

SUMMARY
Sepsis is a life-threatening inflammatory syndrome accompanying a bloodstream infection. Frequently secondary to pathogenic bacterial infections, sepsis remains difficult to treat as a singular disease mechanism. We compared the pathogenesis of murine sepsis experimentally elicited by five bacterial pathogens and report similarities among host responses to Gram-negative Salmonella and E. coli. We observed that a host protective mechanism involving de-toxification of lipopolysaccharide by circulating alkaline phosphatase (AP) isozymes was incapacitated during sepsis caused by Salmonella or E. coli through activation of host Toll-like receptor 4, which triggered Neu1 and Neu3 neuraminidase induction. Elevated neuraminidase activity accelerated the molecular aging and clearance of AP isozymes, thereby intensifying disease. Mice deficient in the sialyltransferase ST3Gal6 displayed increased disease severity, while deficiency of the endocytic lectin hepatic Ashwell-Morell receptor was protective. AP augmentation or neuraminidase inhibition diminished inflammation and promoted host survival. This study illuminates distinct routes of sepsis pathogenesis, which may inform therapeutic development.
In Brief
Yang et al. develop a comparative protocol to identify mechanisms in the pathogenesis of experimental sepsis. Discrete Gram-negative pathogens elicit a TLR4-dependent host response that disrupts the homeostatic regulation of alkaline phosphatase (AP) isozymes through neuraminidase activation. AP augmentation and neuraminidase inhibition are therapeutic by maintaining the de-toxification of LPS-phosphate.
GRAPHICAL ABSTRACT
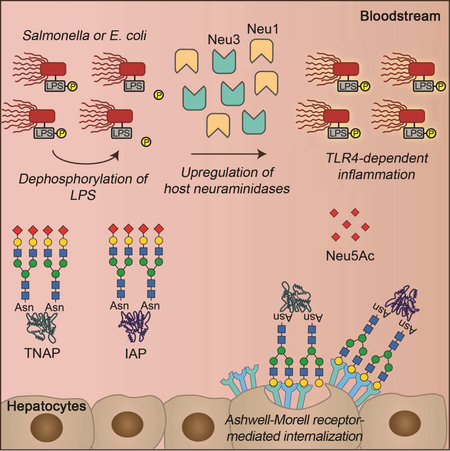
INTRODUCTION
Sepsis is a life-threatening bloodstream infection accompanied by pathological inflammation and organ system dysfunction. The leading cause of death in non-cardiac intensive care units, sepsis is increasing in incidence while no new effective therapies have been developed in decades (Fleischmann et al., 2016; Gaieski et al., 2013; Marshall, 2014; Orban et al., 2017; Stevenson et al., 2014). Among patients with severe sepsis or septic shock, mortality averages 25% with many survivors experiencing long term disabilities from tissue and organ damage caused by thrombosis, hypoperfusion, and hyperinflammation (Chang et al., 2010; Hawiger and Musser, 2011; Iwashyna et al., 2010; Stevenson et al., 2014). The development of more effective treatments for sepsis likely requires additional knowledge of host responses and pathogenic mechanisms activated at earlier stages of disease onset.
At later disease stages, sepsis can appear as a stereotypical disease process of uncontrolled inflammation and coagulopathy. Several identified host factors and cytokines drive inflammatory tissue and vascular injury, but their targeting has yet to lead to safe and effective therapies (Chaudhry et al., 2013; D’Elia et al., 2013; Schulte et al., 2013). Sentinel events in the pathogenesis of human sepsis are not easily identified, as diverse patient groups are infected at unknown times by pathogens that sometimes evade clinical detection. Gram-positive bacterial pathogens commonly identified in sepsis patients include Staphylococcus aureus (SA) and Streptococcus pneumoniae (SPN), while sepsis due to Gram-negative bacterial pathogens such as Escherichia coli (EC) and Salmonella spp. has increased in recent years accompanied by worrisome antibiotic resistance (Hartman et al., 2013; Vincent et al., 2009).
We developed a protocol for comparative studies of experimental sepsis in mice at early and later disease stages calibrated by specific post-infection times and blood pathogen colony-forming unit (cfu) thresholds. This approach provided a reproducible data platform from which cross-comparisons were made using multiple Gram-positive and Gram-negative bacterial pathogens and different routes of infection. Our analysis revealed a common mechanism of host protection unique to the Gram-negative infections studied, involving the regulated half-lives of circulating enzymes capable of detoxifying bacterial lipopolysaccharide (LPS). However, this host-protective mechanism stands at risk of pathogen subversion, resulting in severe inflammation and increased mortality in established sepsis. Efforts to bolster this protective mechanism reduced disease pathophysiology and mortality, suggesting a pharmacological target in a subset of infections to counteract the pathogenesis of sepsis at its early stages.
RESULTS
Acquired and selective deficiency of alkaline phosphatase in the pathogenesis of murine sepsis
Experimental analysis of bacterial sepsis was undertaken in the murine model, where genomic signatures of inflammatory responses were recently shown to closely correlate to those of humans (Takao and Miyakawa, 2015). Although cecal ligation and puncture (CLP) is the most frequently used experimental sepsis model, we chose not to use CLP for multiple reasons. CLP generates polymicrobial infections, which represent a small subset of human sepsis cases, typically much less than 10% among populations surveyed (Lin et al., 2010; Pammi et al., 2014). CLP can also generate variable results depending on ligation length, quantity and quality of intestinal perforations, and different numbers of Gram-negative and Gram-positive bacteria released from the lumen of the intestine (Dejager et al., 2011; Singleton and Wischmeyer, 2003). Sepsis resulting from CLP can evolve to become either Gram-positive or Gram-negative, while the precise identification and titers of the pathogens involved are rarely possible to obtain and monitor. We therefore compared host response patterns and disease outcomes separately among five different bacterial pathogen isolates identified from invasive human infections. These included two common Gram-negative bacterial pathogens, EC and Salmonella enterica Typhimurium (ST), and two preeminent Gram-positive bacterial pathogens, SPN and SA; for the latter, both methicillin-sensitive (MSSA) and methicillin-resistant (MRSA) strains were tested. For all bacterial strains, disease severity and mortality were directly proportional to increased bacterial cfu in the bloodstream. We thus applied criteria for data inclusion in comparative sepsis pathophysiology analyses involving attainment of minimum and maximum thresholds of blood cfu at specified times post-infection.
Sepsis caused by the Gram-negative bacterial pathogens EC and ST was linked to an unexpected reduction of alkaline phosphatase (AP) activity measured at approximately 50% of normal by blood chemistry analyses. This reduction was associated with similarly decreased abundance of both tissue nonspecific alkaline phosphatase (TNAP) and intestinal alkaline phosphatase (IAP) (Figures 1A–1D). TNAP has been found to control bone mineralization and IAP functions in fat absorption through the intestinal epithelium as well as protection against colitis (Narisawa et al., 1997 and 2003; Parlato et al., 2018; Yang et al., 2017). TNAP is produced by a variety of cell types including those in bone, liver, and kidney, while IAP is secreted exclusively by enterocytes of the small intestine (Millán, 2006). Alkaline phosphatase isozymes further include Placental alkaline phosphatase (PLAP) and Germ cell alkaline phosphatase (GCLP) in human and Embryonic alkaline phosphatase (EAP) in mouse (Millán, 2006); however, these other isozymes are not found in the blood stream and were not analyzed in our studies. In contrast to sepsis caused by EC or ST pathogens, there was no change to blood AP activity and AP isozyme abundance in sepsis resulting from infections of Gram-positive Streptococcal or Staphylococcal bacterial pathogens SPN or MRSA (Figures 1E–1H); as well as the related Staphylococcal isolate MSSA (Figures S1A and S1B).
Figure 1. Pathogen-selective reduction of host anti-inflammatory alkaline phosphatase.
