

Abstract
Two synthetic methods were developed for the synthesis of PDE3A inhibitor ORG9935. The first one proceeds in six steps and 34% overall yield and the second one in five steps and an overall yield of 69% starting from commercially available starting material 5,6-dimethoxybenzo[b]thiophene-2-carboxylic acid (6). The enantiomers of ORG9935 were separated by chiral column chromatography and the absolute stereochemistry of the (+)-enantiomer, ORG20865 was determined by X-ray crystallography to possess the (S)-configuration. The (−)-enantiomer, ORG20864, was therefore assigned the (R)-stereochemistry. The biologically less active (+)-isomer ORG20865 was converted to racemic ORG9935 under basic conditions, which then can be separated again into the enantiomers. The crystal structure of ORG20865 is notable for having the highest Z′ for any known pharmaceutical substance.
Keywords: Synthesis, PDE3A inhibitor, ORG9935, female contraception, enantiomer separation, structure determination
Graphical abstract

INTRODUCTION
The cyclic nucleotide phosphodiesterase (PDE) families,1,2 are associated with a variety biological processes such as inflammation, ion channel function, muscle contraction, differentiation, apoptosis, lipogenesis, glycogenolysis, and gluconeogenesis.3 PDEs play a critical role in regulating biological responses to second messengers including cAMP and cGMP by controlling the intracellular levels of cAMP and cGMP by catalyzing their enzymatic hydrolysis.4 PDEs have specific distributions at the cellular and subcellular level. PDE3A, for example, is expressed in the heart, platelets, vascular smooth muscle, airway smooth muscle, and the oocyte.5,6,7
In the oocyte, PDE3A is responsible for downregulating cAMP before germinal vesicle-breakdown and the resumption of meiosis.8–10 PDE3A female knock-out mice, which are viable and ovulate a normal number of oocytes, are completely infertile.11 The mice did not resume oocyte meiosis indicating the major role of PDA3 in resumption of meiosis of oocytes. The observed infertility or impaired fertility indicated that PDA3 could be a target for female contraception.8,9,11,12 Although female hormonal contraceptives have been known for some time, side effects make it desirable to find alternatives that are non-hormonal.13 Wiersma et al.8 and Jensen and coworkers have reported that PDE3A inhibitor ORG9935 prevents oocyte maturation and fertilization.14–17 These results suggested that ORG9935 could be a probe or lead compound for non-hormonal female contraception. For further studies with ORG9935 we developed a scalable method to provide sufficient amounts for fertility studies in the Jensen group. A second goal was to obtain enantiomerically pure (−)-ORG9935, which is known as ORG20864. It was reported that the two isomers (−)-ORG20864 and (+)-ORG20865 differ in their ability to suppress mouse oocyte maturation and have different potencies for the in vitro inhibition of PDA3 in oocytes and cardiac cells.8 The (−)-enantiomer ORG20864 is more potent for mouse oocytes suppression than the (+)-enantiomer and it is also more selective for mouse oocyte PDE3 inhibition compared to cardiac cells. A more selective compound is expected to have fewer side effects and also the amount of compound needed to elicit a contraceptive effect would be lower than for the racemic compound.
A previous report for a five-step very efficient synthesis of ORG9935 from a patent by Redpath et al.18 is shown in Scheme 1 that provided the target compound in 69% overall yield.
Scheme 1.

The synthesis started with a Strecker reaction between aldehyde 1, morpholine and potassium cyanide to form α-aminonitrile 2. Subsequent Michael addition of intermediate 2 to 2-butenenitrile provided addition product 3, which was converted to cyanoketone 4 under refluxing acidic conditions. After converting 4 to carboxylic acid 5, subsequent reaction with hydrazine furnished ORG9935. While this synthesis is short and efficient we were concerned about the large-scale use of potassium cyanide for the Strecker reaction. A further concern was the release of HCN during the conversion of iminonitrile 3 to ketone 4. HCN is a dangerous gas that is challenging to trap on large scale.
RESULTS AND DISCUSSION
We developed alternative safer methods for the synthesis of ORG 9935 that do not require purification by column chromatography (Schemes 2 and 3). Reaction of commercially available acid 6 with N,O-dimethylhydroxylamine and EDCI provided the Weinreb amide 7, which after Grignard reaction with ethylmagnesium bromide gave ketone 8 in excellent yield. Bromination of ketone 8 with pyridinium tribromide furnished the α-ketobromide 9 in 86% yield. Based on reported methods,16, 17 the reaction between ketobromide 9 and diethyl malonate provided intermediate 10 in 90% yield. Diethyl malonate ester 10 was hydrolyzed and decarboxylated using sodium hydroxide. The mono acid 5 (not isolated) was directly condensed with hydrazine hydrate to afford the target compound ORG 9935 in six steps and 34% overall yield.
