

Abstract
Background
A formal single ascending and multiple dose pharmacokinetic (PK) trial of cannabidiol (CBD) oral solution was required to determine the safety and tolerability of CBD, the maximum tolerated dose, and to examine the effect of food on CBD PK parameters.
Objective
This trial assessed the safety, tolerability and PK of CBD oral solution in healthy adult volunteers, as well as the effect of food on CBD PK parameters.
Methods
The study consisted of three arms: single ascending dose (1500, 3000, 4500 or 6000 mg CBD [n = 6 per group]/placebo [n = 8; 2 per CBD dose group]), multiple dose (750 or 1500 mg CBD [n = 9 per group]/placebo [n = 6; 3 per CBD dose group] twice daily), and food effect (1500 mg CBD single dose [n = 12]). All subjects completed all trial arms and were analyzed as planned.
Results
CBD was generally well tolerated. Diarrhea, nausea, headache, and somnolence were the most common adverse events (AEs) across all trial arms, with an increased incidence of some gastrointestinal and nervous system disorder AEs (most notably diarrhea and headache) apparent in subjects taking CBD compared with placebo. All AEs were of mild or moderate severity; none were severe or serious. There were no deaths or discontinuations in the trial. After single oral doses, CBD appeared rapidly in plasma; time to maximum plasma concentration (tmax) was approximately 4–5 h. The major circulating metabolite was 7-carboxy-CBD, then parent CBD, 7-hydroxy-CBD (active metabolite), and 6-hydroxy-CBD (a relatively minor metabolite). Plasma exposure to CBD [maximum plasma concentration (Cmax) and area under the plasma concentration-time curve from time zero to time t (AUCt)] increased in a less than dose-proportional manner (Cmax slope 0.73; AUCt slope 0.64). Oral clearance of CBD was high (1111–1909 L/h) and apparent volume of distribution was large (20,963–42,849 L). CBD reached steady state after approximately 2 days, with moderate accumulation (1.8- to 2.6-fold) after 750 and 1500 mg CBD twice daily. After 7 days, a twofold increase in CBD dose resulted in 1.6- and 1.9-fold increases in geometric mean Cmax and area under the plasma concentration-time curve over a dosing interval (AUCτ), respectively. CBD elimination was multiphasic; the terminal elimination half-life was approximately 60 h after 750 and 1500 mg CBD twice daily; and effective half-life estimates ranged from 10 to 17 h. Cmax was 541.2 ng/mL and AUCτ was 3236 ng·h/mL after 1500 mg CBD twice daily. A high-fat meal increased CBD plasma exposure (Cmax and AUCt) by 4.85- and 4.2-fold, respectively; there was no effect of food on tmax or terminal half-life.
Conclusion
CBD was generally well tolerated. Most AEs were mild in severity; none were severe or serious. The safety and PK profile support twice-daily administration of CBD.
Key Points
| The results of the trial support twice-daily administration of cannabidiol (CBD). |
| When CBD was taken with a high-fat meal, its bioavailability increased by approximately four- to fivefold and, as such, administering CBD with food would maximize bioavailability and likely reduce within-day fluctuation in systemic exposure to drug. |
| All adverse events were of mild or moderate severity. |
| Safety data are consistent with the currently established safety profile of CBD. |
Introduction
GW Pharmaceuticals’ plant-derived highly purified cannabidiol (CBD; Epidiolex®) was FDA-approved in the US in June 2018 for seizures associated with Lennox–Gastaut syndrome (LGS) or Dravet syndrome (DS) in patients ≥ 2 years of age, but is not yet approved outside of the US. The efficacy and safety of CBD in these severe and treatment-refractory epilepsies were demonstrated in three randomized, placebo-controlled trials [1–4]. It is likely that CBD exerts multimodal anticonvulsant effects, reducing neuronal excitability through transient receptor potential vanilloid channels, antagonism of orphan G protein-coupled receptor 55, and modulation of adenosine [5–7]. Importantly, CBD does not cause the high associated with Δ9-tetrahydrocannabinol (THC) due to limited or no interaction with the cannabinoid receptors CB1 and CB2 [8].
CBD is extensively metabolized in the liver, mainly by cytochrome P450 (CYP) 2C19 and, to a lesser extent, CYP3A4 [9, 10]. CBD may also be glucuronidated by several UDP-glucuronosyltransferase (UGT) isoforms, including UGT1A9, UGT2B7 and UGT2B17 [11]. In vitro data suggest that CBD can also inhibit the CYP2C family of isozymes and CYP3A4 [10, 12], as well as UGT1A9 and UGT2B7 [12]. It will therefore be important to understand if any of these in vitro signals translate to drug–drug interactions in vivo based on specific probe substrate studies. Reports of potential in vivo drug–drug interactions with some commonly prescribed epilepsy drugs are beginning to emerge [2, 13, 14]. In patients, concomitant administration of CBD with clobazam (CLB) resulted in increased plasma concentrations of its major active metabolite, N-desmethylclobazam, likely mediated by CYP2C19 inhibition by CBD. This is often associated with increased sedation, an adverse event (AE) commonly observed with CLB administration [13, 14].
There are no previously published studies investigating the pharmacokinetics (PK) of this formulation of CBD in healthy volunteers, and there are no data on the effects that food may have on this drug. In the recent pivotal trials conducted with CBD, the timing of dosing in relation to food was not stipulated [1, 3, 4]. This trial aimed to investigate the safety, tolerability and PK of CBD when administered as single ascending and multiple oral doses to healthy subjects, to determine the maximum tolerated dose. The trial also examined whether food affected the PK of CBD and its major metabolites.
Methods
Compliance with Ethical Standards
This trial was conducted in accordance with International Conference on Harmonisation Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki. Protocols were approved by an Independent Ethics Committee (Foundation Evaluation of Ethics in Biomedical Research, Assen, The Netherlands) before eligibility screening. Written informed consent was obtained from each subject before any trial-related procedures were performed.
Subjects
Healthy adult male or female subjects between 18 and 45 years of age with a body mass index between 18 and 28.0 kg/m2 were enrolled. Female subjects were of non-childbearing potential or were non-pregnant and non-lactating at each visit.
Trial Design
The trial was conducted between 27 May 2015 and 2 October 2015 at one site in The Netherlands. There were three arms to this trial: a single ascending dose (SAD) arm, a multiple dose (MD) arm, and an integrated food effect (FE) arm. Each arm investigated the safety, tolerability, and PK of a pharmaceutical formulation of highly purified CBD derived from Cannabis sativa L. plant in oral solution (100 mg/mL; Epidiolex® in the US; GW Research Ltd, Cambridge, UK), supplied as 100 mL in 105 mL amber glass bottles, and delivered orally using a syringe.
Single Ascending Dose Arm
The SAD arm was double-blind, randomized, and placebo-controlled. Four groups of eight healthy subjects were enrolled and received a single oral dose of 1500, 3000, 4500, or 6000 mg CBD (Groups 1–4, respectively; n = 6 per group) or matching placebo (Groups 1–4; n = 2 per group) after an overnight fast. Administration of each successive dose depended on the safety and tolerability of previous doses.
Multiple Dose Arm
The MD arm was double-blind, randomized, and placebo-controlled. A separate cohort of 24 subjects was enrolled, and two groups of 12 healthy subjects received multiple oral doses of 750 or 1500 mg CBD (n = 9 per group) or matching placebo (n = 3 per group) twice daily under fasted conditions. Morning doses were administered after an overnight fast of at least 10 h, and evening doses were administered after a fast of at least 2 h. Subjects received CBD or placebo twice daily for 6 days, with a single dose on the morning of day 7.
Food Effect Arm
The integrated FE arm was randomized (to a period; fed then fasted, or fasted then fed) and open-label. Twelve randomly selected subjects from the SAD arm, irrespective of group (excluding subjects randomized to the 6000 mg group) or investigational medicinal product (IMP) taken in the SAD arm (CBD or placebo), crossed over into the FE arm to receive a single oral dose of 1500 mg CBD in either the fed (high-fat breakfast) or fasted state before crossing over. The washout period between IMP administration in the SAD and integrated FE arms was ≥ 7 days.