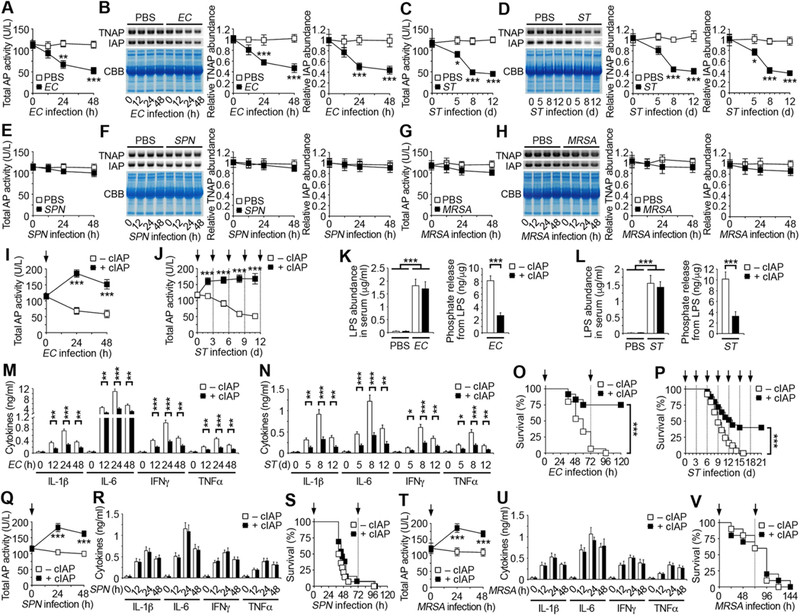
(A, C, E, G) Total alkaline phosphatase (AP) activity measured in wild-type C57BL/6 (WT) mouse serum after intraperitoneal (i.p.) infection with EC (107 cfu), oral infection with ST (107 cfu), i.p. infection with SPN (104 cfu), or intravenous (i.v.) infection with MRSA (108 cfu).
(B, D, F, H) Identical amounts of WT mouse serum protein (20 µg) were separated by SDS-PAGE and analyzed by protein staining with Coomassie brilliant blue (CBB) or by Western blotting with TNAP– and IAP–specific antibodies after infection with EC, ST, SPN, or MRSA. Quantification of the relative abundance of each AP isoform is plotted.
(I, J, Q, T) Serum AP activity of WT mice receiving i.v. injections of cIAP (75 U/kg) at indicated times (arrows) following infection with EC, ST, SPN, or MRSA.
(K and L) LPS abundance and phosphate amount released from LPS in serum of WT mice 24 h after EC infection or 8 d after ST infection in the presence or absence of cIAP.
(M, N, R, U) Serum inflammatory cytokine expression of WT mice receiving i.v. injections of cIAP following infection with EC, ST, SPN, or MRSA.
(O, P, S, V) Survival of WT mice receiving i.v. injections of cIAP at indicated times (arrows) following infection with EC, ST, SPN, or MRSA.
(A–N, Q, R, T, U) n = 6 per condition. (O and S) n = 12–15 per condition. (P) n = 25 per condition. (V) n = 10 per condition. Data are presented as means ± SEM from two independent experiments.
The impact of reduced AP levels in the host during sepsis caused by EC or ST infections was probed by intravenous (i.v.) pharmacological restoration of AP activity using calf IAP (cIAP). AP activity is known to de-toxify the Gram-negative bacterial endotoxin lipopolysaccharide (LPS) through de-phosphorylation of the lipid A moiety (Bates et al., 2007; Beumer et al., 2003; Koyama et al., 2002; Poelstra et al., 1997; Tuin et al., 2006). The toxic form of LPS contains two phosphate groups coupled to glucosamines; removal of a single phosphate group by AP activity is sufficient to generate a monophosphoryl lipid A that is a 100-fold less toxic than fully phosphorylated LPS (Bentala et al., 2002; Park et al., 2009; Schromm et al., 1998). Both TNAP and IAP can de-phosphorylate and de-toxify LPS (Pettengill et al., 2017); however, the specific phosphate(s) hydrolyzed are not currently defined. We measured LPS-phosphate levels using the malachite green phosphate assay and compared findings to total LPS in the contexts of AP reduction and cIAP treatment. Our findings revealed that decreased LPS-phosphate was linked to increased AP activity provided by either endogenous or exogenous sources (Figures 1I–1L). cIAP administration further reduced blood inflammatory cytokine levels and markedly improved mouse survival following infection with the EC and ST pathogens (Figures 1M–1P). In contrast, cIAP treatment did not alter inflammatory cytokine expression or frequencies of mortality during sepsis caused by Gram-positive SPN, MRSA, or MSSA pathogens (Figures 1Q–1V; Figures S1C–S1E). The reduction of AP activity was not dependent upon the route of infection as ST infection elicited by direct intraperitoneal (i.p.) infection resulted in a more rapid disease course than orogastric challenge while yielding similar but earlier reductions of AP levels requiring more rapid cIAP replacement therapy (Figures S1F–S1K).
Accelerated molecular aging of AP isozymes and clearance by the Ashwell-Morell Receptor
Given the importance of AP abundance and activity in protecting the host during sepsis caused by Gram-negative EC and ST pathogens, we sought to identify the mechanism(s) governing TNAP and IAP regulation. Reductions of TNAP and IAP protein abundance occurred without diminished mRNA levels in tissues known to produce these isozymes (Figures S2A and S2B). Rather, the half-lives of both TNAP and IAP in blood circulation were significantly reduced (Figures 2A and 2B). By contrast, TNAP and IAP half-lives were unaltered in sepsis caused by SPN and MRSA (Figures 2C and 2D) consistent with retention of normal AP activity. TNAP and IAP are glycoprotein enzymes synthesized in the secretory pathway of cells and glycosylated prior to secretion (Millán, 2006). Sepsis caused b y EC and ST reduced MAL-II lectin binding to a subset of total sialic acids, coincident with increased exposure of underlying endocytic galactose linkages detected by ECA and RCA lectins (Figures 2E and 2F). No changes involving a subset of α2–6-linked sialic acids or core 1 O-glycan sialylation were detected using the SNA and PNA lectins, respectively. Neither TNAP nor IAP underwent similar glycan remodeling during sepsis caused by SPN or MRSA (Figures S2C and S2D) with retention of normal AP isozyme half-lives and abundance.
Figure 2. Accelerated aging and turnover of AP and the role of the AMR in maintaining AP levels.

(A–D) Half-life analyses of TNAP and IAP proteins in circulation of WT mouse serum 24 h after i.p. infection with EC (107 cfu), day 8 after oral infection with ST (107 cfu), 24 h after i.p. infection with SPN (104 cfu), 24 h after i.v. infection with MRSA (108 cfu) following biotinylation.
(E and F) Lectin blotting analyses are presented from identical amounts of TNAP and IAP isolated from WT mouse serum after i.p. infection with EC (107 cfu) or oral infection with ST (107 cfu).
(G and H) Total AP activity measured in AMR-deficient mouse serum after i.p. infection with EC or oral infection with ST. AMR is encoded by the Asgr1 and Asgr2 genes.
(I and J) Half-life analyses of TNAP and IAP proteins in circulation of AMR-deficient mouse serum 24 h after infection with EC or day 8 after infection with ST following biotinylation.
(K and L) TNAP and IAP abundance in AMR-deficient mouse serum after i.p. infection with EC or oral infection with ST.
(M and N) LPS abundance and phosphate amount released from LPS in serum of AMR-deficient mice 24 h after EC infection or day 8 after ST infection.
(O and P) Serum inflammatory cytokine expression in AMR-deficient mouse serum following i.p. infection with EC or oral infection with ST.
(Q and R) Survival of AMR-deficient mice following i.p. infection with EC or oral infection with ST.
(A–D, I, J) n = 8 per condition. (E–H, K–P) n = 6 per condition. (Q) n = 20 per condition. (R) n = 40 per condition. Data are presented as means ± SEM from two independent experiments.
