

Learning to extinguish traumatic memory depends on fine-tuning of hippocampal-prefrontal connections via the ASIC1a channel.
Abstract
Extinction of conditioned fear necessitates the dynamic involvement of hippocampus, medial prefrontal cortex (mPFC), and basolateral amygdala (BLA), but key molecular players that regulate these circuits to achieve fear extinction remain largely unknown. Here, we report that acid-sensing ion channel 1a (ASIC1a) is a crucial molecular regulator of fear extinction, and that this function requires ASIC1a in ventral hippocampus (vHPC), but not dorsal hippocampus, mPFC, or BLA. While genetic disruption or pharmacological inhibition of ASIC1a in vHPC attenuated the extinction of conditioned fear, overexpression of the channel in this area promoted fear extinction. Channelrhodopsin-2–assisted circuit mapping revealed that fear extinction involved an ASIC1a-dependent modification of the long-range hippocampal-prefrontal correlates in a projection-specific manner. Gene expression profiling analysis and validating experiments identified several neuronal activity–regulated and memory-related genes, including Fos, Npas4, and Bdnf, as the potential mediators of ASIC1a regulation of fear extinction. Mechanistically, genetic overexpression of brain-derived neurotrophic factor (BDNF) in vHPC or supplement of BDNF protein in mPFC both rescued the deficiency in fear extinction and the deficits on extinction-driven adaptations of hippocampal-prefrontal correlates caused by the Asic1a gene inactivation in vHPC. Together, these results establish ASIC1a as a critical constituent in fear extinction circuits and thus a promising target for managing adaptive behaviors.
INTRODUCTION
As a crucial form of learning and memory, extinction learning is necessary for adaptation of the organism to the constantly changing environment. Many brain diseases, especially anxiety disorders, are closely related to deficiencies in extinction learning (1, 2). Because anxiety disorders are characterized by persistence of learned fear, understanding the substrates of fear extinction will ultimately bring benefits to treatment of anxiety disorders (1). In auditory fear conditioning, a tone [conditioned stimulus (CS)] is associated with an aversive foot shock [unconditioned stimulus (US)]. Thereafter, presentation of the tone (CS) alone elicits conditioned fear responses, such as freezing in the rodents. However, repetitive tone presentation to the conditioned subject without foot shock causes the fear response to decrease, which is referred to as fear extinction (1, 2). In general, fear extinction is believed to represent a new learning (i.e., extinction learning) process that leads to the formation of extinction memory. This process involves strength changes of synaptic connections in particular neuronal circuits (2), typically composed of hippocampus, medial prefrontal cortex (mPFC), and basolateral amygdala (BLA).
Notably, the infralimbic subdivision of mPFC (IL/mPFC) plays a central role in the acquisition of fear extinction memories (3–5), whereas the adjacent prelimbic subdivision (PL/mPFC) is essential for sustained fear expression and resistance to extinction (6, 7). After fear extinction learning, synaptic efficacy is reduced in glutamatergic projections from mPFC to the principal neurons but not interneurons of BLA, but synaptic transmission at the mPFC inputs to intercalated neurons remains unchanged (8). Moreover, activation of mPFC projections could repress the transmission from auditory cortical inputs to BLA heterosynaptically (8). The hippocampus is widely considered to play a pivotal part in contextual regulation of fear memory (9, 10). As one of the main inputs to mPFC, the hippocampus also promotes fear extinction through brain-derived neurotrophic factor (BDNF), which is thought to be released from ventral hippocampal projections and act at IL/mPFC neurons (11). On the other hand, although progress on understanding fear extinction at the circuit level is increasing and encouraging, not much attention has been paid to decoding the underlying molecular substrates that coordinate the dynamics of synaptic connections in the above circuits. Identification of these molecular substrates is particularly important in developing therapeutic treatments for extinction-related brain disorders.
The acid-sensing ion channel 1a (ASIC1a) represents one of the potential regulators of fear extinction circuits. ASIC1a is a member of the degenerin/epithelial Na+ channel family (12). It forms a cation-selective H+-gated channel abundantly expressed in the central nervous system (13). ASIC1a has been implicated in many pathophysiological (14–16) and physiological processes including learning and memory (17–21). Mechanistically, ASIC1a is thought to regulate synaptic plasticity through activation by protons released from presynaptic vesicles during synaptic transmission (19, 20). Clinical studies suggest a correlation between ASIC1a polymorphisms and a risk of panic disorder (22–24), although the precise causality remains mysterious (25). Studies in animal models showed that ASIC1a is required for acquisition of conditioned fear (18), in which amygdala serves a key role (26). Mechanically, ASIC1a has been implicated of amygdala synaptic plasticity (19). In stark contrast, it remains unknown whether and how ASIC1a plays a role in other key areas such as hippocampus and mPFC, which are known to be involved in fear extinction.
In the current study, we combined genetic and pharmacological approaches to examine the roles of ASIC1a in the circuit that underlies fear extinction, composed of hippocampus, BLA, and mPFC. We identified a crucial and region-specific role of ASIC1a in ventral hippocampus (vHPC), but not dorsal hippocampus (dHPC), BLA, or mPFC, for fear extinction. We further demonstrated that ASIC1a in vHPC exerts critically a circuit-specific regulation of hippocampal-prefrontal correlates through up-regulation of BDNF.
RESULTS
Brain region–specific roles of ASIC1a in fear acquisition or extinction
The global Asic1a-null mice were previously characterized by substantial deficits in cue and context fear conditioning (18). To verify these characteristics in our system, we subjected Asic1a+/+, Asic1a−/−, and Asic1a+/− mice to a cued fear conditioning protocol (Fig. 1A). During fear conditioning (day 1), animals were placed individually in a conditioning chamber equipped with stainless steel shocking grids at the bottom and walls decorated with black and white checkered wallpapers (context A) and exposed to five pure tones (CS; 4 kHz, 76 dB, 30 s each) delivered at variable intervals (20 to 180 s). A foot shock (US; 0.5 mA, 2 s) was given in the last 2 s of each tone. On day 2, the conditioned mice were individually tested in a test chamber with gray plexiglass floor and walls decorated with dark gray wallpapers (context B) and exposed to tone (CS) only without foot shock for 2 (for retrieval) or 20 (for extinction) times. For each cued tone stimulation, the amount of time spent freezing was measured. Consistent with the previous finding (18), the Asic1a−/− mice exhibited lower freezing responses to CS during fear conditioning (Cond.) and retrieval (Retr.) compared with their Asic1a+/+ littermate controls. No difference was found between the Asic1a heterozygote (Asic1a+/−) and Asic1a+/+ mice in cued fear acquisition (Fig. 1B). These results indicate that ASIC1a plays a critical role in the acquisition of cued fear. However, because ASIC1a is globally absent throughout the life span of the Asic1a−/− mice, these experiments could not inform where in the brain ASIC1a contributes to cued fear acquisition and whether ASIC1a is also involved in the extinction of such fear.