In the fasted state, subjects were fasted overnight for ≥ 10 h prior to CBD administration. In the fed state, subjects were fasted overnight for ≥ 10 h then received a standardized high-fat breakfast of 918 kcal consisting of two fried eggs in 15 g butter (approximately 100 g), one portion of bacon (40 g), one portion of fried potatoes (115 g), two slices of toasted wheat bread with 15 g butter, and one glass (240 mL) of high-fat milk. The meal was consumed within 20 min, and subjects were dosed 30 min after starting breakfast, after which they fasted for 4 h. The washout period between CBD doses in the fed and fasted periods of the FE arm was 10 days.
All Trial Arms
Blood sampling for PK purposes was performed from pre-dose up to 72 h after IMP administration. Safety assessments were performed throughout the trial. Subjects were discharged after 48 (SAD and FE arms) or 72 h (MD arm) following the final IMP administration, and follow-up visits occurred 5–10 days after the last PK blood sample had been taken.
Determination of Sample Size
No prospective calculations of statistical power were made. The sample size was selected to provide information on safety, tolerability, and PK.
Materials
As THC is present as a trace impurity in the CBD formulation under development, plasma concentrations of THC and its metabolites, 11-hydroxy-Δ9-tetrahydrocannabinol (11-OH-THC) and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (11-COOH-THC), were also determined. Reference and internal standards for 6-hydroxy-cannabidiol (6-OH-CBD), 7-hydroxy-cannabidiol (7-OH-CBD), 7-carboxy-cannabidiol (7-COOH-CBD), THC, 11-OH-THC, and 11-COOH-THC bioanalysis were supplied by GW Pharma Ltd (Cambridge, UK), Cerilliant (Round Rock, TX, USA) or BDG Synthesis (CA, USA).
Plasma Sample Preparations
CBD and metabolite samples were extracted from plasma by protein precipitation with isopropyl alcohol and acetonitrile. THC and metabolite samples were extracted by liquid–liquid extraction.
It was discovered that there may have been an overestimation of 7-OH-CBD (30–50% based on comparison with new standards) concentrations in SAD samples due to technical issues with reference standard material. As a result, data obtained for 7-OH-CBD in the SAD arm should be considered with a degree of caution.
Bioanalysis and Pharmacokinetic (PK) Assessment
High-performance liquid chromatography (HPLC) with tandem mass spectrometry was performed using a Waters Acquity (Waters Corporation, Milford, MA, USA) system for HPLC, and Sciex API5000 (AB Sciex Pte. Ltd, Singapore [for analysis of CBD and metabolites]) or Waters Xevo TQS (Waters Corporation) and Sciex API5000 (AB Sciex Pte. Ltd [for analysis of THC and metabolites]) systems for mass spectrometry. These were used to quantify concentrations of CBD, THC and their metabolites 6-OH-CBD, 7-OH-CBD, 7-COOH-CBD, 11-OH-THC and 11-COOH-THC in human plasma using 200 µL sample volumes. Chromatographic separations were performed on an Acquity BEH Phenyl column (internal diameter 1.7 μm, 2.1 × 100 mm). The selectivity of the HPLC method was checked by comparing chromatograms from blank plasma samples with chromatograms from spiked samples to ensure that there were no potential interfering peaks with any analyte under investigation. Selectivity was ensured at the lower limit of quantification (LLOQ) for each analyte. Cross-interference between analytes was also assessed with samples spiked at the analytes’ upper level of quantification. The lower and upper limits of quantification for each analyte are given in Table 1.
Table 1.
Lower and upper limits of quantification of all analytes
| Analyte | LLOQ (ng/mL) | ULOQ (ng/mL) |
|---|---|---|
| CBD | 2 | 10,000 |
| 6-OH-CBD | 0.250 | 250 |
| 7-OH-CBD | 0.250 | 1250 |
| 7-COOH-CBD | 0.250 | 20,000 |
| THC | 0.125 | 62.5 |
| 11-OH-THC | 0.250 | 125 |
| 11-COOH-THC | 0.250 | 125 |
7-COOH-CBD 7-carboxy-cannabidiol, 6-OH-CBD 6-hydroxy-cannabidiol, 7-OH-CBD 7-hydroxy-cannabidiol, CBD cannabidiol, LLOQ lower limit of quantification, 11-COOH-THC 11-nor-9-carboxy-Δ9-tetrahydrocannabinol, 11-OH-THC 11-hydroxy-Δ9-tetrahydrocannabinol, THC Δ9-tetrahydrocannabinol, ULOQ upper limit of quantification
The precision [coefficient of variation (%CV)] and accuracy [relative error (RE%)/mean % different (Bias%)] of the HPLC method was acceptable for all analytes [≤ 15% (20% at the LLOQ)]. Recovery was > 90% for CBD, 6-OH-CBD, 7-OH-CBD and 7-COOH-CBD, and was considered acceptable. The recovery of THC, 11-OH-THC and 11-COOH-THC was considered adequate (between 56.3 and 124%).
It should be noted that of the 1808 samples analyzed, 45 (2.5%) for 6-OH-CBD and 20 (1.1%) for 7-OH-CBD may have been affected by a hemolysis matrix effect, but this would not have any notable effect on the results shown.
Blood samples were taken from subjects via an indwelling intravenous catheter or direct venepuncture, and centrifuged for 10 min at 2600 g, at 18°C. The resultant plasma was stored in a freezer at − 80°C. Blood samples for PK analysis were taken at the following time points:
SAD arm: pre-dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 24 and 48 h post-dose.
- MD arm, as below:
- Day 1: pre-morning dose, and 0.5, 1, 2, 2.5, 3, 4, 5, 6, 8 and 12 h post-morning dose, then at 0.5, 1, 2, 2.5, 3, 4, 5 and 12 h post-evening dose.
- Days 3, 4, 5 and 6: pre-morning dose.
- Day 7: pre-dose and 0.5, 1, 2, 2.5, 3, 4, 5, 6, 8, 12, 24, 48 and 72 h post-dose.
- FE arm: pre-dose and 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 12, 24, 48, and 72 h post-dose.
PK parameters were derived by non-compartmental analysis using WinNonlin® version 6.3. PK parameters evaluated included area under the plasma concentration-time curve from time zero to time t (AUCt), area under the concentration-time curve from time zero to infinity (AUC∞), area under the plasma concentration-time curve over a dosing interval (AUCτ; where τ is the dosing interval), maximum measured plasma concentration (Cmax), time to maximum plasma concentration (tmax), terminal elimination half-life (t½,z), oral clearance of drug from plasma (CL/F), apparent volume of distribution (Vz/F), elimination rate constant (from the central compartment; Kel), and accumulation ratio (Rac), based on AUCτ of day 7 versus day 1 (MD part only), measured using non-compartmental analysis.
Safety Assessments
The safety and tolerability of CBD were evaluated by recording the incidence and severity of AEs throughout the trial, review of clinical laboratory tests, vital signs, electrocardiogram (ECG), physical examination, sleep disruption 0–10 numerical rating scale (NRS) score, Epworth Sleepiness Scale (ESS) score, Cannabis Withdrawal Scale (CWS) scores, and Columbia-Suicide Severity Rating Scale (C-SSRS) questionnaire (MD part of the trial only). Safety data were recorded in case report forms by contract research organization staff at the clinical trial site.
Statistical Analysis
The primary objective of this study was to evaluate the safety and tolerability of a SAD and MD of CBD. Descriptive statistics of subject demographics and outcomes were based on the safety analysis set (all subjects who received one or more doses of CBD).
Secondary objectives were to assess the PK of SAD and MD regimens of CBD, as well as the effect of food on CBD PK. Secondary safety assessments included sleep disruption 0–10 NRS, ESS, CWS, and C-SSRS, which were summarized descriptively or listed.