We suspected that one or more host endocytic lectin receptor(s), likely to include the hepatic Ashwell-Morell receptor (AMR) (Ashwell and Morell, 1974), may be involved in the accelerated clearance of de-sialylated TNAP and IAP enzymes in the context of sepsis. In normal uninfected animals, the multimeric AMR binds to de-sialylated multivalent glycan ligands bearing terminal galactose linkages, including those on TNAP and IAP, thereby regulating their half-lives and abundance by this homeostatic and clearance mechanism (Yang et al., 2015). We observed that mice with a genetic deficiency of either the Asgr1 or Asgr2 chain of the AMR maintained elevated circulating AP activity and half-lives even during sepsis caused by EC or ST (Figures 2G–2J) and which was linked to the elevated abundance of circulating TNAP and IAP (Figures 2K and 2L). In the normal liver parenchyma, TNAP and IAP are mostly co-localized with Asgr1 and Asgr2 chains of the AMR, while their abundance in the liver is increased in sepsis caused by ST infection as well as being diminished in the absence of either AMR chain (Figure S3). This is consistent with the increased half-lives and abundance of de-sialylated TNAP and IAP isozymes circulating in AMR deficiency.
Elevated AP isozymes in AMR deficiency protect the host during sepsis caused by Gram-negative EC and ST pathogens
Elevated bloodstream AP activity in AMR deficiency increased the ratio of de-phosphorylated to phosphorylated LPS during EC or ST sepsis concurrent with reduced inflammatory cytokine levels in circulation (Figures 2M–2P). In stark contrast to a protective role for host AMR function in SPN sepsis (Grewal et al., 2008 and 2013), AMR deficiency reduced animal mortality in sepsis caused by EC or ST (Figures 2Q and 2R). Selective inhibition of TNAP activity using the pharmacological inhibitor SBI-425 (Dahl et al., 2009; Pinkerton et al., 2018; Sheen et al., 2015) reduced total serum AP activity levels by over 70% on average among wild-type mice and their Asgr-null littermates, consistent with higher TNAP levels in circulation compared with IAP (Figures 3A and 3B). In sepsis caused by EC or ST pathogens, SBI-425 treatment of AMR-deficient mice similarly increased disease signs in measurements of LPS-phosphate levels, inflammation markers, and frequencies of survival (Figures 3C–3H), indicating a strong contribution of the TNAP isozyme to host protection. Among WT mice, there was a trend of increased disease signs measured with TNAP inhibition that did not acquire statistical significance. This likely reflects an overlapping role of IAP in normal host protection, which although expressed at much lower levels has a higher specific activity towards LPS (Kiffer-Moreira et al., 2014). We investigated further using Akp3-null mice (Narisawa et al., 2003) and observed the greatest loss of AP activity in IAP deficiency occurred in the presence of SBI-425, which combined to promote disease and mortality to an extent that Asgr-null mice were unable to survive sepsis caused by the ST pathogen (Figure 3I–3L). AP activity levels among normal uninfected mice and their persistent elevation obtained by AMR deficiency conferred host protection in our sepsis protocols, while further AP augmentation among Asgr-null mice resulted in marginal improvement (Figures 3M and 3N). In sepsis caused by SPN or MRSA, AMR deficiency had no protective effect and instead appeared to shorten survival times similar to that reported previously using SPN (Grewal et al., 2008), suggesting that further studies may find AMR function to be protective to the host in Staphylococcal sepsis (Figures 3O and 3P). These findings together show that elevated AP levels in AMR deficiency are protective in sepsis caused by Gram–negative EC and ST, but not in sepsis caused by Gram–positive SPN or MRSA, and that both TNAP and IAP enzymes have host–protective roles in the former context.
Figure 3. Effects of AP inhibition in AMR deficiency and sepsis.
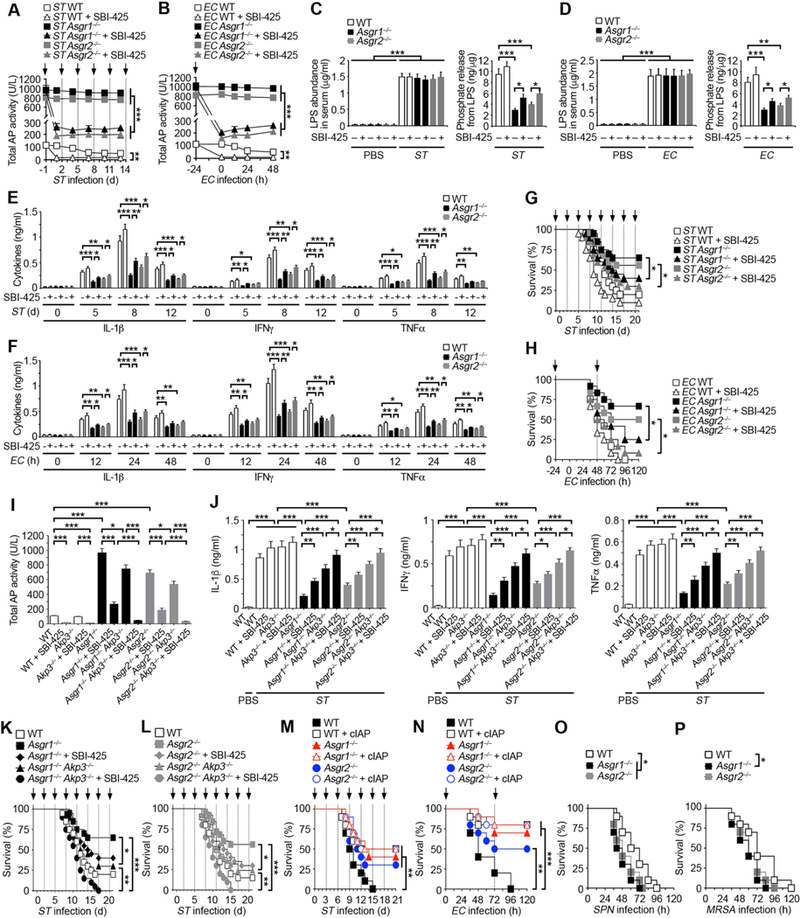
(A and B) Total AP activity measured in the serum of indicated genotypes after oral infection with ST (107 cfu) or i.p. infection with EC (107 cfu) in the absence and presence of TNAP inhibitor SBI-425 (10 mg/kg) at indicated times (arrows).
(C and D) LPS abundance and phosphate amount released from LPS in serum of indicated genotypes 8 d after ST infection or 24 h after EC infection in the presence or absence of SBI-425.
(E and F) Serum inflammatory cytokine expression of indicated genotypes after oral infection with ST (107 cfu) or i.p. infection with EC (107 cfu) in the absence and presence of SBI-425.
(G) Survival of indicated genotypes following oral infection with ST (106 cfu) or i.p. infection with EC (107 cfu) in the absence and presence of SBI-425 (arrows).
(I) Total AP activity measured in the serum of indicated genotypes 24 h after i.p. injection with SBI-425.
(J) Serum inflammatory cytokine expression of indicated genotypes 8 d after oral infection with ST (107 cfu) in the absence and presence of SBI-425.
(K and L) Survival of indicated genotypes following oral infection with ST (106 cfu) in the absence and presence of SBI-425 at indicated times (arrows).
(M and N) Survival of AMR-deficient mice receiving i.v. injections of cIAP (75 U/kg) at indicated times (arrows) following oral infection with ST (107 cfu) or i.p. infection with EC (107 cfu).
(O and P) Survival of AMR-deficient mice following i.p. infection with SPN (104 cfu) or i.v. infection with MRSA (108 cfu).
(A–F) n = 6 per condition. (G, H, K, L) n = 20–25 per condition. (I) n = 10 per condition. (J) n = 8 per condition. (M–P) n = 10–12 per condition. Data are presented as means ± SEM from two independent experiments.
