptotic Cell Death. J Neurol Neuromed 2016; 1(4): 5–10 [Google Scholar]
- 34.Rossiter JP, Anderson LL, Yang F, Cole GM. Caspase-3 activation and caspase-like proteolytic activity in human perinatal hypoxic-ischemic brain injury. Acta neuropathologica 2002; 103:66–73. [DOI] [PubMed] [Google Scholar]
- 35.Ferriero DM, Miller SP. Imaging selective vulnerability in the developing nervous system. Journal of anatomy 2010; 217:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hattori I, Takagi Y, Nozaki K, et al. Hypoxia-ischemia induces thioredoxin expression and nitrotyrosine formation in new-born rat brain. Redox Rep. 2002;7(5):256–9. [DOI] [PubMed] [Google Scholar]
- 37.Chehaibi K, le Maire L, Bradoni S, et al. Effect of PPAR-β/δ agonist GW0742 treatment in the acute phase response and blood-brain barrier permeability following brain injury. Transl Res. 2017;182:27–48. [DOI] [PubMed] [Google Scholar]
- 38.Teng Z, Jiang L, Hu Q, et al. Peroxisome Proliferator-Activated Receptor β/δ Alleviates Early Brain Injury After Subarachnoid Hemorrhage in Rats. Stroke. 2016;47(1):196–205. [DOI] [PubMed] [Google Scholar]
- 39.Strange BA, Witter MP, Lein ES, Moser EI. Functional organization of the hippocampal longitudinal axis. Nat Rev Neurosci. 2014;15(10):655–69. [DOI] [PubMed] [Google Scholar]
- 40.Challet E, Denis I, Rochet V, et al. The role of PPARβ/δ in the regulation of glutamatergic signaling in the hamster suprachiasmatic nucleus. Cell Mol Life Sci. 2013;70(11):2003–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44(D1):D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ibabe A, Bilbao E, Cajaraville MP. Expression of peroxisome proliferator-activated receptors in zebrafish (Danio rerio) depending on gender and developmental stage. Histochem Cell Biol. 2005;123(1):75–87. [DOI] [PubMed] [Google Scholar]
- 43.Ibabe A, Grabenbauer M, Baumgart E, et al. Expression of peroxisome proliferator-activated receptors in the liver of gray mullet (Mugil cephalus). Acta Histochem. 2004. ;106(1):11–9. [DOI] [PubMed] [Google Scholar]
- 44.Mammalian Gene Collection Program, T. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proceedings of the National Academy of Sciences of the United States of America 99(26), 2002; 16899–16903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin CM, Tsai JT, Chang CK, et al. Development of telmisartan in the therapy of spinal cord injury: pre-clinical study in rats. Drug Des Devel Ther. 2015;9:4709–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsai CC, Lee KS, Chen SH, et al. Decrease of PPARδ in Type-1-Like Diabetic Rat for Higher Mortality after Spinal Cord Injury. PPAR Res. 2014:456386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lundell K, Thulin P, Hamsten A, et al. Alternative splicing of human peroxisome proliferator-activated receptor delta (PPAR delta): effects on translation efficiency and trans-activation ability. BMC Mol Biol. 2007;8:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saitoh M, Nishitoh H, Fujii M, et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17(9):2596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song J, Cheon SY, Lee WT, et al. The effect of ASK1 on vascular permeability and edema formation in cerebral ischemia. Brain Res. 2015;1595:143–55. [DOI] [PubMed] [Google Scholar]
- 50.Fang X, Fang L, Liu A, et al. Activation of PPAR-δ induces microRNA-100 and decreases the uptake of very low-density lipoprotein in endothelial cells. British Journal of Pharmacology 2015;172(15):3728–3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dellago H, Bobbili MR, Grillari J. MicroRNA-17-5p: At the Crossroads of Cancer and Aging - A Mini-Review. Gerontology 2017;63(1):20–28. [DOI] [PubMed] [Google Scholar]
- 52.Jadhav VS, Krause KH, Singh SK. HIV-1 Tat C modulates NOX2 and NOX4 expressions through miR-17 in a human microglial cell line. J Neurochem. 2014;131(6):803–15. [DOI] [PubMed] [Google Scholar]
- 53.Chen D, Dixon BJ, Doycheva DM, et al. IREIα inhibition decreased TXNIP/NLRP3 inflammasome activation through miR-17-5p after neonatal hypoxic-ischemic brain injury in rats. J Neuroinflammation. 2018;15(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haskova Z, Hoang B, Luo G, et al. Modulation of LPS induced pulmonary neutrophil infiltration and cytokine production by the selective PPAR β/δ ligand GW0742. Inflamm Res 2008;57:314–321. [DOI] [PubMed] [Google Scholar]
- 55.Kapoor A, Collino M, Castiglia S, et al. Activation of peroxisome proliferator-activated receptor-beta/delta attenuates myocardial ischemia/reperfusion injury in the rat. Shock 2010;34:117–124. [DOI] [PubMed] [Google Scholar]
- 56.Kilgore KS, Billin AN. PPARbeta/delta ligands as modulators of the inflammatory response. Curr Opin Investig Drugs 2008;9(5):463–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.