The PK parameters of CBD, THC and its metabolites were calculated for the PK analysis set (all subjects who received one or more doses of CBD and provided sufficient bioanalytical assessments to calculate reliable estimates of the PK parameters), using Phoenix® WinNonlin® version 6.3 or higher (Pharsight Corporation, Princeton, NJ, USA). Statistical testing was two-sided and used the 5% significance level. Non-compartmental methods estimated PK parameters for all analytes with sufficient data above LLOQ from the concentration-time profiles for the PK analysis set. A regression power model, relating log-transformed Cmax and area under the curve parameters to log-transformed dose, was used to investigate dose proportionality. Individual and geometric mean dose-normalized Cmax, AUCt and AUC∞ were plotted against dose level. To analyze the FE, an analysis of variance, with a model including fixed effects for treatment, period, and sequence, and a random effect for subject within sequence, was performed on natural log-transformed Cmax, AUCt and AUC∞ data. The back-transformed least-squares means for each treatment, and the ratio of least-squares geometric means between the test treatment and the reference, was calculated (fed over fasted). The FE arm was not powered to demonstrate statistical significance. Nevertheless, the potential for an FE would be suggested if the 90% confidence interval (CI) did not contain 1.
Results
Baseline Characteristics
In total, 56 subjects enrolled into the three trial arms; 32 and 24 into the SAD and MD arms, respectively; 12 subjects from the SAD arm then took part in the integrated FE arm. The demographics of subjects in the three trial arms are summarized in Table 2. All 56 subjects completed the trial through to the final follow-up visit.
Table 2.
Subject demographics for the three arms of the trial
| Group/CBD dose | Sex [n (%)] | Race [n (%)] | Age, years [mean (SD)] | BMI, kg/m2 [mean (SD)] | |||
|---|---|---|---|---|---|---|---|
| Male | Female | White | White/Asian | White/Black/African American | |||
| Single ascending dose arm | |||||||
| Placebo (n = 8) | 3 (37.5) | 5 (62.5) | 7 (87.5) | 0 | 1 (12.5) | 23.1 (1.4) | 23.65 (2.31) |
| 1500 mg (n = 6) | 1 (16.7) | 5 (83.3) | 6 (100.0) | 0 | 0 | 26.0 (3.2) | 23.73 (2.45) |
| 3000 mg (n = 6) | 3 (50.0) | 3 (50.0) | 6 (100.0) | 0 | 0 | 25.0 (4.7) | 23.15 (2.33) |
| 4500 mg (n = 6) | 0 | 6 (100.0) | 6 (100.0) | 0 | 0 | 25.8 (7.9) | 21.38 (1.71) |
| 6000 mg (n = 6) | 2 (33.3) | 4 (66.7) | 4 (66.7) | 1 (16.7) | 1 (16.7) | 22.8 (3.2) | 23.60 (2.53) |
| Multiple dose arm | |||||||
| Placebo (n = 6) | 1 (16.7) | 5 (83.3) | 4 (66.7) | 1 (16.7) | 1 (16.7) | 23.3 (1.9) | 22.48 (2.31) |
| 750 mg (n = 9) | 2 (22.2) | 7 (77.8) | 9 (100.0) | 0 | 0 | 28.6 (8.5) | 22.28 (3.03) |
| 1500 mg (n = 9) | 5 (55.6) | 4 (44.4) | 8 (88.9) | 0 | 1 (11.1) | 25.1 (4.8) | 22.23 (2.33) |
| Food effect arma | |||||||
| 750 mg (n = 12) | 4 (33.3) | 8 (66.7) | 12 (100) | 0 | 0 | 25.1 (6.2) | 23.34 (1.90) |
BMI body mass index, CBD cannabidiol, n number of subjects, SD standard deviation
aSubjects in the food effect arm had previously completed the single ascending dose arm of the trial in either the 1500, 3000 or 4500 mg dose groups
Safety and Tolerability of Cannabidiol (CBD)
Single doses of 1500, 3000, 4500 or 6000 mg CBD were well tolerated, as were multiple doses of 750 and 1500 mg twice daily for 6 days, with a single dose on day 7.
Overall, in the SAD and MD arms of the trial, 24 (75%) and 23 (96%) subjects, respectively, reported one or more treatment-emergent AEs (TEAEs) [Tables 3 and 4]. In the FE arm, 9 (75%) and 3 (25%) subjects reported one or more TEAEs in the fed and fasted states, respectively (Table 5). Diarrhea, nausea, headache, and somnolence were the most common TEAEs reported across all arms of the trial. All TEAEs were of mild or moderate severity. There were no deaths, no subjects discontinued the trial due to AEs, and there were no other serious AEs or pregnancies.
Table 3.
All-causality treatment-emergent adverse events experienced by more than one subject per treatment group, by MedDRA preferred term, in the single ascending dose arm of the trial (safety analysis set)
| SOC (MedDRA PT) | Placebo (n = 8) | Cannabidiol, mg | |||
|---|---|---|---|---|---|
| 1500 (n = 6) | 3000 (n = 6) | 4500 (n = 6) | 6000 (n = 6) | ||
| Subjects experiencing any TEAE | 5 (62.5) | 4 (66.7) | 5 (83.3) | 5 (83.3) | 5 (83.3) |
| Gastrointestinal disorders | 4 (50.0) | 1 (16.7) | 5 (83.3) | 4 (66.7) | 4 (66.7) |
| Diarrhea | 2 (25.0) | 0 | 5 (83.3) | 3 (50.0) | 4 (66.7) |
| Abdominal discomfort | 3 (37.5) | 0 | 2 (33.3) | 0 | 0 |
| Nausea | 2 (25.0) | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) |
| Nervous system disorders | 3 (37.5) | 2 (33.3) | 2 (33.3) | 5 (83.3) | 5 (83.3) |
| Somnolence | 3 (37.5) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 4 (66.7) |
| Headache | 0 | 1 (16.7) | 1 (16.7) | 4 (66.7) | 2 (33.3) |
| Dizziness | 0 | 1 (16.7) | 0 | 1 (16.7) | 2 (33.3) |
| Disturbance in attention | 0 | 2 (33.3) | 0 | 0 | 0 |
Data are expressed as n (%)
MedDRA PT Medical Dictionary for Regulatory Activities preferred term, SOC system organ class, TEAE treatment-emergent adverse event
Table 4.
All-causality treatment-emergent adverse events experienced by more than one subject per treatment group, by MedDRA preferred term, in the multiple dose arm of the trial (safety analysis set)
| SOC (MedDRA PT) | Placebo (n = 6) | Cannabidiol, mg | |
|---|---|---|---|
| 750 (n = 9) | 1500 (n = 9) | ||
| Subjects experiencing any TEAE | 5 (83.3) | 9 (100.0) | 9 (100.0) |
| Gastrointestinal disorders | 3 (50.0) | 6 (66.7) | 9 (100.0) |
| Diarrhea | 0 | 4 (44.4) | 8 (88.9) |
| Nausea | 1 (16.7) | 3 (33.3) | 5 (55.6) |
| Abdominal discomfort | 1 (16.7) | 3 (33.3) | 1 (11.1) |
| Abdominal pain | 0 | 2 (22.2) | 2 (22.2) |
| Flatulence | 1 (16.7) | 0 | 2 (22.2) |
| Nervous system disorders | 2 (33.3) | 6 (66.7) | 9 (100.0) |
| Headache | 0 | 4 (44.4) | 4 (44.4) |
| Somnolence | 2 (33.3) | 2 (22.2) | 4 (44.4) |
| Dizziness | 0 | 0 | 3 (33.3) |
| Presyncope | 0 | 2 (22.2) | 1 (11.1) |
| Dizziness, postural | 0 | 0 | 2 (22.2) |
| General disorders and administration site conditions | 3 (50.0) | 2 (22.2) | 6 (66.7) |
| Fatigue | 2 (33.3) | 1 (11.1) | 1 (11.1) |
| Feeling cold | 0 | 0 | 2 (22.2) |
| Skin and subcutaneous tissue disorders | 1 (16.7) | 2 (2.2) | 3 (33.3) |
| Dermatitis contact | 1 (16.7) | 2 (22.2) | 0 |
| Rash | 0 | 0 | 2 (22.2) |
| Musculoskeletal and connective tissue disorders | 1 (16.7) | 0 | 4 (44.4) |
| Myalgia | 1 (16.7) | 0 | 3 (33.3) |
| Infections and infestations | 0 | 2 (22.2) | 0 |
| Nasopharyngitis | 0 | 2 (22.2) | 0 |
| Psychiatric disorders | 0 | 0 | 2 (22.2) |
| Insomnia | 0 | 0 | 2 (22.2) |
Data are expressed as n (%)
MedDRA PT Medical Dictionary for Regulatory Activities preferred term, SOC system organ class, TEAE treatment-emergent adverse event
Table 5.