Scheme 2.

Scheme 3.

The second synthesis is shown in Scheme 3. According to a known method,14, 15 ethylketone 8 was reacted with LiHMDS and ethyl bromoacetate to produce ketoester 11, which was used in the following reaction without purification. Ester 11 was converted to the corresponding acid 5 by hydrolysis with sodium hydroxide and then used in the next step without purification. The target compound ORG9935 was obtained by reaction of ketoacid 5 with hydrazine hydrate in 85% combined yield for the last three steps. Starting with acid 6 (see Scheme 2 for the conversion of acid 6 to ketone 8) provides ORG9935 in five steps and an overall yield of 69%.
Acid 6 is commercially available but expensive and therefore we prepared this compounds using a published procedure (Scheme 4).19 Reaction between 6-bromoveratraldehyde and ethyl 2-mercaptoacetate formed ethyl 5,6-dimethoxybenzo[b]thiophene-2-carboxylate (12) in 52% yield, which was hydrolyzed to furnish the corresponding acid 6 in 90% yield. Intermediate 12 is also commercially available but costly.
Scheme 4.

After we had sufficiently optimized the synthesis, ORG9935 was separated into its two enantiomers by chiral column chromatography. The enantiomers were separated using a preparative CHIRALPAK AS-H 30 column, using a 1:1 CO2:methanol mobile phase. The retention time for (+)-ORG20865 was 0.77 min and 1.27 min for (−)-ORG20864. The enantiomeric purity was >99 ee for both enantiomers. In a previous publications CHIRALCEL OC and related columns had been used for the separation of the ORG9935 enantiomers.20 However, the optical rotations or assignments of stereochemistry were not reported.
We subsequently converted the undesired (+)-ORG20865 to racemic ORG9935 by refluxing for 24 h under basic conditions (KOH in methanol), which then can be separated into its enantiomers again (Scheme 5).
Scheme 5.
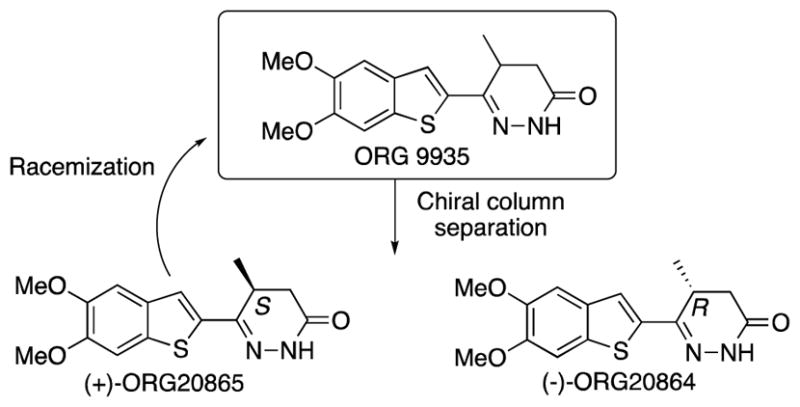
The absolute stereochemistries of the enantiomers were determined by obtaining an X-ray crystal structure of (+)-ORG20865, which demonstrated that the (+)-enantiomer has the (S)-configuration and therefore the (−)-enantiomer ORG20864 has the (R)-stereochemistry.