Fig. 1. Brain region–specific roles of ASIC1a in fear acquisition or extinction.
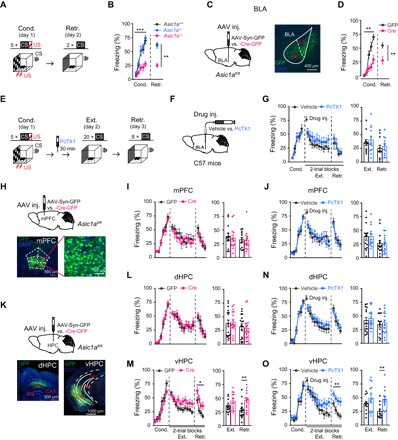
(A to D) Effects of global or BLA-specific KO of Asic1a on cued fear acquisition. (A) Experimental scheme. (C) Left: Schematics of AAV injection. Right: Representative image of GFP expression (green) in a mouse that received AAV-Syn-Cre-GFP injection in BLA. 4′,6-Diamidino-2-phenylindole (DAPI) (blue) was used to label nuclei. (B and D) Time course of freezing response to CS during fear conditioning (Cond.) and retrieval (Retr.). (B) Left: Fear conditioning, two-way repeated-measures analysis of variance (ANOVA), main effect of genotype: F2,29 = 18.043, P < 0.001. Right: Fear retrieval, one-way factorial ANOVA for average freezing during CS of two trials, main effect of genotype, F2,29 = 25.856, P < 0.001. Asic1a+/+, n = 9; Asic1a+/−, n = 12; Asic1a−/−, n = 11. (D) Left: Fear conditioning, two-way repeated-measures ANOVA, main effect of AAV, F1,18 = 14.260, P = 0.001. Right: Fear retrieval, the average freezing during CS of two trials. n = 10 per group. **P < 0.01, unpaired Student’s t test. (E to G, J, N, and O) Effects of pharmacological inhibition of ASIC1a in BLA (G), mPFC (J), dHPC (N), or vHPC (O) on fear extinction. (E) Experimental scheme. (F) Schematics of cannula implantation and drug injection. (G, J, N, and O) Left: Time course of freezing response to CS during fear conditioning, extinction, and retrieval. Two-way repeated-measures ANOVA, main effect of drug, (G) BLA: fear extinction, F1,21 = 0.813, P = 0.377; fear retrieval, F1,21 = 0.236, P = 0.632; (J) mPFC: fear extinction, F1,26 = 0.002, P = 0.964; fear retrieval, F1,26 = 0.162, P = 0.690; (N) dHPC: fear extinction, F1,31 = 0.529, P = 0.472; fear retrieval, F1,31 = 0.143, P = 0.708; and (O) vHPC: fear extinction, F1,24 = 1.574, P = 0.222; fear retrieval, F1,24 = 12.659, P = 0.002. Right: Average freezing response to CS for all trials during fear extinction and retrieval. **P < 0.01, unpaired Student’s t test. n = 11 and 12, 14 and 14, 16 and 17, and 12 and 14 for vehicle and PcTX1 administered in BLA (G), mPFC (J), dHPC (N), and vHPC (O), respectively. (H, I, and K to M) Effects of genetic inactivation of Asic1a in mPFC (I), dHPC (L), or vHPC (M) on fear extinction. (H and K) Schematics of Asic1a gene inactivation in mPFC (H, upper), dHPC, or vHPC (K, upper). Representative images are shown for GFP expression (green) in mPFC (H, bottom), dHPC (K, bottom left), and vHPC (K, bottom right) of AAV-Syn-Cre-GFP–injected mice. DAPI (blue) was used to label nuclei. (I, L, and M) Left: Time course of freezing response to CS during fear conditioning, extinction, and retrieval. Two-way repeated-measures ANOVA, main effect of drug, (I) mPFC: fear conditioning, F1,24 = 0.188, P = 0.668; fear extinction, F1,24 = 0.172, P = 0.682; fear retrieval, F1,24 = 0.020, P = 0.889; (L) dHPC: fear conditioning, F1,32 = 0.029, P = 0.866; fear extinction, F1,32 = 0.079, P = 0.781; fear retrieval, F1,32 = 0.157, P = 0.695; (M) vHPC: fear conditioning, F1,26 = 2.293, P = 0.142; fear extinction, F1,26 = 4.533, P = 0.043; fear retrieval, F1,26 = 6.426, P = 0.018. Right: Average freezing response to CS for all trials during fear extinction and retrieval. **P < 0.01, unpaired Student’s t test. n = 13 and 13, 17 and 17, and 15 and 13 for GFP and Cre in mPFC (J), dHPC (N), and vHPC (O), respectively.
Restoration of ASIC1a expression in the amygdala of Asic1a−/− mice rescued the acquisition of context-dependent fear memory, suggesting that ASIC1a in amygdala is critical and sufficient for conditioned fear acquisition (26). To provide further evidence for the significance of amygdala ASIC1a in the acquisition of conditioned fear including cued fear, we selectively inactivated the Asic1a gene in BLA using floxed-Asic1a+/+ (Asic1aflox/flox) mice, in which Asic1a gene is flanked by loxP (27). The region-specific knockdown of ASIC1a was achieved by the injection of adeno-associated virus (AAV) that simultaneously expressed Cre recombinase and green fluorescent protein (GFP) under the control of human synapsin promoter (AAV-Syn-Cre-GFP, Cre) into both sides of BLA of Asic1aflox/flox mice (Fig. 1C, left). The expression of GFP was detected in BLA 4 weeks after the viral injection (Fig. 1C, right). The AAV-Syn-Cre-GFP–injected (Cre) mice exhibited impaired cued fear acquisition (Fig. 1D) just like the global Asic1a knockouts (KOs) (Fig. 1B), demonstrating that ASIC1a expression in BLA is critical for cued fear acquisition.
To probe whether BLA ASIC1a is also required for cued fear extinction, we specifically inhibited ASIC1a during extinction training (Ext.) through bilateral cannula-aided injection of a specific ASIC1a inhibitor, PcTX1 (10 μM; 0.5 μl administered 30 min before extinction training), at BLA of C57BL/6J mice (Fig. 1, E and F). However, the PcTX1 treatment at BLA did not notably affect fear extinction (Fig. 1G), as reflected by CS-induced freezing responses during extinction training (Ext.; 20 CS on day 2) and extinction retrieval test (Retr.; 8 CS on day 3). These results suggest that ASIC1a in BLA is crucial for cued fear acquisition, but not fear extinction.