All-causality treatment-emergent adverse events experienced by more than one subject per treatment group, by MedDRA preferred term, in the food effect arm of the trial (safety analysis set)
| SOC (MedDRA PT) | Cannabidiol, mg | |
|---|---|---|
| 1500, fasted (n = 12) |
1500, fed (n = 12) |
|
| Subjects experiencing any TEAE | 3 (25.0) | 9 (75.0) |
| Gastrointestinal disorders | 3 (25.0) | 6 (50.0) |
| Nausea | 0 | 4 (33.3) |
| Diarrhea | 3 (25.0) | 0 |
| Abdominal discomfort | 0 | 2 (16.7) |
| Nervous system disorders | 1 (8.3) | 6 (50.0) |
| Headache | 1 (8.3) | 4 (33.3) |
| Somnolence | 1 (8.3) | 3 (25.0) |
| Infections and infestations | 0 | 2 (16.7) |
| Nasopharyngitis | 0 | 2 (16.7) |
Data are expressed as n (%)
MedDRA PT Medical Dictionary for Regulatory Activities preferred term, SOC system organ class, TEAE treatment-emergent adverse event
In the SAD arm, TEAEs in the nervous system disorders system organ class (SOC) affected a greater proportion of subjects taking CBD than placebo. Most notably, headache and dizziness affected 8 (33%) and 4 (17%) subjects taking CBD, respectively, compared with no subjects taking placebo. In the gastrointestinal disorders SOC, diarrhea also affected a greater proportion of subjects taking CBD (12 [50%]) than those taking placebo (2 [25%]). Otherwise, the TEAE incidence was similar across the treatment groups (Table 3).
In the MD arm, a similar proportion of subjects experienced TEAEs in the CBD and placebo groups (Table 4). Most TEAEs in the gastrointestinal disorders SOC affected a greater proportion of subjects taking CBD than placebo, most notably diarrhea [12 (67%) subjects taking CBD vs. 0 subjects taking placebo] and nausea [8 (44%) subjects taking CBD vs. 1 (17%) subject taking placebo]. In addition, more subjects taking CBD were affected by TEAEs in the nervous system disorders SOC than subjects taking placebo, most notably headache [8 (44%) subjects taking CBD vs. 0 subjects taking placebo], dizziness [3 (33%) subjects taking CBD vs. 0 subjects taking placebo] and presyncope [3 (33%) subjects taking CBD vs. 0 subjects taking placebo]. Three subjects had TEAEs of interest. All three subjects were in the 1500 mg CBD group in the MD arm, and all experienced mild TEAEs of rash (preferred terms: two rash and one rash maculopapular; none with mucosal involvement), all of which started after the last CBD dose (range 5–70 h) and recovered within 6–12 days from date of onset. In the FE arm, there were more subjects with TEAEs in the fed group than in the fasted group (Table 5).
There were no clinically significant findings for laboratory parameters, physical examination, vital signs, ECG or body weight in any arm of the trial. Furthermore, there was no consistent effect of CBD on sleep or sleepiness (as assessed by sleep disruption 0–10 NRS score and the ESS), and no clear evidence of any drug withdrawal syndrome during the trial. According to C-SSRS responses, no subject reported any suicide-related thoughts or behaviors in the MD part of the trial.
Single Ascending Dose PK
Figure 1 shows the geometric mean plasma concentration-time profiles, and Table 6 summarizes the PK parameters of CBD and its metabolites after single CBD doses.
Fig. 1.

Single ascending CBD dose (1500, 3000, 4500 or 6000 mg) geometric mean plasma concentration-time profiles for CBD and its major metabolites (semi-logarithmic scale; pharmacokinetic set). 6-OH-CBD 6-hydroxy-cannabidiol, 7-OH-CBD 7-hydroxy-cannabidiol, 7-COOH-CBD 7-carboxy-cannabidiol, CBD cannabidiol
Table 6.
Pharmacokinetic parameters of cannabidiol and metabolites for the single ascending dose arm of the trial
| Pharmacokinetic parameter (unit) | Cannabidiol, mg | |||
|---|---|---|---|---|
| 1500 (n = 6) | 3000 (n = 6) | 4500 (n = 6) | 6000 (n = 6) | |
| CBD | ||||
| Cmax (ng/mL)a | 292.4 (87.9) | 533.0 (35.1) | 722.1 (52.3) | 782 (83.0) |
| AUCt (ng·h/mL)a | 1517 (78.2) | 2669 (36.4) | 3215 (50.3) | 3696 (79.9) |
| AUC∞ (ng·h/mL)a | 1618 (74.6) | 2802 (35.5) | 3426 (48.3) | 3900 (79.3) |
| tmax (h)b | 4.00 (3.00–5.00) | 5.00 (3.00–5.00) | 5.00 (5.00–5.00) | 5.00 (3.00–5.02) |
| t½,z (h)c | 14.43 (36.1) | 14.39 (14.9) | 16.61 (18.7) | 15.42 (29.0) |
| CL/F (L/h)c | 1111 (67.2) | 1121 (30.5) | 1445 (52.6) | 1909 (77.3) |
| Vz/F (L/h)c | 20,963 (55.3) | 23,357 (32.9) | 36,575 (66.8) | 42,849 (75.5) |
| 6-OH-CBD | ||||
| Cmax (ng/mL)a | 10.7 (65.7) | 14.4 (28.6) | 14.5 (36.5) | 23.5 (68.6) |
| AUCt (ng·h/mL)a | 91.23 (66.7) | 121.1 (14.7) | 127.1 (53.1) | 160.7 (91.4) |
| AUC∞ (ng·h/mL)a | 132.5 (93.7)d | 126.0 (18.2)e | 127.1 (50.3)e | 215.0 (92.0)f |
| tmax (h)b | 4.00 (2.50–4.00) | 4.50 (2.50–5.00) | 5.00 (4.00–5.00) | 5.00 (3.00–5.02) |
| t½,z (h)c | 40.75 (73.6) | 22.78 (81.9)d | 33.92 (55.9) | 28.67 (104.5) |
| 7-OH-CBD | ||||
| Cmax (ng/mL)a | 238.7 (84.0) | 332.2 (30.2) | 404.8 (37.3) | 515.8 (42.3) |
| AUCt (ng·h/mL)a | 1616 (76.9) | 1959 (24.1) | 2810 (40.3) | 3299 (63.5) |
| AUC∞ (ng·h/mL)a | 1826 (74.6) | 2143 (23.2) | 3039 (42.2) | 3531 (63.1) |
| tmax (h)b | 3.50 (2.50–4.00) | 4.50 (3.00–5.00) | 5.00 (4.00–5.00) | 5.00 (3.00–5.02) |
| t½,z (h)c | 18.70 (11.4) | 15.42 (32.5) | 14.89 (22.4) | 14.46 (23.1) |
| 7-COOH-CBD | ||||
| Cmax (ng/mL)a | 3060 (62.4) | 3557 (34.7) | 5120 (19.7) | 4591 (44.3) |
| AUCt (ng·h/mL)a | 60,467 (85.4) | 74,660 (40.2) | 111,025 (39.7) | 107,022 (74.1) |
| AUC∞ (ng·h/mL)a | 75,869 (95.1)e | 90,818 (47.5)d | 132,731 (51.1)d | 113,026 (42.3)e |
| tmax (h)b | 4.00 (4.00–5.00) | 5.00 (4.00–5.00) | 5.00 (4.00–8.00) | 5.01 (4.00–8.00) |
| t½,z (h)c | 25.98 (26.5) | 23.88 (40.5) | 25.18 (25.3) | 30.24 (52.7) |
AUCt area under the plasma concentration-time curve from time zero to time t, AUC∞ area under the concentration-time curve from time zero to infinity, 7-COOH-CBD 7-carboxy–cannabidiol, 6-OH-CBD 6–hydroxy-cannabidiol, 7-OH-CBD 7–hydroxy-cannabidiol, CBD cannabidiol, CL/F oral clearance of drug from plasma, Cmax maximum measured plasma concentration, CV% percentage coefficient of variation, tmax time to maximum plasma concentration, t½,z terminal elimination half-life, Vz/F apparent volume of distribution
aGeometric mean (geometric CV%)
bMedian (range)
cArithmetic mean (arithmetic CV%)
dn = 4
en = 3
fn = 5
Over the CBD dose range investigated (1500–6000 mg), CBD Cmax increased with a trend to less than dose proportionality (slope 0.73; 90% CI 0.35–1.12), as did AUCt (slope 0.64; 90% CI 0.27–1.01). The Cmax and AUCs of all metabolites of CBD increased in a less than dose-related manner. CBD appeared rapidly in plasma, with a tmax of 4–5 h, which was independent of dose. The tmax for the CBD metabolites were similar, at approximately 3.5–5 h. The t½,z was in the range of 14–17 h (independent of dose) for CBD, and between 23 and 41 h for 6-OH-CBD, 14–19 h for 7-OH-CBD, and 25–30 h for 7-COOH-CBD. Analysis of the terminal slope suggested that the t½,z for CBD and its metabolites may not have been fully characterized over the 48-h sampling period, thus likely underestimating the terminal slope.