A crystal structure determination was obtained21,22 at room temperature on a Bruker-AXS VENTURE PHOTON-II diffractometer with a micro-focus Mo-anode source. The stereochemistry of ORG20865 at C4 is (S) and is enantiopure. This crystal structure is exceedingly complex in its three-dimensional form being in space group P1 with Z=24 and Z′=24 (Figure 1). One of the 24 unique molecules is depicted in Figure 1A. All bond distances and angles in individual molecules are found within normal ranges. Each molecule has a hydrogen bond donor at N2 and a hydrogen bond acceptor at O1. The molecules form tetramers through a set of four hydrogen bonds with an overall R1644 ring structure.23,24 One such tetramer is depicted in Figure 1B. The unit cell contains six tetramers ascending along the c-axis, as shown in Figure 1C. The symmetry is truly triclinic, however the unit cell is suspiciously close to an alternative setting of a hexagonal unit cell with the a- and b-axes nearly the same length, the α- and β-angles approaching 90°, and γ ~ 60°. These tetramers form layers of molecules perpendicular to the c-axis related to adjacent unit cells by translation only. The layers are rotated approximately 60° to those above and below. The specimen selected was also determined to be a twin by reticular pseudo-merohedry through a simulated C-centered monoclinic twin lattice (TLQS). The twin law was calculated based on this twin symmetry element and used throughout (by rows −1 0 0/−1 1 0/0 0 −1) with the twin individual mass ratio being approximately 0.72:0.28. This crystal structure is notable in having the highest Z′ for any known pharmaceutical substance.25,26 Additional information for this crystal structure determination can be found in the supplemental materials.
Figure 1.

(A) Thermal ellipsoid drawing of molecule A with 50% probability ellipsoids; (B) a tetramer of molecules hydrogen bonded together in a R1644 ring structure drawn with 50% probability ellipsoids; (C) unit cell drawing containing six unique hydrogen bonded tetramers drawn with 50% probability ellipsoids.
In summary, two methods for the synthesis of ORG9935 were developed that avoid the use of hazardous reagents and do not require chromatographic separations and are therefore suitable for scale-up syntheses.
EXPERIMENTAL SECTION
All chemicals and reagents were purchased from commercial sources and used directly without further purification. Solvents were dried using standard procedures. All non-aqueous reactions were performed under an atmosphere of nitrogen in oven-dried glassware. Reaction progress was monitored by thin-layer chromatography using silica gel plates (silica gel 60 F254), and eluted TLC plates were visualized with UV light (254 nm). Compound 12 was purified by flash column chromatography using the Combiflash® Rf Teledyne ISCO system. NMR experiments were performed on a 400/100 MHz instrument. NMR spectra were processed with the MestReNova program. Chemical shifts were reported as ppm relative to acetone-d6 (2.05 ppm for 1H, 206.68 and 29.92 ppm for 13C), and DMSO-d6 (2.50 ppm for 1H, 39.5 ppm for 13C). 1H NMR coupling constants (J) are expressed in hertz (Hz), and multiplicity is described as follows: s = singlet; d = doublet; t = triplet; q = quartet; p = pentet; br = broad; m = multiplet. High-resolution mass spectra and electrospray (ESI) experiments were recorded with electron-spray ionization. Melting points were determined with a BI Barnstead Electrothermal 9100 melting point apparatus and are uncorrected. ORG 9935 was pure (≥99%) by LC-MS. Optical rotations were measured with an Autopol V Polarimeter.
N,5,6-Trimethoxy-N-methylbenzo[b]thiophene-2-carboxamide (7)
To a stirred solution of acid 6 (1.07 g, 4.49 mmol) in CH2Cl2 (60 mL) were added N,O-dimethylhydroxylamine hydrochloride (0.478 g, 4.9 mmol), triethylamine (0.80 mL, 5.74 mmol), and EDCI·HCl (1.29 g, 6.72 mmol). After stirring for 18 h at room temperature under an inert atmosphere, the reaction was quenched with sat. NaHCO3 solution. The resulting mixture was extracted with EtOAc (3 × 50 mL). The organic layer was washed with water followed by brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was crystallized from EtOAc and hexanes to yield 7 as a white solid (1.17 g, 93%) MP 131 – 133 °C.1H NMR (400 MHz, DMSO-d6) δ 8.05 (s, 1H), 7.55 (s, 1H), 7.49 (s, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.80 (s, 3H), 3.31 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 161.5, 150.1, 148.3, 135.3, 131.6, 131.2, 130.3, 106.2, 103.6, 61.6, 55.8, 55.5, 32.9.