Because mPFC and hippocampus are two other brain regions typically implicated in fear extinction, we then tested ASIC1a in these areas (Fig. 1, H to O, and fig. S1). First, Cre-dependent Asic1a gene inactivation (Fig. 1H and fig. S1A) or PcTX1 injection (fig. S1B) before extinction training in mPFC had no significant effect on cued fear extinction (Fig. 1, I and J). We next focused on ASIC1a in hippocampus (Fig. 1, K to O). The hippocampus is structurally and functionally divided into ventral and dorsal parts (28). Local delivery of AAV-Syn-Cre-GFP into dHPC or vHPC of Asic1aflox/flox mice resulted in GFP expression (Fig. 1K) and a marked decrease in ASIC1a protein levels in the injected site (fig. S1, C and E). AAV-Syn-Cre-GFP injection in dHPC affected neither acquisition nor extinction of the cued fear (Fig. 1L). By contrast, while Asic1a gene inactivation in vHPC failed to alter cued fear acquisition, it significantly impaired fear extinction, as shown by the much slower decrease than the GFP-injected control mice in CS-induced freezing responses during retrieval test 1 day after extinction training (Fig. 1M).
To validate the above results from genetic manipulations, we delivered PcTX1 into dHPC or vHPC of C57BL/6J mice 30 min before extinction training. The sites of cannula implantation and drug/vehicle delivery were validated by Nissl staining after the experiments (fig. S1, D and F). While administration of PcTX1 at dHPC did not affect fear extinction (Fig. 1N), that at vHPC significantly slowed down the CS-induced freezing responses during retrieval test (Fig. 1O), supporting a specific role of vHPC ASIC1a in cued fear extinction. Therefore, in the interconnected circuit composed of BLA, mPFC, and hippocampus, ASIC1a exerts a region-specific role in regulation of fear learning and extinction; while ASIC1a in BLA is necessary for fear learning, that in vHPC is critical for cued fear extinction.
ASIC1a contributes to extinction-driven hippocampal-prefrontal plasticity
Given that cellular consequences of learning and memory often include adaptive changes of synaptic connectivity, we searched for synaptic plasticity within the fear extinction circuit that might be regulated by vHPC ASIC1a. We focused on adaptive changes associated with extinction learning in mPFC because it is central for coding and expression of fear extinction memory (3–5), and asked whether these changes depended on vHPC ASIC1a. To address this question, Asic1aflox/flox mice were injected with either AAV-Syn-Cre-GFP or AAV-Syn-GFP at the vHPC region 4 to 6 weeks before the experiment. Before electrophysiological recording, the animals were subjected to fear conditioning and then received either fear extinction training (20 × CS, Ext.) or retrieval without extinction (2 × CS, No Ext. as control) on the second day 2 hours before the preparation of brain slices containing the mPFC region. The recordings of pyramidal neurons in layer II/III of IL/mPFC and PL/mPFC subdivisions and further data analysis revealed that extinction learning differentially modified vHPC→IL/mPFC and vHPC→PL/mPFC synapses, which greatly depended on vHPC ASIC1a.
First, the frequency of spontaneous excitatory postsynaptic current (sEPSC) was significantly increased in IL/mPFC neurons, but decreased in PL/mPFC neurons, in AAV-Syn-GFP–injected mice after the Ext. treatment. However, these extinction-induced adaptive changes were not detected in the Asic1aflox/flox mice that received AAV-Syn-Cre-GFP injection in vHPC (fig. S2). Meanwhile, the amplitudes of sEPSCs were not affected by the Ext. treatment in both IL/mPFC and PL/mPFC neurons (fig. S2). Moreover, the sEPSC frequencies of IL/mPFC and PL/mPFC neurons from mice subjected to US (foot shock) only did not differ from that of naïve mice, regardless of the animals injected by AAV-Syn-GFP or AAV-Syn-Cre-GFP (fig. S3), supporting that the effects of ASIC1a in vHPC neurons on extinction-driven increase (in IL/mPFC neurons) or decrease (in PL/mPFC neurons) in the frequency of sEPSCs are paradigm specific. Therefore, fear extinction is associated with opposite changes in the overall synaptic drive onto IL/mPFC and PL/mPFC neurons, and vHPC ASIC1a plays a pivotal role in the formation of this synaptic plasticity in mPFC.
Second, because Asic1a gene inactivation in vHPC abolished fear extinction–associated frequency changes in sEPSCs of mPFC neurons, vHPC inputs must be important for the synaptic adaptations in mPFC. vHPC has been found to form monosynaptic and glutamatergic projections onto mPFC neurons (29). However, the plasticity of vHPC→mPFC projections associated with fear extinction and the underlying mechanisms are not yet established. To specifically examine the plasticity of vHPC→mPFC projections, we applied an optogenetic approach to record light-induced excitatory inputs from vHPC onto mPFC neurons. AAV expressing channelrhodopsin-2 (ChR2-E123T/T159C, referred to as ChR2), driven by the CaMKIIα promoter for projection neuron–specific expression (AAV-CaMKIIα-ChR2-mCherry), was coinjected with AAV-Syn-Cre-GFP or AAV-Syn-GFP bilaterally into vHPC of Asic1aflox/flox mice. After 4 to 6 weeks, animals were subjected to fear conditioning and Ext. or No Ext. treatment as described above and used to prepare brain slices. Ex vivo whole-cell recordings from mPFC neurons combined with light-induced stimulation of vHPC fibers were used to examine optically evoked EPSCs (oEPSCs) (Fig. 2). Using paired light pulses to stimulate vHPC afferents with varying interstimulus intervals, we calculated the paired-pulse ratios (PPRs) for oEPSCs, which are inversely related to the presynaptic release probability (30). In AAV-Syn-GFP–injected mice, the PPRs were decreased at the vHPC→IL/mPFC projection (Fig. 2, B and C) but increased at the vHPC→PL/mPFC projection (Fig. 2, E and F) in the Ext. as compared to the No Ext. group. However, these extinction-associated PPR changes were not detected in AAV-Syn-Cre-GFP–injected animals (Fig. 2, B, C, E, and F). These suggest that fear extinction training enhances presynaptic release probability at vHPC→IL/mPFC synapses but causes the opposite at vHPC→PL/mPFC synapses in a vHPC-ASIC1a–dependent manner. These results echo the frequency changes of sEPSCs in these neurons after Ext. training (fig. S2).
Fig. 2. ASIC1a in vHPC is important for extinction learning–driven presynaptic changes at the vHPC→mPFC projections.