The CL/F of CBD was high (1111–1909 L/h), with a large Vz/F (20,963–42,849 L), suggesting extensive biotransformation of CBD and a wide distribution in the body. Both CL/F and Vz/F increased with dose. The 7-COOH-CBD metabolite was the major circulating product, followed by parent drug CBD, 7-OH-CBD, and 6-OH-CBD.
In all trial arms, THC was detected in plasma, generally at trace levels observed only around tmax, with most subjects presenting with less than the LLOQ (< 0.125 ng/mL) at most time points. As such, no PK parameters could be estimated. These data were consistent with the low levels of THC in the formulation and demonstrate a lack of conversion in vivo of CBD to THC.
Multiple Dose PK
Figure 2 shows the geometric mean plasma concentration-time profiles, and Table 7 summarizes the PK parameters of CBD and its metabolites after single and multiple CBD doses (twice daily on days 1–6, and a single dose on day 7).
Fig. 2.

Multiple CBD dose (1500 mg) geometric mean plasma concentration-time profiles for CBD and its major metabolites (semi-logarithmic scale; pharmacokinetic set). 6-OH-CBD 6-hydroxy-cannabidiol, 7-OH-CBD 7-hydroxy-cannabidiol, 7-COOH-CBD 7-carboxy-cannabidiol, CBD cannabidiol
Table 7.
Pharmacokinetic parameters of cannabidiol and metabolites for multiple dose arm of the trial
| Pharmacokinetic parameter (unit) | 750 mg CBD (n = 9) | 1500 mg CBD (n = 9) | ||||
|---|---|---|---|---|---|---|
| Day 1, am | Day 1, pm | Day 7, am | Day 1, am | Day 1, pm | Day 7, am | |
| CBD | ||||||
| Cmax (ng/mL)a | 290.8 (86.3) | 732.4 (39.4) | 330.3 (40.8) | 361.8 (105.8) | 1385 (52.4) | 541.2 (53.7) |
| AUCτ (ng·h/mL)a | 1070 (74.6) | 2683 (33.4)d | 1745 (38.4) | 1444 (101.4) | 9819 (32.3)e | 3236 (44.0) |
| tmax (h)b | 5.00 (2.50–5.00) | 2.50 (2.00–5.00) | 3.00 (2.50–5.00) | 5.00 (2.50–5.00) | 4.00 (2.50–5.00) | 3.00 (2.00–4.00) |
| t½,z (h)c | – | – | 56.41 (32.6) | – | – | 60.54 (20.2) |
| Racc | – | 2.914 (44.7) | 1.795 (45.4) | – | 6.319 (62.8) | 2.618 (59.3) |
| 6-OH-CBD | ||||||
| Cmax (ng/mL)a | 8.2 (82.6) | 15.3 (17.0) | 12.8 (43.6) | 9.0 (96.1) | 21.4 (53.4) | 16.3 (82.8) |
| AUCτ (ng·h/mL)a | 47.60 (34.4)f | 71.90 (28.2)g | 93.35 (38.6)d | 30.50 (187.3)h | 96.41 (104.5)e | 104.3 (82.3) |
| tmax (h)b | 5.00 (2.50–6.00) | 2.50 (2.00–5.00) | 2.50 (2.00–5.00) | 4.03 (3.00–5.00) | 3.00 (2.07–5.00) | 3.00 (2.50–5.00) |
| t½,z (h)c | – | – | 21.54 (25.4)d | – | – | 82.21 (198.7) |
| Racc | – | 1.828 (31.3)g | 2.185 (55.4)g | – | 3.580 (45.4) | 3.330 (83.8)d |
| 7-OH-CBD | ||||||
| Cmax (ng/mL)a | 123.0 (90.1) | 197.2 (21.7) | 152.6 (33.9) | 139.5 (117.0) | 305.2 (77.7) | 187.9 (57.7) |
| AUCτ (ng·h/mL)a | 699.0 (59.6)f | 958.7 (33.3)g | 976.3 (27.9) | 457.4 (207.8)h | – | 1246 (58.8) |
| tmax (h)b | 4.00 (2.50–6.00) | 2.50 (2.00–5.00) | 2.50 (2.00–5.00) | 4.03 (3.00–5.00) | 4.00 (2.50–5.00) | 3.00 (2.50–5.00) |
| t½,z (h)c | – | – | 24.73 (24.6) | – | – | 31.70 (19.4) |
| Racc | – | 1.59 (42.7)g | 1.61 (55.8)g | – | 3.19 (47.3)d | 2.46 (70.3) |
| 7-COOH-CBD | ||||||
| Cmax (ng/mL)a | 2785 (56.0) | 5307 (32.5) | 9824 (57.1) | 2748 (120.8) | 9776 (50.0) | 16,306 (61.3) |
| AUCτ (ng·h/mL)a | 20,807 (60.5) | 41,656 (60.8)f | 86,179 (60.0) | 20,526 (135.7) | 86,076 (74.2)i | 151,336 (66.4) |
| tmax (h)b | 5.00 (4.00–5.00) | 4.00 (3.00–12.00) | 4.00 (3.00–5.00) | 5.00 (4.00–12.00) | 12.00 (5.00–12.00) | 3.00 (0.53–5.00) |
| t½,z (h)c | – | – | 21.32 (14.1) | – | – | 22.00 (16.9) |
| Racc | – | 2.52 (29.3) | 4.54 (47.0) | – | 4.73 (62.3) | 9.59 (79.4) |
AUCτ area under the plasma concentration-time curve over a dosing interval, where τ is the dosing interval, 7-COOH-CBD 7-carboxy-cannabidiol, 6-OH-CBD 6-hydroxy-cannabidiol, 7-OH-CBD 7-hydroxy-cannabidiol, CBD cannabidiol, Cmax maximum measured plasma concentration, CV% percentage coefficient of variation, Rac accumulation ratio, tmax time to maximum plasma concentration, t½,z terminal elimination half-life
aGeometric mean (geometric CV%)
bMedian (range)
cArithmetic mean (arithmetic CV%)
dn = 8
en = 2
fn = 4
gn = 7
hn = 5
in = 6
Visual inspection of the Ctrough data (not presented) indicated that steady-state CBD, 6-OH-CBD and 7-OH-CBD plasma concentrations were reached at approximately 2 days, and at approximately 4 days for 7-COOH-CBD. At steady state, there was an almost doubling in exposure for a twofold increase in dose (750–1500 mg); Cmax increased by approximately 1.6-fold (330 and 541 ng/mL after 750 and 1500 mg CBD, respectively, on day 7) and AUCτ increased by approximately 1.9-fold (1745 and 3236 ng·h/mL for 750 and 1500 mg CBD, respectively, on day 7). At steady state, the tmax of CBD and its metabolites occurred at approximately 3 h, independent of dose. Plasma concentrations declined for CBD on day 7 in multiphasic manner, with a mean t½,z of 56 and 61 h for the 750 and 1500 mg CBD doses, respectively.