1-(5,6-Dimethoxybenzo[b]thiophen-2-yl)propan-1-one (8)
To a stirred solution of Weinreb amide 7 (1.1 g, 3.91 mmol) in anhydrous THF (50 mL) was slowly added EtMgBr (1 mol/L in THF, 12 mL, 12 mmol) at −10 °C under an argon atmosphere. After stirring for 3.5 h at room temperature, the reaction was cooled to 0 °C and quenched with HCl (1N, 100 mL). The resulting mixture was extracted with EtOAc (3 × 50 mL), then the organic layer was washed with water followed by brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was crystallized from EtOAc to afforded 8 (0.849 g, 87%). Cream colored solid. MP 159 – 161 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (s, 1H), 7.58 (s, 1H), 7.46 (s, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.04 (q, J = 7.3 Hz, 2H), 1.12 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 194.5, 150.7, 148.5, 140.8, 135.5, 132.5, 130.0, 106.5, 104.2, 55.8, 55.6, 31.3, 8.5. HRMS calcd for C13H14O3S (M+Na)+ 273.0556, found 273.0555.
2-Bromo-1-(5,6-dimethoxybenzo[b]thiophen-2-yl)propan-1-one (9)
To a solution of ethylketone 8 (5.0 g, 20 mmol) in chloroform (250 mL) cooled to 10 °C, pyridinium tribromide (7.1 g, 22 mmol) was added in two batches. After the reaction mixture was stirred overnight at room temperature, the solvent was distilled off under reduced pressure. Water (250 mL) was added and stirring was continued for 10 min at room temperature. The resultant mixture was filtered and washed with water. After drying, the compound was crystallized from ethyl acetate to afford 9 (5.63 g, 86%) as a light yellow solid. MP 167 – 169 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.32 (s, 1H), 7.62 (s, 1H), 7.48 (s, 1H), 5.76 (q, J = 6.5 Hz, 1H), 3.87 (s, 3H), 3.84 (s, 3H), 1.79 (d, J = 6.5 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 187.9, 151.2, 148.7, 137.4, 136.5, 132.3, 131.7, 106.6, 104.2, 55.9, 55.6, 43.2, 20.3. HRMS calcd for C13H1379BrO3S (M+Na)+ 350.9661, found 350.9657. HRMS calcd for C13H1381BrO3S (M+Na)+ 352.9646, found 352.9640.
Diethyl 2-(1-(5,6-Dimethoxybenzo[b]thiophen-2-yl)-1-oxopropan-2-yl)malonate (10)
To a suspension of anhydrous potassium carbonate (1.36 g, 9.8 mmol) in dry DMF (60 mL) at room temperature, diethyl malonate (2.58 g, 16 mmol) was added. After the reaction mixture stirred for 3 h at room temperature, bromoketone 9 (5.0 g, 15.2 mmol) was added and the reaction was heated at 110 °C overnight. After cooling, the reaction was quenched into water (250 mL) and extracted with ethyl acetate (3 × 50 mL). The combined organic layer was washed with water, dried over sodium sulfate and concentrated under reduced pressure to afford 10 as a brown oily compound. 1H NMR (400 MHz, acetone-d6) δ 8.25 (s, 1H), 7.55 (s, 1H), 7.52 (s, 1H), 4.27 (q, J = 7.1 Hz, 2H), 4.18 – 4.02 (m, 3H), 3.96 (s, 3H), 3.92 (s, 3H), 3.85 (d, J = 10.8 Hz, 1H), 1.35 – 1.27 (m, 6H), 1.15 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, acetone-d6) δ 195.6, 169.0, 168.6, 152.6, 150.3, 140.8, 137.7, 133.9, 131.1, 107.5, 104.8, 62.2, 62.0, 56.4, 56.2, 55.7, 42.3, 17.0, 14.4, 14.2. HRMS calcd for C20H24O7S (M+Na)+ 431.1135, found 431.1144.
6-(5,6-Dimethoxybenzo[b]thiophen-2-yl)-5-methyl-4,5-dihydropyridazin-3(2H)-one (ORG9935)
The crude diethyl malonate 10 (2.28 g, 5.58 mmol), was dissolved in methanol (50 mL) and water (10 mL) and sodium hydroxide (0.720 g, 18 mmol) was added. After the reaction was refluxed overnight the methanol was removed under reduced pressure. The reaction mixture was extracted with ethyl acetate (3 × 50 mL), washed with brine and dried over sodium sulfate. The ethyl acetate was distilled off under reduced pressure, ethanol was (60 mL) was added and then hydrazine hydrate (85%, 6.25 mL). The reaction was heated at reflux overnight, cooled to 40 °C and filtered. The solid was washed with plenty of water and dried at 40 °C under reduced pressure to afford pure ORG9935 (0.935 g) as a cream colored solid in 55% yield over three steps. MP 223 – 225 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.96 (s, 1H), 7.64 (s, 1H), 7.49 (s, 1H), 7.33 (s, 1H), 3.82 (s, 3H), 3.81 (s, 3H), 3.45 – 3.38 (m, 1H), 2.76 (dd, J = 16.8, 6.9 Hz, 1H), 2.27 (d, J = 16.7 Hz, 1H), 1.14 (d, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.2, 149.8, 149.1, 147.9, 137.9, 132.8, 132.8, 124.0, 105.7, 104.4, 55.7, 55.5, 33.4, 28.0, 16.4. HRMS calcd for C15H16N2O3S (M+Na)+ 327.0774, found 327.0774.