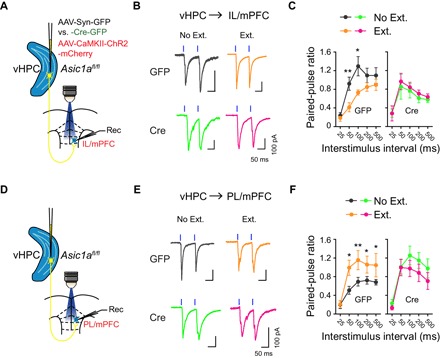
Effects of fear extinction learning on the PPRs for oEPSCs at vHPC→IL/mPFC (A to C) and vHPC→PL/mPFC (D to F) projections and the dependence of these effects on ASIC1a in vHPC. (A and D) Experimental schemes. (B and E) Representative traces of oEPSCs at vHPC→IL/mPFC (B) or vHPC→PL/mPFC (E) synapses in No Ext. and Ext. groups of GFP (top) and Cre (bottom) mice induced by paired photostimulations (blue vertical bars) with a 100-ms interval. (C and F) Summary of PPRs of oEPSCs plotted against interstimulus intervals at the vHPC→IL/mPFC (C) or vHPC→PL/mPFC (F) synapses. Two-way repeated-measures ANOVA, main effect of behavior, (C) vHPC→IL/mPFC: GFP, F1,118 = 15.33, P < 0.001 (left); Cre, F1,148 = 0.541, P = 0.463 (right); (F) vHPC→PL/mPFC: GFP, F1,178 = 23.17, P < 0.001 (left); Cre, F1,133 = 4.478, P = 0.036 (right). *P < 0.05, **P < 0.01, Ext. versus No Ext., unpaired Student’s t test. (C) vHPC→IL/mPFC: GFP, No Ext., n = 13 neurons of 10 mice; GFP, Ext., n = 11 neurons of 7 mice; Cre, No Ext., n = 18 neurons of 8 mice; Cre Ext., n = 14 neurons of 8 mice. (F) vHPC→PL/mPFC: GFP, No Ext., n = 19 neurons of 11 mice; GFP, Ext., n = 17 neurons of 11 mice; Cre, No Ext., n = 16 neurons of 7 mice; Cre, Ext., n = 12 neurons of 8 mice.
Third, we examined whether the presynaptic adaptive changes seen above in the vHPC→mPFC pathway were also accompanied by postsynaptic alterations. By holding the cell at either +40 or −70 mV, we measured oEPSCs mediated by N-methyl-d-aspartate (NMDA) receptors (NMDARs) or AMPA receptors (AMPARs), respectively (Fig. 3, A to D). In AAV-Syn-GFP–injected mice, the IL/mPFC neurons exhibited a marked increase (Fig. 3, A and B), whereas the PL/mPFC ones displayed a clear decrease (Fig. 3, C and D) in the NMDAR/AMPAR oEPSC ratios in the Ext. as compared to the No Ext. group. These changes were not detected in AAV-Syn-Cre-GFP–injected mice (Fig. 3, A to D). Given that the AMPAR function of mPFC neurons is mostly unaffected by fear extinction, as shown by the lack of changes in the amplitudes of sEPSCs in both AAV-Syn-GFP– and AAV-Syn-Cre-GFP–injected mice (fig. S2), the extinction-induced changes in NMDAR/AMPAR oEPSC ratios may be largely ascribed to alterations of NMDAR activities. Plotting the NMDAR current amplitudes against different voltages revealed a significant increase of NMDAR-mediated synaptic responses specifically at the vHPC→IL/mPFC projection in the Ext. as compared to the No Ext. group in AAV-Syn-GFP– but not AAV-Syn-Cre-GFP–injected mice (fig. S4). Therefore, fear extinction training could be associated with increased and decreased postsynaptic NMDAR (over AMPAR) functions at the vHPC→IL/mPFC and vHPC→PL/mPFC projections, respectively, and these alterations are dependent on vHPC ASIC1a.
Fig. 3. ASIC1a in vHPC is required for extinction learning–driven postsynaptic changes at the vHPC→mPFC projections and mPFC neuronal activity pattern.
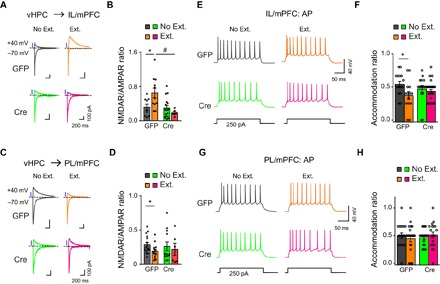
(A to D) Effects of fear extinction learning on the NMDAR/AMPAR ratios of vHPC→IL/mPFC (A and B) and vHPC→PL/mPFC (C and D) projections and the dependence of these effects on ASIC1a in vHPC. (A and C) Representative traces of AMPAR- and NMDAR-mediated currents in response to photostimulation of vHPC fibers at vHPC→IL/mPFC (A) or vHPC→PL/mPFC (C) synapses in No Ext. and Ext. groups of GFP (left) and Cre (right) mice. (B and D) Ratios of peak NMDAR- to AMPAR-mediated currents at vHPC→IL/mPFC (B) or vHPC→PL/mPFC (D) synapses for individual neurons and the summary. *P < 0.05, Ext. versus No Ext.; #P < 0.05, GFP versus Cre, unpaired Student’s t test. (B) vHPC→IL/mPFC: GFP, No Ext., n = 9 neurons of 7 mice; GFP, Ext., n = 11 neurons of 8 mice; Cre, No Ext., n = 16 neurons of 8 mice; Cre, Ext., n = 6 neurons of 4 mice. (D) vHPC→PL/mPFC: GFP, No Ext., n = 16 neurons of 9 mice; GFP, Ext., n = 13 neurons of 9 mice; Cre, No Ext., n = 12 neurons of 7 mice; Cre, Ext., n = 6 neurons of 6 mice. (E to H) Action potential firing of pyramidal neurons in layer II/III of IL/mPFC (E and F) or PL/mPFC (G and H) to a depolarizing current injection (+250 pA) recorded ex vivo. (E and G) Representative traces of action potential firing recorded from the IL/mPFC (E) or PL/mPFC (G) neurons. (F and H) Accommodation ratios of the action potential number in IL/mPFC (F) or PL/mPFC (H) neurons during the last 100-ms current injection to that during the first 100-ms current injection for individual neurons and the summary. *P < 0.05, Ext. versus No Ext, unpaired Student’s t test. (F) IL/mPFC: GFP, No Ext., n = 16 neurons of 7 mice; GFP, Ext., n = 14 neurons of 5 mice; Cre, No Ext., n = 16 neurons of 6 mice; Cre, Ext., n = 25 neurons of 9 mice. (H) PL/mPFC: GFP, No Ext., n = 19 neurons of 6 mice; GFP, Ext., n = 22 neurons of 6 mice; Cre, No Ext., n = 20 neurons of 6 mice; Cre, Ext., n = 19 neurons of 7 mice.