There was moderate accumulation with multiple CBD doses; on the morning of day 7, compared with the morning of day 1, Rac increased by 1.8- and 2.6-fold after 750 and 1500 mg CBD doses, respectively. The extent of accumulation was similar for CBD metabolites, except 7-COOH-CBD, for which Rac was 4.5- and 9.6-fold after 750 and 1500 mg CBD, respectively. There was also accumulation between the initial morning and evening CBD doses; on the evening of day 1, versus the morning of day 1, Rac increased by 2.9- and 6.3-fold after 750 and 1500 mg CBD, respectively. This within-day effect was also observed for the CBD metabolites.
At steady state, the 7-COOH-CBD metabolite was the major circulating product (47-fold higher than CBD after 1500 mg CBD, and 97% of total drug material measured), followed by parent drug CBD, 7-OH-CBD and 6-OH-CBD.
Effect of Food on CBD PK
In the fed state, all subjects had increased exposure to CBD (Cmax and AUC) compared with the fasted state (Fig. 3a and b). Table 8 summarizes the PK parameters of CBD and its metabolites in the fasted and fed state.
Fig. 3.
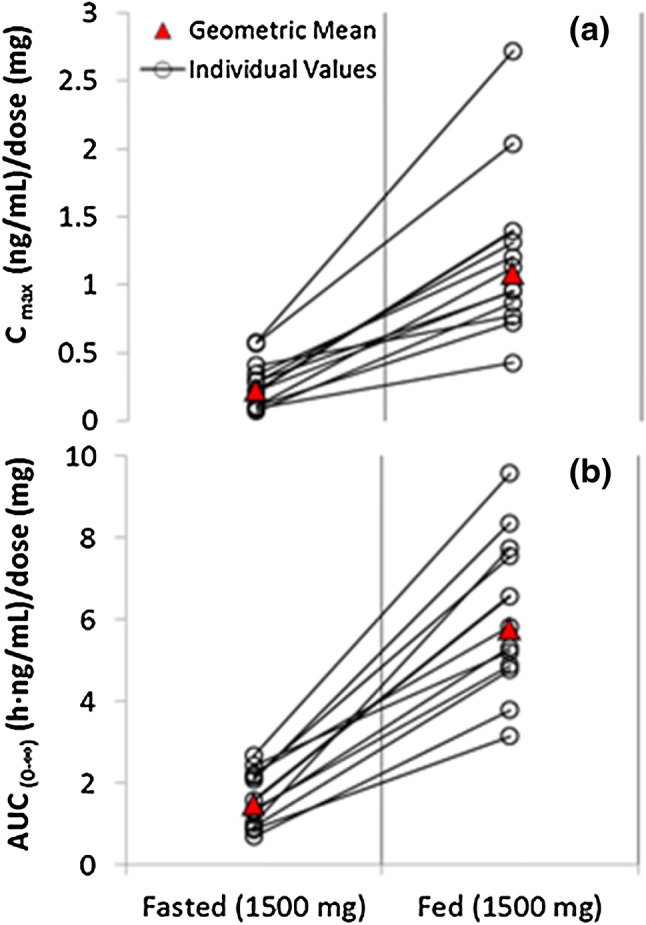
Geometric mean and individual (expressed as dose normalized) Cmax (a) and AUC∞ (b) of CBD for the fasted and fed state following a single 1500 mg dose of CBD (pharmacokinetic set). AUC∞ area under the concentration-time curve from time zero to infinity, CBD cannabidiol, Cmax maximum measured plasma concentration
Table 8.
Pharmacokinetic parameters of cannabidiol and metabolites for the food effect arm of the trial
| Pharmacokinetic parameter (unit) | 1500 mg CBD, fasted (n = 12) | 1500 mg CBD, fed (n = 12) |
|---|---|---|
| CBD | ||
| Cmax (ng/mL)a | 335.4 (81.3) | 1628 (51.4) |
| AUCt (ng·h/mL)a | 1987 (53.6) | 8347 (34.1) |
| AUC∞ (ng·h/mL)a | 2198 (48.2) | 8669 (33.9) |
| tmax (h)b | 3.50 (2.50–5.03) | 3.00 (1.50–5.00) |
| t½,z (h)c | 30.33 (28.5) | 24.40 (16.0) |
| CL/F (L/h)c | 751.8 (46.6) | 182.0 (34.2) |
| Vz/F (L/h)c | 34,101 (60.5) | 6349 (31.6) |
| 6-OH-CBD | ||
| Cmax (ng/mL)a | 9.6 (47.7) | 26.8 (47.0) |
| AUCt (ng·h/mL)a | 96.93 (44.0) | 255.6 (40.2) |
| AUC∞ (ng·h/mL)a | 113.3 (34.0)d | 279.1 (39.7) |
| tmax (h)b | 2.50 (1.00–5.00) | 3.00 (0.50–5.00) |
| t½,z (h)c | 20.31 (30.4)d | 22.80 (53.3) |
| 7-OH-CBD | ||
| Cmax (ng/mL)a | 135.2 (54.7) | 393.1 (35.8) |
| AUCt (ng·h/mL)a | 1065 (43.2) | 3365 (32.7) |
| AUC∞ (ng·h/mL)a | 1051 (37.0)e | 3467 (32.5) |
| tmax (h)b | 2.75 (1.50–5.03) | 3.00 (1.50–5.00) |
| t½,z (h)c | 18.74 (18.7)e | 15.88 (17.8) |
| 7-COOH-CBD | ||
| Cmax (ng/mL)a | 2426 (61.0) | 5044 (33.8) |
| AUCt (ng·h/mL)a | 56,865 (56.6) | 137,421 (32.9) |
| AUC∞ (ng·h/mL)a | 65,496 (54.2) | 154,160 (34.1) |
| tmax (h)b | 4.00 (3.00–5.03) | 5.00 (3.00–6.02) |
| t½,z (h)c | 23.51 (26.0) | 21.20 (20.9) |
AUCt area under the plasma concentration-time curve from time zero to time t, AUC∞ area under the concentration-time curve from time zero to infinity, 7-COOH-CBD 7-carboxy-cannabidiol, 6-OH-CBD 6-hydroxy-cannabidiol, 7-OH-CBD 7-hydroxy-cannabidiol, CBD cannabidiol, CL/F oral clearance of drug from plasma, Cmax maximum measured plasma concentration, CV% percentage coefficient of variation, tmax time to maximum plasma concentration, t½,z terminal elimination half-life, Vz/F apparent volume of distribution
aGeometric mean (geometric CV%)
bMedian (range)
cArithmetic mean (arithmetic CV%)
dn = 10
en = 11
Following a high-fat breakfast, there was a significant increase in Cmax for CBD of 4.85-fold (90% CI 4.01–5.87), and a similar increase in AUCt of 4.2-fold (90% CI 3.63–4.85). The extent of the FE was also reflected in the exposure to the CBD metabolites. Statistical analysis revealed a period effect (for Cmax, AUCt and AUC∞), but no sequence effect for CBD, suggesting that there was a residual level of drug after a 10-day washout. These results demonstrate that the bioavailability of CBD is increased by food.