ST3Gal6 maintains AP abundance in providing host protection
Sialylation of TNAP and IAP occurs during their transit through the secretory pathway. Secreted proteins in the blood lose sialic acid linkages at distinct and measurable rates as they age in the presence of neuraminidase activity, resulting in their endocytic clearance by lectins such as the AMR (Yang et al., 2015). Among various sialyltransferase-deficient mice studied, those lacking the ST3Gal6 sialyltransferase had diminished blood AP activity with corresponding deficits of TNAP and IAP abundance (Figures 4A and 4B), while TNAP or IAP mRNA levels were unaltered (Figure S4A). Reduced AP activity in ST3Gal6 deficiency was further linked to a reduction of sialic acid linkages on TNAP and IAP, coincident with increased exposure of underlying galactose linkages and diminished half-lives of both isozymes (Figures 4C and 4D).
Figure 4. Role of AP sialylation by ST3Gal6 sialyltransferase in isozyme homeostasis.

(A and B) Total AP activity and TNAP and IAP abundance measured in ST3Gal6-deficient mouse serum. ST3Gal6 is encoded by the St3gal6 gene.
(C) Lectin blotting analyses are presented from identical amounts of TNAP and IAP isolated from sera of ST3Gal6-deficient mice.
(D) Half-life analyses of TNAP and IAP glycoproteins in circulation of ST3Gal6-deficient mouse serum following biotinylation.
(E–H) Total AP activity and TNAP and IAP abundance measured in ST3Gal6-deficient mouse serum after i.p. infection with EC (107 cfu) or oral infection with ST (107 cfu).
(I and J) Lectin binding on TNAP and IAP proteins isolated from ST3Gal6-deficient mouse serum after infection with EC (107 cfu) or ST (107 cfu).
(K and L) Half-life analyses of TNAP and IAP glycoproteins in circulation of ST3Gal6-deficient mouse serum 24 h after infection with EC (107 cfu) or day 8 after infection with ST (107 cfu) following biotinylation.
(M and N) Serum AP activity of ST3Gal6-deficient mice receiving i.v. injections of cIAP (75 U/kg) at indicated times (arrows) following infection with EC (107 cfu) or ST (107 cfu).
(O and P) LPS abundance and phosphate amount released from LPS in serum of ST3Gal6-deficient mice 24 h after EC infection (107 cfu) or day 8 after ST infection (107 cfu) in the presence or absence of cIAP.
(Q and R) Serum inflammatory cytokine expression in ST3Gal6-deficient mouse serum following infection with EC (107 cfu) or ST (107 cfu).
(S and T) Survival of ST3Gal6-deficient mice receiving i.v. injections of cIAP at indicated times (arrows) following infection with EC (107 cfu) or ST (5 × 10 5 cfu).
(A, C, E–J, M–R) n = 6 per condition. (B, D, K, L) n = 8 per condition. (S) n = 12 per condition. (T) n = 20 per condition. Data are presented as means ± SEM from two independent experiments.
Sepsis caused by EC or ST infection further diminished AP activity and TNAP and IAP abundance in ST3Gal6 deficiency, wherein glycan remodeling evidenced by diminished sialic acid linkages and unmasked galactose linkages was further amplified (Figures 4E–4J). Moreover, ST3Gal6 deficiency contributed to marked reductions of TNAP and IAP half-lives during sepsis (Figures 4K and 4L). Augmentation with cIAP boosted blood AP activity levels among St3gal6-null mice and lowered the phosphorylation level of LPS during sepsis with reduced inflammatory cytokine levels and improved host survival (Figures 4M–4T). In contrast, frequencies of death were unaltered among St3gal6-null mice in sepsis caused by Gram-positive SPN or SA (Figures S4B and S4C). The opposing effects of ST3Gal6 (glycoprotein sialylation) and the AMR (endocytic clearance of de-sialylated glycoproteins) were further investigated in mice genetically deficient in both components. The AMR effect was dominant as expected with continued elevation of AP activity and increased abundance of TNAP and IAP bearing diminished sialic acid linkages in ST3Gal6 co-deficiency, coincident with increased frequencies of host survival (Figures S4D–S4I). These findings indicated an interaction between ST3Gal6 and the AMR in controlling AP homeostasis and host protection in sepsis caused by Gram-negative EC and ST pathogens. How EC and ST trigger a reduction of sialic acid linkages on circulating TNAP and IAP in compromising this mechanism of host protection was next investigated.
Toll-like receptor-4 (TLR4)-dependent induction of host neuraminidases in sepsis caused by Gram-negative EC and ST pathogens
Neuraminidase (Neu) activity cleaves sialic acid linkages thereby exposing underlying galactose ligands of the AMR among host glycoproteins. Some neuraminidases are encoded by pathogens as virulence factors; however, no neuraminidases reside in the genomes of the EC and ST isolates used herein (Fueroa-Bossi et al., 2001; Russo et al., 1990; Vimr and Troy, 1985). The de-siaylation of TNAP and IAP during sepsis therefore implied the presence and induction of host Neu activity with consequent decreased AP isozyme abundance leading to increases of LPS-phosphate and resulting Toll-like receptor-4 (TLR4) activation of innate immune responses causing inflammation (Beutler, 2000; Poltorak et al., 1998).
We detected the presence of a TLR4-dependent mechanism that elevated blood Neu activity during sepsis caused by ST and EC (Figures 5A and 5B). Among the four Neu isozymes encoded by mammalian genomes (Neu1–4), only Neu1 and Neu3 have been detected in blood circulation (Yang et al., 2015). Both Neu1 and Neu3 are widely expressed among cell types and may be secreted into circulation by multiple mechanisms including lysosomal, exosomal, or proteolytic mechanisms. In sepsis caused by EC or ST infections, the expression of both Neu1 and Neu3 in circulation were induced in a TLR4 dependent manner (Figures 5C and 5D). In contrast, elevated blood Neu activity detected in SPN sepsis was independent of TLR4 function and occurred without changes in the abundance of Neu1 and Neu3, consistent with expression of the pathogen-encoded neuraminidase, NanA (Grewal et al., 2008) (Figures 5E and 5F). No changes were found in overall Neu activity or the abundance of circulating Neu1 and Neu3 in similar studies with MRSA (ikkj ). TLR4-dependent reductions of AP activity and abundance in sepsis caused by Gram-negative EC and ST pathogens were linked to diminished TNAP and IAP half-lives in circulation, concurrent with reductions of sialic acid linkages and exposure of underlying galactose ligands (Figures 5I and 5J; Figures S5A– S5D). TLR4 deficiency was further associated with reductions of both phosphate levels on circulating LPS and inflammatory cytokine expression (Figures 5K–5N). Moreover, the frequency of survival was significantly increased in the absence of TLR4 function (Figures 5O and 5P) while cIAP treatment of the TLR4-deficient mice had no additional protective effect (Figures S5E and S5F). These findings reveal a TLR4-dependent mechanism that elevates host Neu1 and Neu3 levels in circulation during sepsis caused by Gram-negative EC or ST infections, consequently diminishing AP activity, reducing TNAP and IAP abundance, promoting inflammation, and increasing mortality.
Figure 5. Inductions of host neuraminidases by TLR4 and LPS control AP sialylation and abundance in the pathophysiology of sepsis.