Ethyl 4-(5,6-Dimethoxybenzo[b]thiophen-2-yl)-3-methyl-4-oxobutanoate (11)
To a stirred solution of intermediate 8 (30 g, 120 mmol) in anhydrous THF (200 mL) and HMPA (75 mL) was added LiHMDS in THF (1.0 mol/L, 132 mL) at −70 °C under an inert atmosphere. After stirring for 20 min at the same temperature, ethyl bromoacetate (25.5 g, 152.7 mmol) was added dropwise to the reaction mixture at −70 °C, and then the reaction mixture was stirred overnight at room temperature. After the reaction mixture was cooled to −20 °C, saturated NH4Cl solution (50 mL) was added slowly and then water (300 mL). After extraction with ethyl acetate (3 × 350 mL), the combined organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The target compound 11 was obtained as cream colored solid and used in the next step without further purification.
4-(5,6-Dimethoxybenzo[b]thiophen-2-yl)-3-methyl-4-oxobutanoic Acid (5)
To a stirred solution of intermediate 11 (40 g, crude) in ethanol (250 mL) and water (100 mL) was added a NaOH solution (6.18 g, 154 mmol/50 mL water) dropwise at room temperature. The resulting mixture was stirred at room temperature overnight. The ethanol was distilled off under reduced pressure and the reaction was diluted with water (200 mL), followed by extraction with diethyl ether (2 × 100 mL) to remove organic impurities. The reaction mixture was cooled to 10 °C and the pH value was adjusted to pH = 2 with dilute hydrochloric acid. The resulting suspension was extracted with ethyl acetate (3 × 350 mL). The combined organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to afforded acid 5 as a cream colored solid (36.5 g). The compound was used in the next step without further purification. MP 164–166° C. 1H NMR (400 MHz, DMSO-d6) δ 12.18 (s, 1H), 8.23 (s, 1H), 7.59 (s, 1H), 7.48 (s, 1H), 3.86 (s, 3H), 3.83 (s, 3H), 3.81-3.79 (m, 1H), 2.72 (dd, J = 16.9, 9.4 Hz, 1H), 2.43 (dd, J = 16.9, 5.0 Hz, 1H), 1.19 (d, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 196.53, 172.95, 150.80, 148.48, 139.88, 135.82, 132.57, 130.33, 106.61, 104.20, 55.82, 55.55, 37.62, 37.11, 18.31. HRMS calcd for C15H16O5S (M+Na)+ 331.0611, found 331.0608.
6-(5,6-Dimethoxybenzo[b]thiophen-2-yl)-5-methyl-4,5-dihydropyridazin-3(2H)-one (ORG9935)
Acid 5 (36.5 g, 120 mmol) was dissolved in ethanol (800 mL) and then hydrazine hydrate (85%, 120 mL) was added while stirring. The reaction mixture was heated to reflux and maintained at reflux for 3 h and then cooled to 40 °C. The solids were collected by filtration and washed thoroughly with water and dried at 40 °C under reduced pressure. Recrystallized from methanol afforded ORG9935 (31 g) as a cream colored solid. The overall yield for the three steps from intermediate 8 was 85%.