There was no obvious effect of food on tmax of CBD (fasted 3.5 h; fed 3.0 h) or its metabolites. There was no obvious effect of food on the t½,z of CBD or its metabolites. The differences in CBD t½,z observed in the FE arm (fasted 30 h; fed 24 h) of the trial compared with the SAD arm (14–17 h, independent of dose) reflect the longer sampling to 72 h in the FE arm versus 48 h in the SAD arm. As such, the t½,z values in the FE arm are likely to represent a better estimate of the single-dose t½,z.
Discussion
This trial was the first to investigate the single (1500, 3000, 4500 or 6000 mg) and multiple (750 or 1500 mg twice daily for 6 days, with a single dose on the morning of day 7) dose PK of this CBD formulation, as well as the effects of food on CBD PK using a standard fed–fasted crossover design. CBD is an oral formulation recently approved by the FDA for the treatment of two severe and refractory pediatric-onset epilepsy syndromes—LGS and DS. The PK profile of single and multiple doses is therefore of clinical interest.
Safety
Historically, doses up to 1500 mg CBD per day (different formulations) for 4 weeks were reported to be well tolerated in humans [15, 16]. In recent studies in epilepsy and tuberous sclerosis complex patients, open-label use of the current CBD formulation for up to 3 months was well tolerated at doses up to 50 mg/kg/day (equivalent to 3500 mg/day in a 70 kg person) [1–4, 17, 18].
In the current trial, demographic and baseline characteristics were similar across all arms, and single doses up to 6000 mg and multiple doses up to 1500 mg twice daily were well tolerated. Only mild and moderate AEs were reported during the trial. There were no deaths or severe or serious AEs, no discontinuations due to AEs, or any clinically significant laboratory, vital sign, ECG or physical examination findings. The most common TEAEs in all trial arms were diarrhea, nausea, headache, and somnolence. Some TEAEs in the gastrointestinal disorders and nervous system disorders SOCs were more common in subjects taking CBD compared with placebo, most notably diarrhea and headache. In patients with epilepsy taking up to 50 mg/kg/day CBD, the most common side effects were somnolence, decreased appetite, diarrhea, pyrexia and fatigue [1, 2, 4, 18]. In tuberous sclerosis complex patients taking up to 50 mg/kg/day CBD, the most common side effects were drowsiness, ataxia and diarrhea [17].
Three AEs of mild rash experienced by three subjects were considered events of interest; however, all three cases started after the last CBD dose was administered, none were considered related to CBD treatment, and all recovered within 6–12 days following AE onset. In the FE arm, there were more subjects with TEAEs under fed conditions than under fasted conditions, which coincided with a higher exposure to CBD and all its metabolites when subjects were fed compared with fasted. However, there was no worsening in the severity of TEAEs in the fed state, hence there is no appreciable safety concern. Of note, a high-fat meal is not a typical diet that would be consumed by most patients, but was designed to explore a worst-case scenario.
Single and Multiple Dose CBD PK
After both single and multiple dosing, and irrespective of dose, CBD and its metabolites appeared rapidly in plasma. The most abundant CBD metabolite was 7-COOH-CBD, followed by 7-OH-CBD, with 6-OH-CBD being the least abundant. Of note, recent non-clinical data showed that 7-OH-CBD was an active metabolite, exerting significant anticonvulsant activity in an animal model of epilepsy. A separate study showed no anticonvulsant effect of 7-COOH-CBD [19].
After single dosing, there was a trend to a less than dose-proportional increase in exposure to CBD and its metabolites. CL/F and Vz/F increased with dose. As there were no important differences in t½,z with CBD dose, these observations most likely reflect a decrease in bioavailability with rising dose. Given the high lipophilicity of CBD (log p value > 5), solubility-limited absorption at higher doses is a possible explanation for these observations. However, in the MD arm, for a twofold increase in dose (750 or 1500 mg) there was an almost doubling in exposure. With multiple CBD doses, t½,z of CBD was estimated to be 61 h with 1500 mg CBD, compared with 14 h after a single 1500 mg dose in the SAD arm. This difference may partly reflect the longer PK blood sampling in MD compared with SAD (72 vs. 48 h), and may also be suggestive of a degree of time-dependency in the elimination of CBD upon multiple dosing.
Based on visual inspection of Ctrough data, steady-state plasma concentrations were reached at approximately 2 days for CBD, 6-OH-CBD and 7-OH-CBD, and at approximately 4 days for 7-COOH-CBD, earlier than would be predicted from t½,z alone. For most drugs with simple, linear elimination kinetics, use of t½,z may be appropriate to help construct dosing regimens and to predict systemic accumulation. However, for drugs such as CBD that display relatively slower oral absorption and apparently wide tissue distribution, resulting in a multi-exponential decline in plasma concentration, the commonly calculated t½,z may be somewhat misleading. The most probable explanation for the PK observed in this trial is that the CBD terminal elimination phase accounts for a very small portion of the overall AUC, and thereby contributes negligibly to accumulation of the drug upon multiple dosing. To explain this disconnect between terminal t½,z and accumulation, other terms have been sought, such as effective half-life (t½,eff), which better describes the rate of drug accumulation, and systemic removal across the entire dosing interval [20, 21]. Using t½,eff better allows us to gauge CBD clearance at steady state.
Unlike t½,z, which is calculated using the slope of the last drug elimination phase following dose cessation, t½,eff takes into consideration the entire concentration-time profile of a drug. The t½,eff is calculated based on both the drug-dosing interval and drug accumulation over time following multiple-dose administration, as shown in Eq. 1.
| 1 |
Applying Eq. 1 to CBD (using mean Rac of 1.80 and 2.62 for the 750 and 1500 mg doses, respectively) gave a t½,eff estimate in the range of 10–17 h, which most likely describes the majority of CBD elimination from the body.
The extent of accumulation was similar for CBD metabolites, except for 7-COOH-CBD. There was a within-day effect with regard to exposure to CBD on day 1 after the evening dose, compared with the morning dose (Rac 2.9- and 6.3-fold after 750 and 1500 mg CBD, respectively). This within-day effect was also observed for CBD metabolites, and likely reflects both accumulation and the differences in prandial state between morning, following overnight fasting, and evening administration.
The PK of CBD have favorable characteristics for epilepsy treatment following twice-daily dosing, with a low peak-to-trough ratio, which could diminish the risk for seizure breakthrough in the case of poor compliance or missed doses.
Effect of Food on CBD PK
High-fat meals ingested at the time of dosing can increase bile salt secretion, which solubilizes drugs and enhances absorption via transport through hydrophobic barriers [22]. As CBD is a highly lipophilic product with low aqueous solubility, food appears to increase its bioavailability. When subjects were administered 1500 mg CBD with a high-fat breakfast, CBD exposure (Cmax and AUCt) increased notably. The estimated ratio of geometric least-squares means of Cmax and AUCt for CBD, after administration of CBD during fed/fasting conditions, was 4.85- and 4.2-fold, respectively. The extent of the FE was also reflected in the exposure to the CBD metabolites. There was no obvious FE on tmax or t½,z of CBD or its metabolites. Statistical analysis of the FE revealed a significant period effect for Cmax and AUC (AUCt and AUC∞), but no sequence effect for CBD, suggesting that there was a residual level of drug after a 10-day washout. Food may therefore represent a significant source of variability of CBD. It is likely that patients who take CBD with food would benefit from reduced within-day variability, and the risk of poor bioavailability would be minimized. Taking CBD consistently with food could maximize treatment outcomes.
Δ9-Tetrahydrocannabinol PK
Extracts from the plants used to formulate CBD also contain ≤ 0.1% (w/w) THC as a minor impurity. Due to the psychoactive properties of THC, plasma concentrations of THC and its metabolites were also monitored in this trial. Exposure to THC was low throughout the trial, even at the highest single (6000 mg) and multiple (1500 mg twice daily) CBD doses; plasma concentrations were below the LLOQ in most subjects at most time points.