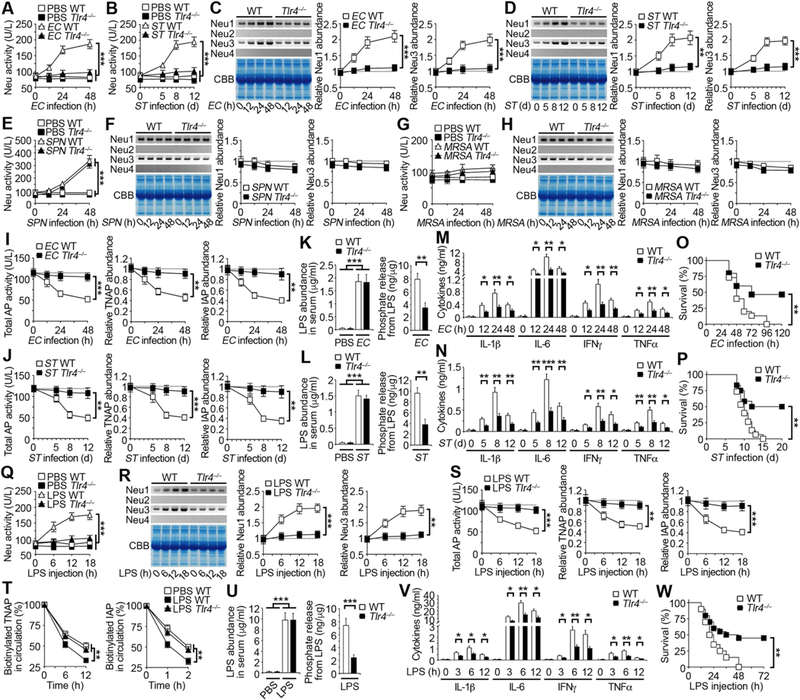
(A, B, E, G) Total neuraminidase (Neu) activity measured in TLR4-deficient mouse serum after i.p. infection with EC (107 cfu), oral infection with ST (107 cfu), i.p. infection with SPN (104 cfu), or i.v. infection with MRSA (108 cfu).
(C, D, F, H) Antibodies specific to Neu1, Neu2, Neu3, or Neu4 were used to detect Neu protein abundance in TLR4-deficient mouse serum after infection with EC, ST, SPN, or MRSA by Western blotting. Quantification of the relative abundance of each protein sample is plotted. (I and J) Total AP activity and TNAP and IAP abundance measured in TLR4-deficient mouse serum after infection with EC or ST. (K and L) LPS abundance and phosphate amount released from LPS in serum of TLR4-deficient mice on 24 h after EC infection or day 8 after ST infection. (M and N) Serum inflammatory cytokine expression in TLR4-deficient mouse serum following infection with EC or ST. (O and P) Survival of TLR4-deficient mice following infection with EC or ST. (Q and R) Total Neu activity and Neu1, 2, 3 and 4 abundance measured in TLR4-deficient mouse serum after i.p. injection with LPS (40 mg/kg; EC 0111:B4).
(S) Total AP activity and TNAP and IAP abundance in TLR4-deficient mouse serum after i.p. injection with LPS.
(T) Half-life analyses of TNAP and IAP glycoproteins in circulation of TLR4-deficient mouse serum 12 h after i.p. injection with LPS following biotinylation.
(U) LPS abundance and phosphate amount released from LPS in serum of TLR4-deficient mice 12 h after LPS injection.
(V) Serum inflammatory cytokine expression of TLR4-deficient mice following i.p. injection with LPS.
(W) Survival of TLR4-deficient mice following i.p. injection with LPS.
(A–N, Q–S, U, V) n = 6 per condition. (O and P) n = 12–15 per condition. (T) n = 8 per condition. (W) n = 20 per condition. Data are presented as means ± SEM from two independent experiments.
LPS recapitulates host TLR4-dependent Neu induction in provoking pathogenesis
The suspected role of LPS produced by Gram-negative EC and ST pathogens in TLR4-dependent induction of host Neu1 and Neu3 was probed. Intraperitoneal administration of E. coli LPS obtained from commercial sources resulted in elevated Neu activity in circulation in a TLR4-dependent manner coincident with increased abundance of Neu1 and Neu3 (Figures 5Q and 5R). LPS administration reduced circulating AP activity and diminished TNAP and IAP levels (Figure 5S) without altering TNAP or IAP transcript abundance, while instead diminishing half-lives of AP isozymes in circulation with exposure of underlying endocytic galactose linkages (Figure 5T; Figures S6A–S6C). Phosphorylated LPS and inflammatory cytokine expression were concurrently increased in the presence of TLR4 function, while TLR4 deficiency resulting in reduced LPS-phosphate levels, diminished inflammatory cytokine induction, and increased frequencies of host survival (Figures 5U–5W). Augmentation of AP activity by cIAP treatment was further therapeutic in the LPS challenge model (Figures S6D–S6G). Similarly, elevated AP levels in AMR deficiency lessened susceptibility to LPS with reduced LPS–phosphate, diminished inflammatory cytokine expression, and improved host survival (Figures S6H–S6L). Correspondingly, ST3Gal6 deficiency increased susceptibility to LPS toxicity, with further reductions of AP activity and isozyme half-lives resulting in increased LPS–phosphate, elevated inflammatory cytokine expression, and reduced host survival, all of which were modulated as expected by cIAP treatment or by AMR co–deficiency (Figures S6M–S6S).
Neu inhibition sustains AP function and limits LPS-mediated pathology in sepsis
The induction of host Neu activity by LPS and TLR4 was linked to increased inflammatory cytokine expression and mortality, implying that Neu inhibitors may be of therapeutic value in sepsis caused by the Gram-negative EC and ST pathogens. We investigated whether Neu inhibitors representing anti-viral compounds and marketed drugs including 2,3-dehydro-2-deoxy-N-acetylneuraminic acid (DANA) and Zanamivir (tradename Relenza) could maintain AP levels and thereby provide host protection.
DANA or Zanamivir were administered immediately following EC or ST infections and every 24 h thereafter. Marked inhibition of blood Neu activity was achieved with either inhibitor, resulting in circulating Neu activity levels comparable to uninfected animals, and which coincided with retentions of basal AP activity, TNAP and IAP abundance, and sialic acid linkages (Figures 6A–6H). Treatment with DANA or Zanamivir also lowered circulating levels of LPS–phosphate during sepsis, blunting inflammatory cytokine expression and markedly improving host survival (Figures 6I–6N). Notably, inhibition of circulating Neu activity during SPN or MRSA sepsis did not significantly affect disease markers including AP levels, inflammatory cytokines, and survival (Figures 6O–6R). Similar studies of Neu inhibition in LPS administration further revealed therapeutic effects linked to the retention of sialic acid linkages, circulating AP levels, LPS de-phosphorylation, reduced inflammatory cytokines, and increased host survival (Figure S7). These findings together indicate the presence of a mechanism of host protection against sepsis caused by Gram-negative EC and ST pathogens that is dysregulated by signals linked to TLR4 function and which can be reinforced by AP augmentation or Neu inhibition.
Figure 6. Effects of neuraminidase inhibitors in sepsis.
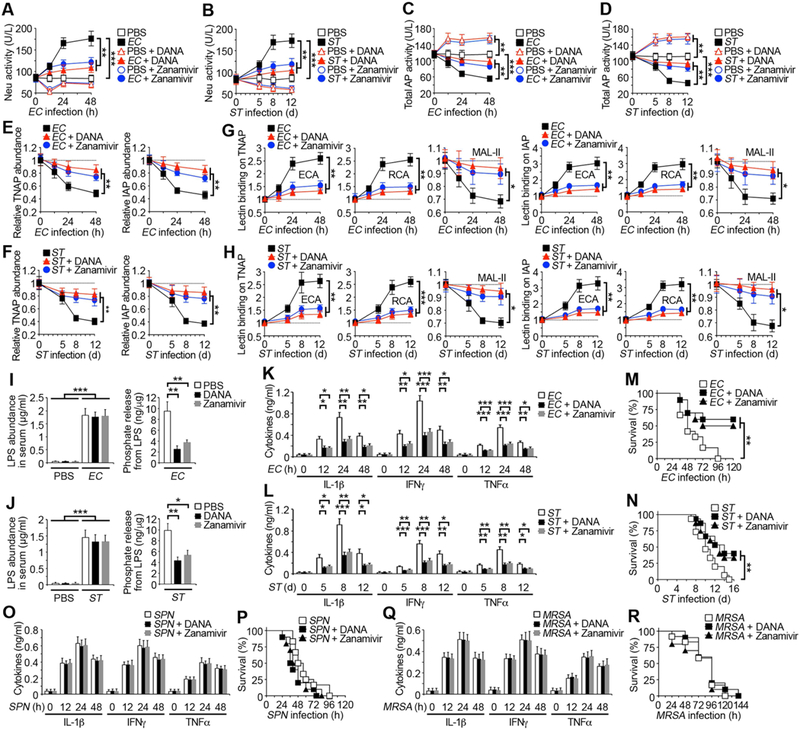
(A and B) Total Neu activity measured in WT mouse serum after i.p. infection with EC (107 cfu) or oral infection with ST (107 cfu) in the absence or presence of the broad-spectrum neuraminidase inhibitor DANA (2,3-dehydro-2-deoxy-N-acetylneuraminic acid, 250 mg/kg, every 24 h) or Zanamivir (250 mg/kg, every 24 h).