Fourth, we tested whether the adaptive changes in synaptic inputs would alter the firing properties of IL/mPFC and PL/mPFC neurons. By whole-cell recordings of pyramidal neuronal firing ex vivo, we found that IL/mPFC neurons from AAV-Syn-GFP–injected mice displayed an accelerated accommodation ratio in response to a depolarizing current injection in the Ext. group (Fig. 3E) compared with the No Ext. group. This observation is in line with the increased firing of action potential bursts in vivo (31) and the reported role of NMDAR-dependent burst firing in neurons of IL/mPFC for consolidation of fear extinction memory (5). Consistent with the observation that vHPC ASIC1a controls fear extinction as well as regulation of extinction-dependent NMDAR enhancement (Fig. 3, A and B), the above changes of action potential firing in IL/mPFC were not detected in AAV-Syn-Cre-GFP–injected animals (Fig. 3, E and F). In addition, likely due to the enhanced accommodation of action potential firing, fear extinction training resulted in a paradoxical decrease of the overall firing rates of IL/mPFC neurons to current injection (fig. S5, A to F) in AAV-Syn-GFP– but not AAV-Syn-Cre-GFP–injected mice. Meanwhile, analysis of the neuronal firing in PL/mPFC did not reveal a significant difference in accommodation ratios (Fig. 3, G and H) or firing rates (fig. S5, G to L) under all conditions tested. Moreover, the changes in accommodation ratios and firing rates associated with fear extinction found in IL/mPFC neurons of AAV-Syn-GFP–injected mice were abolished by the treatment of the NMDAR antagonist D-APV (20 μM) (fig. S6). Thus, extinction training results in region-specific changes in action potential firing in IL/mPFC, rather than in PL/mPFC, such that more burst firings are generated to transmit information more reliably than single spikes (32), in manners that depend on ASIC1a in vHPC and NMDAR in mPFC.
Overall, fear extinction training induced distinct synaptic changes at vHPC→IL/mPFC and vHPC→PL/mPFC projections in a vHPC-ASIC1a–dependent fashion. These synaptic and cellular underpinnings may underlie the mechanism by which ASIC1a in vHPC contributes to fear extinction.
ASIC1a regulates fear extinction–associated gene profile changes in vHPC
To explore the potential mediators of vHPC ASIC1a function in fear extinction and the extinction-associated plasticity of vHPC→mPFC projections, whole-tissue mRNA sequencing was carried out for vHPC of Asic1aflox/flox mice that received the injection of AAV-Syn-Cre-GFP or AAV-Syn-GFP (as control) at the same brain region (Fig. 4A). The vHPC tissues were dissected on day 2 following fear conditioning at 2 hours after either fear extinction training (Ext.) or retrieval without extinction training (No Ext.). The tissues were quickly frozen in liquid nitrogen and stored at −80°C before being used for mRNA sequencing. Consistent with the results shown in Fig. 1M, this cohort of animals exhibited no difference among the four groups in cued fear acquisition, as shown by the similar levels of freezing responses to CS during the first two-trial block of extinction training and fear retrieval (fig. S7).
Fig. 4. ASIC1a regulates extinction-associated gene expression profiles in vHPC.
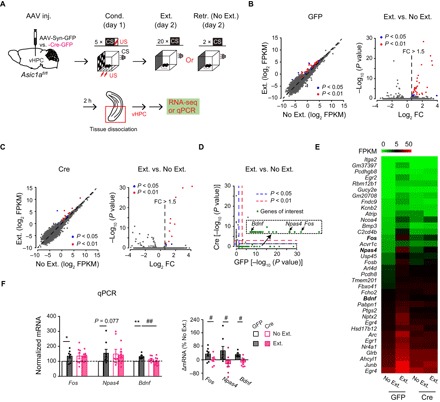
(A) Experimental scheme. (B) Left: Differential enrichment calling of vHPC mRNA of GFP mice, showing 38 genes (blue and red dots) enriched in the Ext. group with P < 0.05, after adjusting for multiple comparisons across the detected gene repertoire (Fisher’s exact test for multiple group comparison with Benjamini-Hochberg FDR correction; n = 2 samples from 8 mice per group). Broken lines represent unit slope. Right: Volcano plot showing vHPC mRNA with absolute fold change (FC) values > 1.5 in the Ext. group. (C) Similar to (B) but for vHPC mRNA of Cre mice, showing 14 genes (blue and red dots) enriched in the Ext. group with P < 0.05 (n = 2 samples from 8 mice per group). (D) Scatter plot comparing P values of enrichment in Ext. samples for Cre versus GFP mice. The red dashed lines indicate P = 0.01, and the blue dashed lines represent P = 0.05. Note the genes in the bottom right corner, shown in the inset, which are enriched in the Ext. samples only for GFP mice. These include activity-induced immediate-early gene, Fos, and plasticity- or memory-related genes, Npas4 and Bdnf. (E) Heatmap of 36 genes enriched by fear extinction in GFP, but not Cre, mice. (F) mRNA levels determined by qPCR. Fos, n = 8; Npas4, n = 8; Bdnf, n = 6 to 8 per group. Left: Gene expression levels relative to the control (GFP, No Ext.). Right: Relative difference between Ext. and No. Ext. of mice injected with the same AAV construct. *P < 0.05, **P < 0.01, Ext. versus No Ext; ##P < 0.01, GFP versus Cre, unpaired Student’s t test. FPKM, fragments per kilobase of transcript per million; RNA-seq, RNA sequencing.
Results of mRNA sequencing showed that for AAV-Syn-GFP–injected mice, although most of the genes were expressed at comparable levels in the Ext. and No Ext. groups, a subset of genes was identified by differential expression analysis to be specially enriched after the extinction training [38 genes with P < 0.05, among which 31 with P < 0.01, false discovery rate (FDR) adjusted; Fig. 4B]. However, for AAV-Syn-Cre-GFP–injected mice, the numbers of the genes enriched in vHPC from the Ext. group were significantly less (14 genes with P < 0.05, among which 10 with P < 0.01, FDR adjusted; Fig. 4C), suggesting that Asic1a gene inactivation in vHPC had resulted in the loss of at least a major transcriptional pathway that responds to fear extinction learning. Moreover, the enriched genes hardly overlapped between AAV-Syn-GFP– and AAV-Syn-Cre-GFP–injected mice; among the 38 genes found to be enriched by fear extinction in vHPC of AAV-Syn-GFP–injected control animals, 36 did not show enrichment in AAV-Syn-Cre-GFP–injected counterparts (Cre) (Fig. 4, D and E), implicating a more profound role of ASIC1a in regulating extinction-associated gene expression profiles in vHPC. Of the 36 genes that failed to enrich following fear extinction due to Asic1a gene inactivation are Fos, neuronal PAS domain protein 4 (Npas4), immediate-early genes that link neuronal activity to memory (33), and Bdnf, which is well known to play key roles in neuronal plasticity (Fig. 4D, inset, and E) (11, 34). Using quantitative polymerase chain reaction (qPCR) assay, we verified that mRNA levels of Fos, Npas4, and Bdnf were increased in vHPC of Ext. versus No Ext. group for control, and the differences were abolished by Asic1a gene inactivation in this brain region (Fig. 4F).