Trial Limitations
Performing the trial in healthy volunteers rather than in patients is a limitation; however, reports of CBD PK in patients, as well as potential drug–drug interactions with some commonly prescribed antiepileptic drugs, are emerging, with some data already available [2, 13, 14]. Another trial limitation was that a comparison of 7-OH-CBD-certified solution data with the original 7-OH-CBD reference material data indicated a decrease in the reference material purity, probably caused by water absorption during storage. This was likely to have caused an overestimation of the SAD sample 7-OH-CBD concentrations. It was calculated that this would have resulted in a potential 30–50% overestimation based on comparison with new standards. Indeed, when these values were compared with other single-dose arms in the study (FE fasted arm, and day 1 in the MD arm), the Cmax values were consistent and lower than those reported in the SAD arm, thus tending to support this observation was due to a bioanalytical bias. Steady-state exposures were unaffected.
Conclusions
The results of this trial show that oral administration of CBD at doses up to 6000 mg was well tolerated. PK analysis showed that Cmax and AUC increased with dose in a slightly less than dose-proportional manner after a single administration (1500–6000 mg). A change in bioavailability is the most likely explanation for this finding. Following multiple dosing, exposures increased in an almost dose-proportional manner (750 or 1500 mg twice daily). CBD was rapidly and extensively metabolized to its active metabolite 7-OH-CBD, and primarily to 7-COOH-CBD, which represented 97% of all drug-related products measured in plasma, indicating low absolute bioavailability of CBD. Steady state was achieved rapidly, with moderate accumulation of CBD after multiple doses, likely reflecting a short t½,eff. These results support twice-daily administration of CBD and were consistent with dosing schedules of other concomitant antiepileptic drugs. Food increased the bioavailability of CBD and, as such, administering CBD with food would maximize bioavailability and likely reduce within-day fluctuation in systemic exposure to drug.
Acknowledgments
The authors would like to thank the volunteers who took part in the trial, as well as the staff who assisted with the trial. They would also like to acknowledge and thank Sue Ditton (Senior Clinical Project Manager at GW Research Ltd) for her contributions to the management of the trial, and Linda White (Senior Clinical Project Manager at GW Research Ltd) for her early review of the manuscript.
Data Availability Statement
The datasets generated and/or analyzed during the current trial are not yet publicly available.
Compliance with Ethical Standards
Funding
This trial was funded by GW Research Ltd. The open-access fee was paid by Greenwich Biosciences, Inc.
Conflicts of Interest
Lesley Taylor, Gilmour Morrison and Bola Tayo are employed by GW Research Ltd and have shares in the company. Graham Blakey and Barry Gidal have received consultancy fees from GW Research Ltd.
Research Involving Human Participants
This trial was conducted in accordance with International Conference on Harmonisation Good Clinical Practice guidelines and ethical principles that have their origin in the Declaration of Helsinki. Protocols were approved by an Independent Ethics Committee (Foundation Evaluation of Ethics in Biomedical Research, Assen, The Netherlands) before eligibility screening.
Informed Consent
Written informed consent was obtained from each subject before any trial-related procedures were performed.
References
- 1.Devinsky O, Cross JH, Laux L, et al. Cannabidiol in Dravet Syndrome Study Group. Trial of Cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011–2020. doi: 10.1056/NEJMoa1611618. [DOI] [PubMed] [Google Scholar]
- 2.Devinsky O, Patel AD, Thiele EA, et al. Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology. 2018;90(14):e1204–e1211. doi: 10.1212/WNL.0000000000005254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox–Gastaut syndrome. N Engl J Med. 2018;378(20):1888–1897. doi: 10.1056/NEJMoa1714631. [DOI] [PubMed] [Google Scholar]
- 4.Thiele EA, Marsh ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10125):1085–1096. doi: 10.1016/S0140-6736(18)30136-3. [DOI] [PubMed] [Google Scholar]
- 5.Ibeas Bih C, Chen T, Nunn AV, Bazelot M, Dallas M, Whalley BJ. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics. 2015;12(4):699–730. doi: 10.1007/s13311-015-0377-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henstridge CM, Balenga NA, Kargl J, et al. Minireview: recent developments in the physiology and pathology of the lysophosphatidylinositol-sensitive receptor GPR55. Mol Endocrinol. 2011;25(11):1835–1848. doi: 10.1210/me.2011-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sylantyev S, Jensen TP, Ross RA, Rusakov DA. Cannabinoid and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc Natl Acad Sci USA. 2013;110(13):5193–5198. doi: 10.1073/pnas.1211204110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McPartland JM, Duncan M, Di Marzo V, Pertwee RG. Are cannabidiol and Δ(9)-tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol. 2015;172(3):737–753. doi: 10.1111/bph.12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011;89(5–6):165–170. doi: 10.1016/j.lfs.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 10.Zendulka O, Dovrtělová G, Nosková K, et al. Cannabinoids and cytochrome P450 interactions. Curr Drug Metab. 2016;17(3):206–226. doi: 10.2174/1389200217666151210142051. [DOI] [PubMed] [Google Scholar]
- 11.Mazur A, Lichti CF, Prather PL, et al. Characterization of human hepatic and extrahepatic UDP-glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos. 2009;37(7):1496–1504. doi: 10.1124/dmd.109.026898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46(1):86–95. doi: 10.3109/03602532.2013.849268. [DOI] [PubMed] [Google Scholar]
- 13.Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. 2015;56(8):1246–1251. doi: 10.1111/epi.13060. [DOI] [PubMed] [Google Scholar]
- 14.Gaston TE, Bebin EM, Cutter GR, Liu Y, Szaflarski JP. UAB CBD Program. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. 2017;58(9):1586–1592. doi: 10.1111/epi.13852. [DOI] [PubMed] [Google Scholar]
- 15.Zuardi AW, Morais SL, Guimarães FS, Mechoulam R. Antipsychotic effect of cannabidiol. J Clin Psychiatry. 1995;56(10):485–486. [PubMed] [Google Scholar]
- 16.Bergamaschi MM, Queiroz RH, Zuardi AW, Crippa JA. Safety and side effects of cannabidiol, a Cannabis sativa constituent. Curr Drug Saf. 2011;6(4):237–249. doi: 10.2174/157488611798280924. [DOI] [PubMed] [Google Scholar]
- 17.Hess EJ, Moody KA, Geffrey AL, et al. Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia. 2016;57(10):1617–1624. doi: 10.1111/epi.13499. [DOI] [PubMed] [Google Scholar]
- 18.Devinsky O, Marsh E, Friedman D, et al. Cannabidiol in patients with treatment resistant epilepsy: an open-label interventional trial. The Lancet Neurology. 2016;15:270–278. doi: 10.1016/S1474-4422(15)00379-8. [DOI] [PubMed] [Google Scholar]
- 19.Whalley BJ, Stott C, Gray RA. The human metabolite of cannabidiol, 7-hydroxy-cannabidiol, but not 7-carboxy-cannabidiol, is anticonvulsant in the maximal electroshock threshold test (MEST) in mouse [abstract no. 1.435]. AES Conference; 2017.
- 20.Boxenbaum H, Battle M. Effective half-life in clinical pharmacology. J Clin Pharmacol. 1995;35(8):763–766. doi: 10.1002/j.1552-4604.1995.tb04117.x. [DOI] [PubMed] [Google Scholar]
- 21.Gidal BE, Clark AM, Anders B, Gilliam F. The application of half-life in clinical decision making: comparison of the pharmacokinetics of extended-release topiramate (USL255) and immediate-release topiramate. Epilepsy Res. 2017;129:26–32. doi: 10.1016/j.eplepsyres.2016.10.020. [DOI] [PubMed] [Google Scholar]
- 22.Moghimipour E, Ameri A, Handali S. Absorption-enhancing effects of bile salts. Molecules. 2015;20(8):14451–14473. doi: 10.3390/molecules200814451. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current trial are not yet publicly available.