(C–F) Total AP activity and TNAP and IAP abundance measured in WT mouse serum after infection with EC or ST in the absence or presence of DANA or Zanamivir.
(G and H) Lectin binding analyses of TNAP and IAP isolated from WT mouse serum after infection with EC or ST in the absence or presence of DANA or Zanamivir.
(I and J) LPS abundance and phosphate amount released from LPS in serum of WT mice 24 h after EC infection or day 8 after ST infection in the presence or absence of DANA or Zanamivir.
(K, L, O, Q) Serum inflammatory cytokine expression in WT mouse serum following infection with EC, ST, SPN, or MRSA in the absence or presence of DANA or Zanamivir.
(M, N, P, R) Survival of WT mice following infection with EC, ST, SPN, or MRSA in the absence or presence of DANA or Zanamivir.
(A–L, O, Q) n = 6 per condition. (M, P, R) n = 12 per condition. (N) n = 15 per condition. Data are presented as means ± SEM from two independent experiments.
DISCUSSION
Originating from infections caused by disparate agents including bacteria, fungi, and viruses, host responses in sepsis commonly elicit excessive inflammation and coagulopathy that determine pathological severity and mortality (Stearns-Kurosawa et al., 2011). Commonalities identified in the pathways driving tissue injury and organ dysfunction have not precluded the potential for distinct pathogenic mechanisms operating at the onset of host responses to specific microbes, the identification of which may provide stratification to guide individualized therapeutic interventions prior to the manifestation of severe disease. From our comparative studies of distinct Gram-positive bacterial pathogens such as SPN and SA and Gram-negative bacterial pathogens such as EC and ST, we have identified a selective mechanism of host protection against LPS toxicity that is targeted by Gram-negative EC and ST pathogens. EC is a common cause of human sepsis worldwide, with increasing problems of antibiotic resistance (Poolman and Wacker, 2016) while nontyphoidal ST is a leading cause of bacteremia in sub-Saharan Africa with an associated case mortality of 25% (de Jong et al., 2012; Feasey et al., 2012). In more developed countries recurrent ST infections are a leading cause of food poisoning and may increase the potential for the development of colitis (Yang et al., 2017). In the present study, we found that ST and EC pathogens selectively target and disrupt a mechanism of host protection by activating TLR4 to increase circulating levels of Neu1 and Neu3, implicating similar pathogenic mechanisms as operating in sepsis and colitis. Elevated Neu activity accelerated the molecular aging and endocytic clearance of circulating anti-inflammatory isozymes TNAP and IAP by the hepatic AMR, diminishing the host capacity for LPS de-phosphorylation and de-toxification, and thereby promoting TLR4-dependent inflammation with reduced likelihood of host survival.
Using pharmacological and genetic approaches to discriminate among AP isozyme function, we determined that both TNAP and IAP contributed substantially to host protection against LPS toxicity in sepsis. Failure of the host to establish or maintain post-translational control of AP isozyme abundance in the blood had severe consequences and could also manifest by ST3Gal6 sialyltransferase deficiency with failure to adequately sialylate AP isozymes during isozyme synthesis, while in contrast, AMR deficiency was strongly protective by maintaining high levels of AP isozymes that would normally have been cleared following de-sialylation with increased molecular age. Notably, this contrasts with AMR function in providing host protection during SPN sepsis (Grewal et al., 2008 and 2013), and together placing the AMR at a nexus of host response pathways influencing the outcomes of sepsis.
Differences detected in responses of the host to sepsis caused by Gram-positive SPN and SA pathogens compared with Gram-negative EC and ST pathogens centered in this study on the regulation of circulating host neuraminidases. The functions of mammalian neuraminidases and other glycosidases present in the bloodstream are under increased scrutiny and have been recently shown to contribute to an intrinsic mechanism of protein aging involving rates of de-sialylation that generate endocytic lectin ligands in determining protein half-lives, abundance, and thereby function (Yang et al., 2015 and 2017). In previous studies of the host response to Gram-positive SPN sepsis, where de-toxification of LPS was not a consideration, the de-sialylation of platelets by the SPN NanA neuraminidase benefitted the host in the context of sepsis. NanA-mediated platelet de-sialylation was detected by the host, resulting in AMR-dependent platelet clearance producing an essential moderate thrombocytopenia that moderated an otherwise invariably lethal form of disseminated intravascular coagulation (Grewal et al., 2008 and 2013). Neither of the Gram-positive SA strains studied herein, which both lack neuraminidase genes, caused an appreciable change in circulating Neu activity in the host. Moreover, AP augmentation had no untoward effects to the host during sepsis caused by Gram-positive SPN and SA pathogens studied. In contrast, the increase in circulating Neu1 and Neu3 caused by Gram-negative ST and EC pathogens in the presence of host TLR4 accelerated the rate of TNAP and IAP de-sialylation increasing AMR-dependent clearance, impairing LPS de-toxification, and thereby elevating TLR4-dependent inflammation. The de-phosphorylation of LPS by AP activity was only therapeutic in the context of TLR4 function and thus AP was beneficial to WT but not TLR4-deficient hosts. Our results including maximal survival frequencies in these protocols support the view that there remain additional unidentified mechanisms unrelated to AP modulation that are also involved in the pathogenesis of Gram–negative sepsis.
Discoveries of dissimilar biomarkers elicited early in host responses to different pathogens may begin to stratify sepsis patients for earlier targeted treatment approaches that block the emergence of common downstream pathologies spanning hyperinflammation, thrombosis, and hypoperfusion. The clinical utility of unique pathogenic biomarkers will likely depend upon advances in patient fluid culture-independent diagnostic modalities to confirm pathogen locations and identities early in the presentation of sepsis. We have identified a post-translational mechanism of AP isozyme regulation that is targeted by Gram-negative EC and ST bacterial pathogens early in the onset and progression of sepsis. The resulting disruption of host protection by acquired AP deficiency increased host sensitivity to the bacterial LPS toxin through TLR4-mediated inflammation. Therapeutic intervention to maintain normal AP abundance by AP augmentation or by Neu inhibition were each highly effective in suppressing TLR4-dependent inflammation and reducing mortality. Clinical trials with AP augmentation in humans have similarly indicated anti-inflammatory effects in the settings of post-cardiopulmonary surgery, ulcerative colitis, and sepsis, while genetic deficiency of IAP in humans has been linked with colitis (Heemskerk et al., 2009; Kats et al., 2009; Lukas et al., 2010; Parlato, et al., 2018; Peters et al., 2015; Pickkers et al., 2012). Clinical approaches to block TLR4 signaling for therapeutic benefit have been unsuccessful thus far. Neither the LPS analog Eritoran nor the small molecule TAK-242 were able to reduce inflammatory cytokine responses and alter outcomes in patients diagnosed with sepsis (Opal et al., 2013; Rice et al., 2010). This may reflect difficulties in the therapeutic delivery of each compound or the possibility that human TLR4 complexes are structurally dissimilar from those in the mouse wherein each compound was primarily tested. Impeding TLR4 signaling in sepsis by AP-mediated de-phosphorylation of LPS represents a cogent therapeutic approach wherein Gram– negative pathogens otherwise dismantle this mechanism of host protection by depleting anti-inflammatory AP isozymes.
Supplementary Material
Highlights.