ASIC1a regulates fear extinction through learning-dependent BDNF expression
Previous studies have implicated BDNF as a key mediator in hippocampal regulation of fear extinction, and this process depends on NMDAR (11). The gene expression profile analysis above suggested that ASIC1a in vHPC may contribute to fear extinction through regulation of extinction learning–dependent BDNF expression. Using enzyme-linked immunosorbent assay (ELISA), we showed that extinction training of AAV-Syn-GFP–injected Asic1aflox/flox mice caused a marked increase in BDNF protein level in vHPC, as compared with the No Ext. group, and this response was significantly attenuated in AAV-Syn-Cre-GFP–injected mice (Fig. 5A). These results further confirm that vHPC ASIC1a regulates fear extinction–associated BDNF expression, which may underlie the contribution of ASIC1a to fear extinction.
Fig. 5. Extinction learning–dependent BDNF expression mediates the effect of vHPC ASIC1a on cued fear extinction.
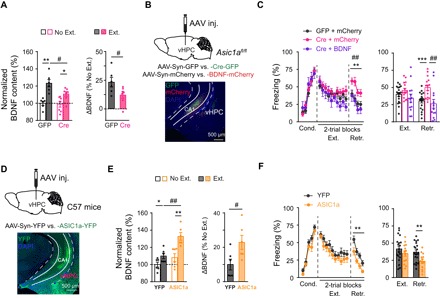
(A) BDNF protein levels determined by ELISA in vHPC of Asic1aflox/flox mice infected with AAV-Syn-GFP (GFP) or AAV-Syn-Cre-GFP (Cre) and subjected to fear extinction (Ext.) or retrieval without extinction (No Ext.) (GFP, n = 6; Cre, n = 7 per group). Individual data points and the summary. Left: BDNF protein levels relative to the control (GFP, No Ext.). Right: Relative difference between Ext. and No. Ext. of mice injected with the same AAV construct. *P < 0.05, **P < 0.01, Ext. versus No Ext; #P < 0.05, GFP versus Cre, unpaired Student’s t test. (B and C) In situ BDNF overexpression rescued the deficiency of cued fear extinction in vHPC Asic1a gene–inactivated mice. (B) Top: Schematics of AAV-mediated genetic manipulation on Asic1aflox/flox mice. Bottom: Representative images are shown for the expression of GFP (green) and mCherry (red) in vHPC. (C) Time course of freezing response to CS during fear conditioning, extinction, and retrieval. Left: Two-way repeated-measures ANOVA, main effect of AAV, fear conditioning, F2,44 = 0.173, P = 0.841; fear extinction, F2,44 = 1.949, P = 0.154; fear retrieval, F2,44 = 8.955, P = 0.001, followed by post hoc Bonferroni, P = 0.005 for Cre + mCherry versus GFP + mCherry, P = 0.001 for Cre + BDNF versus Cre + mCherry, P = 1.000 for Cre + BDNF versus GFP + mCherry. Right: One-way factorial ANOVA for average freezing during CS of all trials, main effect of AAV, fear extinction, F2,44 = 1.946, P = 0.155; fear retrieval, F2,44 = 8.272, P = 0.001, followed by post hoc Bonferroni, P = 0.002 for Cre + mCherry versus GFP + mCherry, P = 0.001 for Cre + BDNF versus Cre + mCherry, P = 0.506 for Cre + BDNF versus GFP + mCherry. GFP + mCherry, n = 9; Cre + mCherry, n = 12; Cre + BDNF, n = 11. (D to F) Asic1a gene overexpression in vHPC increased extinction-associated BDNF expression in vHPC and enhanced fear extinction. (D) Top: Schematics of AAV-mediated genetic manipulation on C57BL/6J mice. Bottom: Representative images are shown for the expression of YFP (green) in vHPC. (E) Effects of Asic1a gene overexpression in vHPC on BDNF protein levels. Left: BDNF protein levels relative to the control (YFP, No Ext.). Right: Relative difference between Ext. and No. Ext. of mice injected with the same AAV construct. *P < 0.05, **P < 0.01, Ext. versus No Ext; #P < 0.05, GFP versus Cre, unpaired Student’s t test. n = 6 per group. (F) Time course of freezing response to CS during fear conditioning, extinction, and retrieval. Left: Two-way repeated-measures ANOVA, main effect of AAV, fear conditioning, F1,30 = 4.213, P = 0.050; fear extinction, F1,30 = 1.348, P = 0.255; fear retrieval, F1,30 = 11.183, P = 0.002. Right: Average freezing responses. YFP, n = 19; ASIC1a, n = 13. **P < 0.01, YFP versus ASIC1a, unpaired Student’s t test.
To verify the above hypothesis, we tested whether in situ BDNF overexpression could rescue the deficiency caused by Asic1a gene inactivation in fear extinction. AAV containing the coding sequence of BDNF (AAV-Syn-BDNF-mCherry) was injected for BDNF overexpression, and AAV-Syn-mCherry was used as the control. The Asic1aflox/flox mice received bilateral vHPC injection of a mixture of AAV-Syn-Cre-GFP and AAV-Syn-BDNF-mCherry for simultaneous Asic1a gene inactivation and Bdnf overexpression. Infection by the two viruses at vHPC was confirmed by the coappearance of GFP and mCherry fluorescence (Fig. 5B). As shown by ELISA, the AAV-Syn-BDNF-mCherry injection greatly increased BDNF protein levels in vHPC (fig. S8A). Consistent with the results shown in Fig. 1M, Asic1a gene inactivation at vHPC by AAV-Syn-Cre-GFP, now in the presence of AAV-Syn-mCherry, impaired fear extinction. However, the coinjection of AAV-Syn-BDNF-mCherry abolished this effect, with the mice exhibiting normal fear extinction as those coinjected with AAV-Syn-GFP and AAV-Syn-mCherry (Fig. 5C). Therefore, BDNF overexpression in vHPC can rescue the deficiency in fear extinction caused by Asic1a gene inactivation in this brain area.
To further strengthen the functional link between ASIC1a and BDNF at vHPC in fear extinction, we overexpressed ASIC1a in vHPC of C57BL/6J mice through bilateral injection of AAV that contains the coding sequences of ASIC1a and yellow fluorescent protein (YFP) (AAV-Syn-ASIC1a-YFP). For control, we used AAV-Syn-YFP. Virus infection at vHPC was confirmed by the YFP fluorescence (Fig. 5D). AAV-Syn-ASIC1a-YFP injection at vHPC not only significantly increased the expression of ASIC1a, as shown by Western blotting (fig. S8B), but also that of BDNF, as assessed by ELISA (Fig. 5E), in the injected area. This manipulation facilitated the extinction of cued fear (Fig. 5F), suggesting that the enhanced ASIC1a expression in vHPC improved fear extinction by increasing learning-dependent BDNF expression. By contrast, ASIC1a overexpression in dHPC produced no detectable effect on cued fear extinction (fig. S9), which is consistent with the fact that genetic inactivation of ASIC1a in dHPC had negligible impacts on fear extinction (Fig. 1L). Together, the above results suggest that vHPC ASIC1a positively contributes to fear extinction by up-regulating the learning-dependent BDNF expression in this brain region.