Ethyl 5,6-Dimethoxybenzo[b]thiophene-2-carboxylate (12).19
A solution of ethyl 2-mercaptoacetate (2.85 g, 23.7 mmol) in anhydrous DMF (40 mL) under a nitrogen atmosphere was cooled to 10 °C. Anhydrous potassium carbonate (3.85 g, 27.9 mmol), CuI (0.10 g, 0.52 mmol), diethylamine (0.060 mL, 0.53 mmol), and 6-bromoveratraldehyde (5.00 g, 20.4 mmol) were added to the reaction and stirred at 10 °C to room temperature for 4 h. The temperature was slowly raised to 110 °C and maintained for 24 h. The reaction was cooled to room temperature and water (150 mL) was added. The reaction was extracted with ethyl acetate (3 × 200 mL). The combined organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to give a reddish brown solid. The product was purified on silica gel column chromatography using ethyl acetate/hexanes (20:80) as the eluent to afford 12 (2.80 g, 52%) as a white solid. The spectral data matched those reported.27
5,6-Dimethoxybenzo[b]thiophene-2-carboxylic acid (6)
To a stirred solution of ester 12 (2.0 g, 7.51 mmol) in ethanol (50 mL) and water (40 ml) was added a KOH solution (0.547 g, 9.76 mmol in 10 mL H2O) dropwise at room temperature. The resulting mixture was stirred at room temperature overnight and then ethanol was removed under reduced pressure. The reaction was diluted with water (200 mL), washed twice with diethyl ether (2 × 30 mL) to remove impurities and then was cooled to 10 °C. The pH was adjusted to pH = 2 with dilute hydrochloric acid and then the resulting suspension was extracted with ethyl acetate (3 × 50 mL). The combined organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to afford 6 as an off white solid (1.61 g, 90%). MP 265–267 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H), 7.94 (s, 1H), 7.58 (s, 1H), 7.48 (s, 1H), 3.86 (s, 3H), 3.83 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 163.6, 150.2, 148.4, 135.1, 132.1, 131.9, 130.0, 106.2, 104.1, 55.8, 55.5.
Racemization Procedure
To (+)-ORG20865 (78 g, 256 mmol), suspended in methanol (2000 mL), was added a KOH solution (66.7 g, 1189 mmol in 312 mL water) at room temperature. The reaction mixture was heated to reflux for 24 h. After some time the solids dissolved and a clear solution formed and after 4 h precipitation began to take place. After 24 h, the reaction mixture was cooled to 40 °C and the solids were collected by filtration and washed thoroughly with water and dried at 40 °C under reduced pressure to afford 64.74 g (83%) ORG9935. (c 1.00, CH2Cl2).
Preparative-SFC conditions
Instrument: SFC-200 (Thar, Waters); Column: CHIRALPAK AS-H 30.250 mm, 10 um (Daicel); Column temperature: 35 °C; Mobile phase: CO2/methanol = 50/50; Flow rate: 120 g/min; Back pressure: 100 bar; Detection wavelength: 214 nm, Cycle time: 8.0 min; Sample solution: 120.92 g dissolved in 5000 mL (methanol/CH2Cl2=1/2, 40 °C); Injection volume: 8.5 mL (loading: 206 mg/injection). Retention time for (+)-ORG20865 was 0.77 min and 1.27 min for (−)-ORG20864 (HPLC traces can be found in the SI). The optical rotation for (−)-ORG20864 was: . (c 0.610, CH2Cl2. The optical rotation for (+)-ORG20865 was: . (c 0.567, CH2Cl2).
LCMS condition for purity analysis of ORG9935, ORG20864, and ORG20865
Instrument: Agilent 1200/6100 LCMS; Mobile phase: A: Water (0.01% TFA), B: Acetonitrile; Gradient: 5% B for 0.2 min, increase to 95% B in 1.5 min, 95% B for 1.5 min, back to 5% B within 0.01min; Flow rate: 2.0 mL/min; Column: Sunfire C18, 4.6 × 50 mm, 3.5 μm; Oven Temperature: 50 °C. MS m/e calc for [M+1]+ C15H17N2O3S 305.3, found 305.3.
Supplementary Material
Acknowledgments
Financial support for this project is gratefully acknowledged from the NICHD: 1U01HD076542 (GIG), HHSN275201300017C (GIG), and U54 HD055744 (Jeffrey Jensen, OHSU). The determination of the ORG9935 purity and the separation of the ORG9935 enantiomers were carried out by Shanghai ChemPartner Co., Ltd. China. Partial support for the purchase of the Bruker-AXS Venture PHOTON-II diffractometer was obtained through the National Science Foundation Major Research Instrumentation grant award #1229400.
Footnotes
Notes
The authors have no competing intersts.