The pathophysiology of sepsis can be stratified by pathogens and host responses
Host neuraminidases induced by LPS activation of TLR4 accelerate AP clearance mechanisms
The Ashwell-Morell receptor and ST3Gal6 antagonistically participate in AP homeostasis
Neuraminidase inhibition and AP augmentation therapeutically diminish TLR4 downstream pathology
ACKNOWLEDGMENTS
This research was funded by NIH grants HL125352 (J.D.M. and V.N.) and HL131474 (J.D.M., M.J.M., and V.N.). Additional support was provided by the Swedish Research Council 2017–00192 (J.S.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no conflicts of interest or financial interests.
REFERENCES
- Ashwell G, and Morell A (1974). The dual role of sialic acid in the hepatic recognition and metabolism of serumglycoproteins. Biochem. Soc. Symp 40, 117–124. [PubMed] [Google Scholar]
- Bates JM, Akerlund J, Mittge E, and Guillemin K (2007). Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentala H, Verweij WR, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, and Poelstra K (2002). Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock 18, 561–566. [DOI] [PubMed] [Google Scholar]
- Beumer C, Wulferink M, Raaben W, Fiechter D, Brands R, and Seinen W (2003). Calf intestinal alkaline phosphatase, a novel therapeutic drug for lipopolysaccharide (LPS)-mediated diseases, attenuates LPS toxicity in mice and piglets. J. Pharmacol. Exp. Ther 307, 737–744. [DOI] [PubMed] [Google Scholar]
- Beutler B (2000). Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr. Opin. Microbiol 3, 23–28. [DOI] [PubMed] [Google Scholar]
- Chang HJ, Lynm C, and Glass RM (2010). JAMA patient page. Sepsis. JAMA 304, 1856. [DOI] [PubMed] [Google Scholar]
- Chaudhry H, Zhou J, Zhong Y, Ali MM, McGuire F, Nagarkatti PS, and Nagarkatti M(2013). Role of cytokines as a double-edged sword in sepsis. In Vivo 27, 669–684. [PMC free article] [PubMed] [Google Scholar]
- Chow OA, von Köckritz-Blickwede M, Bright AT, Hensler ME, Zinkernagel AS, Cogen AL, Gallo RL, Monestier M, Wang Y, Glass CK, et al. (2010). Statins enhance formation of phagocyte extracellular traps. Cell Host & Microbe 8, 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross AS, Hyun SW, Miranda-Ribera A, Feng C, Liu A, Nguyen C, Zhang L, Luzina IG, Atamas SP, Twaddell WS, et al. (2012). NEU1 and NEU3 sialidase activity expressed in human lung microvascular endothelia: NEU1 restrains endothelial cell migration, whereas NEU3 does not. J. Biol. Chem 287, 15966–15980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl R, Sergienko EA, Su Y, Mostofi YS, Yang L, Simao AM, Narisawa S, Brown B, Mangravita-Novo A, Vicchiarelli M, et al. (2009). Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). J. Med. Chem 52, 6919–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MR Jr, and Goldberg JB (2012). Purification and visualization of lipopolysaccharide from Gram-negative bacteria by hot aqueous-phenol extraction. J. Vis. Exp 63, 3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejager L, Pinheiro I, Dejonckheere E, and Libert C (2011). Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol 19, 198–208. [DOI] [PubMed] [Google Scholar]
- de Jong HK, Parry CM, van der Poll T, and Wiersinga WJ (2012). Host-pathogen interaction in invasive Salmonellosis. PLoS Pathog 10, e1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Elia RV, Harrison K, Oyston PC, Lukaszewski RA, and Clark GC (2013). Targeting the “cytokine storm” for therapeutic benefit. Clin. Vaccine Immunol 20, 319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellies LG, Ditto D, Levy GG, Wahrenbrock M, Ginsburg D, Varki A, Le DT, and Marth JD (2002). Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc. Natl. Acad. Sci. USA 99, 10042–10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feasey NA, Dougan G, Kingsley RA, Heyderman RS, and Gordon MA (2012). Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet 379, 2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Bossi N, Uzzau S, Maloriol D, and Bossi L (2001). Variable assortment of prophages provides a transferable repertoire of pathogenic determinants in Salmonella. Mol. Microbiol 39, 260–271. [DOI] [PubMed] [Google Scholar]
- Fleischmann C, Scherag A, Adhikari NK, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, and Reinhart K (2016). Assessment of global incidence and mortality of hospital-treated sepsis current estimates and limitations. Am. J. Respir. Crit. Care Med 193, 259–272. [DOI] [PubMed] [Google Scholar]
- Gaieski DF, Matthew EJ, Kallan MJ, and Carr BG (2013). Benchmarking the incidence and mortality of severe sepsis in the United States. Crit. Care Med 41, 1167–1174. [DOI] [PubMed] [Google Scholar]
- Goldberg RF, Austen WG Jr, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, et al. (2008). Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc. Natl. Acad. Sci. USA 105, 3551–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal PK, Aziz PV, Uchiyama S, Rubio GR, Lardone RD, Le D, Varki NM, Nizet V, Marth JD (2013). Inducing host protection in pneumococcal sepsis by pre-activation of the Ashwell-Morell receptor. Proc. Natl. Acad. Sci. USA 110, 20218–20223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal PK, Uchiyama S, Ditto D, Varki N, Le DT, Nizet V, and Marth JD (2008). The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat. Med 14, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman ME, Linde-Zwirble WT, Angus DC, and Watson RS (2013). Trends in the epidemiology of pediatric severe sepsis. Pediatr. Crit. Care Med 14, 686–693. [DOI] [PubMed] [Google Scholar]
- Hata K, Koseki K, Yamaguchi K, Moriya S, Suzuki Y, Yingsakmongkon S, Hirai G, Sodeoka M, von Itzstein M, and Miyagi T (2008). Limited inhibitory effects of oseltamivir and zanamivir on human sialidases. Antimicrob. Agents Chemother 52, 3484–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawiger J, and Musser JM (2011). How to approach genome wars in sepsis? Crit. Care 15, 1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk S, Masereeuw R, Moesker O, Bouw MP, van der Hoeven JG, Peters WH, Russel FG, and Pickkers P (2009). Alkaline phosphatase treatment improves renal function in severe sepsis or septic shock patients. Crit. Care Med 37, 417–423. [DOI] [PubMed] [Google Scholar]
- Heithoff DM, Shimp WR, House JK, Xie Y, Weimer BC, Sinsheimer RL, and Mahan MJ (2012). Intraspecies variation in the emergence of hyperinfectious bacterial strains in nature. PLoS Pathog 8, e1002647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Hammer RE, and Herz J (1994). Asialoglycoprotein receptor deficiency in mice lacking the minor receptor subunit. J. Biol. Chem 269, 27803–27806. [PubMed] [Google Scholar]
- Iwashyna TJ, Ely EW, Smith DM, and Langa KM (2010). Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 304, 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kats S, Brands R, Seinen W, de Jager W, Bekker MW, Hamad MA, Tan ME, and Schönberger JP (2009). Anti-inflammatory effects of alkaline phosphatase in coronary artery bypass surgery with cardiopulmonary bypass. Recent Pat. Inflamm. Allergy Drug Discov 3, 214–220. [DOI] [PubMed] [Google Scholar]
- Kiffer-Moreira T, Sheen CR, Gasque KC, Bolean M, Ciancaglini P, van Elsas A, Hoylaerts MF, and Millan JL (2014). Catalytic signature of a heat-stable chimeric human alkaline phosphatase with therapeutic potential. PLoS One, 9, e89374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama I, Matsunaga T, Harada T, Hokari S, and Komoda T (2002). Alkaline phosphatases reduce toxicity of lipopolysaccharides in vivo and in vitro through dephosphorylation. Clin. Biochem 35, 455–461. [DOI] [PubMed] [Google Scholar]
- Lee CH, and Tsai CM (1999). Quantification of bacterial lipopolysaccharides by the purpald assay: measuring formaldehyde generated from 2-keto-3-deoxyoctonate and heptose at the inner core by periodate oxidation. Anal. Biochem 267, 161–168. [DOI] [PubMed] [Google Scholar]