ASIC1a regulates fear extinction and the associated circuit plasticity through vHPC→mPFC BDNF signaling
The above results point to the fact that ASIC1a in vHPC regulates fear extinction through BDNF. Hippocampal-derived BDNF has been shown to induce extinction of fear memory in rats through antegrade BDNF signaling that targets the IL/mPFC region (11). We thus examined whether ASIC1a in vHPC regulates fear extinction through BDNF that signals onto vHPC→IL/mPFC projections. To this end, Asic1aflox/flox mice injected with AAV-Syn-GFP or AAV-Syn-Cre-GFP bilaterally in vHPC 4 weeks before were subjected to fear conditioning followed by fear extinction training and retrieval test on the second and third days, respectively, as before, except that BDNF or vehicle (saline) was injected into mPFC 30 min before the fear extinction training (Fig. 6A). Similar to the results shown in Figs. 1M and 5C, Asic1a gene inactivation at vHPC by AAV-Syn-Cre-GFP, and now with saline injection in mPFC, impaired fear extinction. However, BDNF injection in the mPFC prevented this effect, with the mice exhibiting normal fear extinction similar to those injected with AAV-Syn-GFP in vHPC and then saline in mPFC (Fig. 6B). Therefore, BDNF supplement in mPFC rescued the deficiency in fear extinction caused by Asic1a gene inactivation in vHPC, supporting the hypothesis that ASIC1a in vHPC regulates fear extinction through vHPC→IL/mPFC BDNF signaling.
Fig. 6. ASIC1a in vHPC regulates adaptive changes of vHPC-mPFC synapses via BDNF signaling, which underlies cued fear extinction.
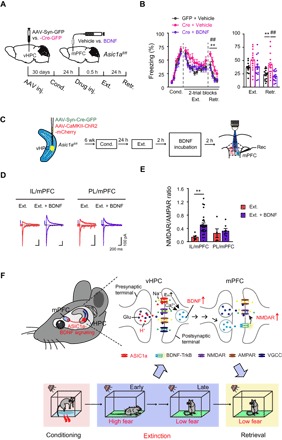
(A) Experimental scheme. (B) Effects of BDNF infusion into mPFC on the cued fear extinction in vHPC Asic1a gene–inactivated mice. Time course of freezing response to CS during fear conditioning, extinction, and retrieval. Left: Two-way repeated-measures ANOVA, main effect of AAV plus drug, fear conditioning, F2,43 = 2.152, P = 0.129; fear extinction, F2,43 = 2.875, P = 0.067; fear retrieval, F2,43 = 9.263, P < 0.001, followed by post hoc Bonferroni, P = 0.005 for Cre + vehicle versus GFP + vehicle, P = 0.001 for Cre + BDNF versus Cre + vehicle, P = 1.000 for Cre + BDNF versus GFP + vehicle. Right: One-way factorial ANOVA for average freezing during CS for all trials, main effect of AAV, fear extinction, F2,43 = 2.883, P = 0.067; fear retrieval, F2,43 = 9.263, P < 0.001, followed by post hoc Bonferroni, P = 0.005 for Cre + vehicle versus GFP + vehicle, P = 0.001 for Cre + BDNF versus Cre + vehicle, P = 1.000 for Cre + BDNF versus GFP + vehicle. GFP + vehicle, n = 15; Cre + vehicle, n = 15; Cre + BDNF, n = 16. (C) Experimental schemes. Animals were treated and brain slides were prepared as in Fig. 2. Brain slices containing mPFC were then incubated with BDNF (200 ng/ml in aCSF) or vehicle (aCSF) for 2 hours before electrophysiological recordings. (D) Representative traces of AMPAR- and NMDAR-mediated oEPSCs at vHPC→IL/mPFC (left) and vHPC→PL/mPFC (right) synapses with or without BDNF pretreatment of mPFC slice. (E) Ratios of peak NMDAR- to AMPAR-mediated oEPSCs at vHPC→IL/mPFC and vHPC→PL/mPFC synapses for individual neurons and the summary. vHPC→IL/mPFC: vehicle, n = 6 neurons of 5 mice; BDNF, n = 28 neurons of 9 mice; vHPC→PL/mPFC: vehicle, n = 5 neurons of 4 mice; BDNF, n = 7 neurons of 5 mice. **P < 0.01, vehicle versus BDNF, unpaired Student’s t test. (F) Working model of vHPC ASIC1a-dependent regulation of fear extinction. ASIC1a activity drives the expression of BDNF in vHPC in response to fear extinction training, which is followed by an antegrade BDNF signaling to modify the synaptic efficacy of vHPC→mPFC projections (see the main text for more details).
Finally, we examined whether BDNF treatment of mPFC slices in vitro could rescue the synaptic deficits caused by Asic1a gene inactivation in vHPC on extinction-associated plasticity of vHPC→mPFC projections (see Figs. 2 and 3). We focused especially on the postsynaptic NMDAR function, as it was previously suggested to be required for BDNF action on IL/mPFC to mediate extinction of conditioned fear (11). To investigate this question, mPFC slices from the Ext. group of AAV-CaMKII-ChR2-mCherry/AAV-Syn-Cre-GFP–coinjected Asic1aflox/flox mice were incubated with recombinant BDNF protein or vehicle [artificial cerebrospinal fluid (aCSF)] for 2 hours before oEPSC recordings (Fig. 6C). The BDNF treatment significantly increased the NMDAR/AMPAR oEPSC ratio (Fig. 6, D and E) as well as the isolated NMDAR response under different voltages (fig. S4) at vHPC→mPFC synapses, supporting the idea that BDNF mediates ASIC1a-dependent extinction-associated postsynaptic adaptation of vHPC-mPFC projections. BDNF may serve as an antegrade signal synthesized in vHPC neurons and released from vHPC projection terminals to finally act on postsynaptic targets in mPFC. Together, we provide strong evidence that ASIC1a in vHPC influences fear extinction behavior and extinction-associated circuit plasticity via the hippocampal-prefrontal BDNF signaling (Fig. 6F), implicating ASIC1a as a promising target for managing adaptive behaviors.