HPLC traces for the separation of enantiomers, proton and carbon NMR spectra, and the crystal structure report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beavo JA, Conti M, Heaslip RJ. Mol Pharmacol. 1994;46:399–405. [PubMed] [Google Scholar]
- 2.Bolger GB. Cell Signal. 1994;6:851–859. doi: 10.1016/0898-6568(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 3.Perry MJ, Higgs GA. Curr Opin Chem Biol. 1998;2:472–481. doi: 10.1016/s1367-5931(98)80123-3. [DOI] [PubMed] [Google Scholar]
- 4.Manganiello VC, Murata T, Taira M, Belfrage P, Degerman E. Arch Biochem Biophys. 1995;322:1–13. doi: 10.1006/abbi.1995.1429. [DOI] [PubMed] [Google Scholar]
- 5.Thompson WJ, Appleman MM. J Biol Chem. 1971;246:3145–3150. [PubMed] [Google Scholar]
- 6.Degerman E, Belfrage P, Manganiello VC. J Biol Chem. 1997;272:6823–6826. doi: 10.1074/jbc.272.11.6823. [DOI] [PubMed] [Google Scholar]
- 7.Tsafriri A, Chun S-Y, Zhang R, Hsueh AJW, Conti M. Dev Biol. 1996;178:393–402. doi: 10.1006/dbio.1996.0226. [DOI] [PubMed] [Google Scholar]
- 8.Wiersma A, Hirsch B, Tsafriri A, Hanssen RG, Van de Kant M, Kloosterboer HJ, Conti M, Hsueh AJ. J Clin Invest. 1998;102:532–537. doi: 10.1172/JCI2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conti M, Andersen CB, Richard FJ, Shitsukawa K, Tsafriri A. Mol Cell Endocrinol. 1998;145:9–14. doi: 10.1016/s0303-7207(98)00187-7. [DOI] [PubMed] [Google Scholar]
- 10.Sasseville M, Cote N, Guillemette C, Richard FJ. BMC Dev Biol. 2006;6:47. doi: 10.1186/1471-213X-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masciarelli S, Horner K, Liu C, Park SH, Hinckley M, Hockman S, Nedachi T, Jin C, Conti M, Manganiello V. J Clin Invest. 2004;114:196–205. doi: 10.1172/JCI21804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richard Francois J, Tsafriri Alex, Conti M. Biol Reprod. 2001;65:1444–1451. doi: 10.1095/biolreprod65.5.1444. [DOI] [PubMed] [Google Scholar]
- 13.Peipert JF, Gutmann J. Obstet Gynecol. 1993;82:112–117. [PubMed] [Google Scholar]
- 14.Jensen JT, Zelinski-Wooten MB, Schwinof KM, Vance JE, Stouffer RL. Contraception. 2005;71:68–73. doi: 10.1016/j.contraception.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Jensen JT, Stouffer RL, Stanley JE, Zelinski MB. Contraception. 2010;81:165–171. doi: 10.1016/j.contraception.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen JT, Zelinski MB, Stanley JE, Fanton JW, Stouffer RL. Contraception. 2008;77:303–307. doi: 10.1016/j.contraception.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jensen JT, Schwinof KM, Zelinski-Wooten MB, Conti M, DePaolo LV, Stouffer RL. Hum Reprod. 2002;17:2079–2084. doi: 10.1093/humrep/17.8.2079. [DOI] [PubMed] [Google Scholar]
- 18.Repath J, Logan RT, Roy RG, McGarry G. 4952571. US. 1990
- 19.Deng H, Fang Y. ACS Med Chem Lett. 2012;3:550–554. doi: 10.1021/ml300076u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maris FA, Vervoort RJM, Hindriks H. J Chromatogr. 1991;547:45–58. [Google Scholar]
- 21.Sheldrick GM. Acta Crystallogr. 2015;A71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheldrick GM. Acta Crystallogr. 2015;C71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Etter MC. Acc Chem Res. 1990;23:120–126. [Google Scholar]
- 24.Etter MC, MacDonald JC, Bernstein J. Acta Crystallogr. 1990;B46:256–262. doi: 10.1107/s0108768189012929. [DOI] [PubMed] [Google Scholar]
- 25.Groom CR, Bruno IJ, Lightfoot MP, Ward SC. Acta Crystallogr. 2016;B72:171–179. doi: 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steed KM, Steed JW. Chem Rev. 2015;115:2895–2933. doi: 10.1021/cr500564z. [DOI] [PubMed] [Google Scholar]
- 27.Guo H, Shao H, Yang Z, Xue S, Li X, Liu Z, He X, Jiang J, Zhang Y, Si S, Li ZJ. Med Chem. 2010;53:1819–1829. doi: 10.1021/jm901685n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.