- Lin JN, Lai CH, Chen YH, Chang LL, Lu PL, Tsai SS, Lin HL, and Lin HH (2010). Characteristics and outcomes of polymicrobial bloodstream infections in the emergency department: A matched case-control study. Acad. Emerg. Med 17, 1072–1079. [DOI] [PubMed] [Google Scholar]
- Lukas M, Drastich P, Konecny M, Gionchetti P, Urban O, Cantoni F, Bortlik M, Duricova D, and Bulitta M (2010). Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm. Bowel Dis 16, 1180–1186. [DOI] [PubMed] [Google Scholar]
- Marshall JC Why have clinical trials in sepsis failed? (2014). Trends Mol. Med 20, 195–203. [DOI] [PubMed] [Google Scholar]
- Millán JL (2006). Mammalian Alkaline Phosphatase s: From Biology to Applications in Medicine and Biotechnology (Weinheim, Chichester: Wiley-VCH; ). [Google Scholar]
- Narisawa S, Fröhlander N, and Millán JL (1997). Inactivation of two mouse alkaline phosphatase genes and establishment of model of infantile hypophosphatasia. Dev. Dyn 208, 432–446. [DOI] [PubMed] [Google Scholar]
- Narisawa S, Huang L, Iwasaki A, Hasegawa H, Alpers DH, and Millán JL (2003). Accelerated fat absorption in intestinal alkaline phosphatase knockout mice. Mol. Cell. Biol 23, 7525–7530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsubo K, Chen MZ, Olefsky JMA, and Marth JD (2011). Pathway to diabetes through attenuation of pancreatic beta cell glycosylation and glucose transport. Nat. Med 17, 1067– 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, et al. (2013). Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309, 1154–1162. [DOI] [PubMed] [Google Scholar]
- Orban JC, Walrave Y, Mongardon N, Allaouchiche B, Argaud L, Aubrun F, Barjon G, Constantin JM, Dhonneur G, Durand-Gasselin J, et al. (2017). Causes and Characteristics of Death in Intensive Care Units: A Prospective Multicenter Study. Anesthesiology 126, 882–889. [DOI] [PubMed] [Google Scholar]
- Parlato M, Charbit-Henrion F, Pan J, Romano C, Duclaux-Loras R, Le Du MH, Warner N, Francalanci P, Bruneau J, Bras M, et al. (2018). Human ALPI deficiency causes inflammatory bowel disease and highlights a key mechanism of gut homeostasis. EMBO Mol. Med 10, pii: e8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pammi M, Zhong D, Johnson Y, Revell P, and Versalovic J (2014). Polymicrobial bloodstream infections in the neonatal intensive care unit are associated with increased mortality: a case-control study. BMC Infect. Dis 14, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, and Lee JO (2009). The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195. [DOI] [PubMed] [Google Scholar]
- Peters E, Stevens J, Arend J, Guan Z, Raaben W, Laverman P, Elsas AV, Masereeuw R, and Pickkers P (2015). Biodistribution and translational pharmacokinetic modeling of a human recombinant alkaline phosphatase. Int. J. Pharm 495, 122–131. [DOI] [PubMed] [Google Scholar]
- Pettengill M, Matute JD, Tresenriter M, Hibbert J, Burgner D, Richmond P, Millán JL, Ozonoff A, Strunk T, Currie A, et al. (2017). Human alkaline phosphatase dephosphorylates microbial products and is elevated in preterm neonates with a history of late-onset sepsis. PLoS One 12, e0175936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickkers P, Heemskerk S, Schouten J, Laterre PF, Vincent JL, Beishuizen A, Jorens PG, Spapen H, Bulitta M, Peters WH, et al. (2012). Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: a prospective randomized double-blind placebo-controlled trial. Crit. Care 16, R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkerton AB, Sergienko E, Bravo Y, Dahl R, Ma CT, Sun Q, Jackson MR, Cosford ND, and Millán JL (2018). Discovery of 5-((5-c hloro-2-methoxyphenyl)sulfonamido) nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorg. Med. Chem. Lett 28, 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelstra k., Bakker WW, Klok PA, Hardonk MJ, and Meijer DK (1997). A physiologic function for alkaline phosphatase: endotoxin detoxification. Lab. Invest 76, 319–327. [PubMed] [Google Scholar]
- Poolman JT, and Wacker M (2016). Extraintestinal pathogenic Escherichia coli, a common human pathogen: challenges for vaccine development and progress in the field. J. Infect. Dis 213, 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, et al. (2010). A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med 38, 1685–1694. [DOI] [PubMed] [Google Scholar]
- Russo TA, Thompson JS, Godoy VG, and Malamy MH (1990). Cloning and expression of the Bacteroides fragilis TAL2480 neuraminidase gene, nanH, in Escherichia coli. J. Bacteriol 172, 2594–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schromm AB, Brandenburg K, Loppnow H, Zähringer U, Rietschel ET, Carroll SF, Koch MH, Kusumoto S, and Seydel U (1998). The charge of endotoxin molecules influences their conformation and IL-6-inducing capacity. J. Immunol 161, 5464–5471. [PubMed] [Google Scholar]
- Schulte W, Bernhagen J, and Bucala R (2013). Cytokines in sepsis: potent immunoregulators and potential therapeutic targets-an updated view. Mediators Inflamm 2013, 165974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheen CR, Kuss P, Narisawa S, Yadav MC, Nigro J, Wang W, Chhea TN, Sergienko EA, Kapoor K, Jackson MR, et al. (2015). Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J. Bone Miner. Res 30, 824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton KD, and Wischmeyer PE (2003). Distance of cecum ligated influences mortality, tumor necrosis factor-alpha and interleukin-6 expression following cecal ligation and puncture in the rat. Eur. Surg. Res 35, 486–491. [DOI] [PubMed] [Google Scholar]
- Soukharev S, Miller JL, and Sauer B (1999). Segmental genomic replacement in embryonic stem cells by double lox targeting. Nucleic Acids Res 27, e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, and Remick DG (2011). The pathogenesis of sepsis. Annu. Rev. Pathol 6, 19–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson EK, Rubenstein AR, Radin GT, Wiener RS, and Walkey AJ (2014). Two decades of mortality trends among patients with severe sepsis: a comparative meta-analysis. Crit. Care Med 42, 625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao K, and Miyakawa T (2015). Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 112, 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuin A, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, and Poelstra K (2006). On the role and fate of LPS-dephosphorylating activity in the rat liver. Am. J. Physiol. Gastrointest. Liver Physiol 290, G377–G385. [DOI] [PubMed] [Google Scholar]
- van Sorge NM, Beasley FC, Gusarov I, Gonzalez DJ, von Köckritz-Blickwede M, Anik S, Borkowski AW, Dorrestein PC, Nudler E, and Nizet V (2013). Methicillin-resistant Staphylococcus aureus bacterial nitric-oxide synthase affects antibiotic sensitivity and skin abscess development. J. Biol. Chem 288, 6417–6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimr ER, and Troy FA (1985). Identification of an inducible catabolic system for sialic acids (nan) in Escherichia coli. J. Bacteriol 164, 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent JL, Martinez EO, and Silva E (2009). Evolving concepts in sepsis definitions. Crit. Care Clin 25, 665–675. [DOI] [PubMed] [Google Scholar]
- Yang WH, Aziz PV, Heithoff DM, Mahan MJ, Smith JW, and Marth JD (2015). An intrinsic mechanism of secreted protein aging and turnover. Proc. Natl. Acad. Sci. USA 112, 13657–13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WH, Heithoff DM, Aziz PV, Sperandio M, Nizet V, Mahan MJ, and Marth JD (2017). Recurrent infection progressively disables host protection against intestinal inflammation. Science 358, pii: eaao5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WH, Nussbaum C, Grewal PK, Marth JD, and Sperandio M (2012). Coordinated roles of ST3Gal-VI and ST3Gal-IV sialyltransferases in the synthesis of selectin ligands. Blood 120, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.