DISCUSSION
Extinction learning is necessary for adaptation of the organism to the constantly changing environment, and identifying essential regulators of fear extinction will aid the treatment of anxiety disorders (1, 2). Here, we identify a critical role of vHPC ASIC1a in fear extinction, which involves regulation of extinction learning–induced hippocampal-prefrontal synaptic plasticity. Mechanistically, this regulation occurs through increasing BDNF expression by ASIC1a in vHPC neurons and an antegrade BDNF signaling at the vHPC→IL/mPFC projections, which, in turn, enhances postsynaptic NMDAR function. Together with well-established roles of mPFC (especially IL/mPFC) and BLA in fear learning and extinction, our data offer important novel insights into the mechanism of fear extinction and reveal ASIC1a in vHPC as a key molecular substrate that regulates fear extinction through BDNF signaling at hippocampal-prefrontal synapses. These findings lay a solid foundation for ASIC1a and its regulation of the BDNF pathway to serve as promising targets for therapeutic development against anxiety disorders.
It is well recognized that fear extinction strongly relies on the neuronal circuits composed of interconnected brain structures (1, 2) including hippocampus, mPFC, and BLA. In addition to the well-documented engagement of mPFC (especially IL/mPFC) (3–5) and BLA (35) in fear extinction, the involvement of hippocampus in fear extinction is also emerging. vHPC projections to amygdala and mPFC are vital for context-dependent control of fear memory retrieval after extinction learning (9, 10). In addition, fear extinction learning causes an increased expression of BDNF in hippocampus, and more specifically in its ventral part. Moreover, infusion of BDNF in hippocampus sufficiently induced extinction of conditioned fear, which was prevented by coadministration of the BDNF-inactivating antibody in IL/mPFC (11), suggesting that the hippocampal-prefrontal BDNF signaling is critical for fear extinction.
Ample evidence has implicated ASIC1a as an important modulator of hippocampal function. Early in situ hybridization analysis indicated a high level of ASIC1a expression particularly in pyramidal neurons of CA1 and CA3 areas of rodent hippocampus (13). Subsequent studies, however, showed higher ASIC current densities in hippocampal γ-aminobutyric acid (GABA)–ergic interneurons than pyramidal neurons (16, 36). In hippocampus, ASIC1a has been shown to be involved in synaptic plasticity including long-term potentiation (17) and metabotropic glutamate receptor–dependent long-term depression (37). In cultured hippocampal neurons, lowering extracellular pH significantly increased cytosolic Ca2+ concentration through activation of ASIC1a (38). By mediating Ca2+ influx, hippocampal ASIC1a is thought to function postsynaptically to contribute to synaptic plasticity and learning and memory. Here, our data revealed a new signaling pathway by which ASIC1a regulates hippocampal output through BDNF signaling. An extinction learning–associated up-regulation of BDNF expression occurs in an ASIC1a-dependent manner in vHPC, and this pathway contributes critically to fear extinction through modulating vHPC-mPFC synaptic strength. In sharp contrast to most previous studies on behavioral relevance of ASIC1a in postsynaptic neurons (17–21), we establish here a clear example of ASIC1a operation in the presynaptic partner of the vHPC→mPFC circuits for control of fear extinction memory.
Our current study offers considerable advancement in the knowledge on function and regulation of extinction circuits. First, we identified region-specific roles of ASIC1a in fear learning and extinction. While ASIC1a in BLA is important for fear acquisition, ASIC1a in vHPC is crucial for behavioral regulation of fear extinction. These findings are consistent with the notion that original and extinction memories of fear are acquired and stored within interacting but distinct neuronal networks. ASIC1a in amygdala was previously reported to control acquisition of contextual fear memory (26). Our new results using conditional region-specific Asic1a KO mice further indicate that amygdala ASIC1a is involved in cued fear learning. However, pharmacological inhibition of amygdala ASIC1a activity after fear acquisition failed to influence extinction of the acquired fear, whereas a similar manipulation at vHPC impaired fear extinction. The selective inactivation of Asic1a gene in vHPC, contrary to amygdala, disrupted fear extinction without an impact on its acquisition. Unexpectedly, applying a similar approach to interfere with ASIC1a in mPFC did not influence fear acquisition or extinction. Therefore, ASIC1a in specific brain areas regulates distinct aspects of fear memory. How ASIC1a, which is widely expressed in many areas in the brains, including those implicated in fear conditioning and extinction, specifically executes distinct cognitive aspects of fear extinction? The answer may lie in the cognitive demand–dependent recruitment and activity-dependent modification of particular region- and/or projection-specific neuronal circuits. The need for vHPC (as well as other potential key areas), but not mPFC or BLA, to act during fear extinction may coincide with the activation mechanism(s) for ASIC1a there, allowing it to serve as a molecular substrate to transform eligibility traces at specific synapses. The regional specificity could also arise from a particular distribution and/or signaling mechanisms of ASIC1a in a specific population of vHPC neurons involved in fear extinction. Moreover, it cannot be ruled out at this point that variations in ASIC subunit compositions and/or even binding partners in different brain regions (i.e., vHPC versus mPFC or BLA) may also contribute to the selective engagement of region-specific ASIC1a regulation in fear acquisition and extinction, respectively, the molecular and cellular details of which warrant further investigations.
Second, we demonstrated an extinction-associated increase in BDNF expression, which relies on the vHPC ASIC1a activity. BDNF has been well recognized as a key mediator of fear extinction (11, 39), and many anxiolytics may exert their effects via interactions with BDNF signaling (1). In the present study, mRNA sequencing analysis revealed an extinction-associated expression of Bdnf and other genes (such as Npas4) in vHPC that is dependent, to a large extent, on ASIC1a. As Npas4 has been shown to directly regulate activity-dependent BDNF expression (33), we focused on BDNF as a potential downstream key molecule regulated by ASIC1a in vHPC for fear extinction. In the mammalian brain, the mRNA levels of BDNF are tightly regulated by neural activities, including both membrane depolarization and Ca2+ influx (34). Because of the low pH inside synaptic vesicles, the release of neurotransmitters in response to presynaptic stimulation is accompanied by a transient pH drop within the synaptic cleft, leading to activation of postsynaptic ASIC1a (19, 20). The membrane depolarization and Ca2+ influx brought about by ASIC1a activation would then enhance BDNF expression in vHPC neurons to levels higher than that induced by glutamate alone, and this mechanism would most likely also involve Npas4 (33).
Third, we characterized a cluster of extinction-associated plasticity features in the hippocampal-prefrontal correlates, including changes in transmitter release probability and postsynaptic NMDAR activities. These adaptive changes are opposite in vHPC→IL/mPFC versus vHPC→PL/mPFC synapses. Although the precise circuits that underlie these opposite effects remain to be studied in the future, most of the extinction-associated changes were shown to be dependent on ASIC1a in vHPC, highlighting the importance of vHPC neurons in shaping the extinction circuits beyond its engagement in contextual control of fear memory after extinction learning (40). The ASIC1a-dependent vHPC→mPFC BDNF signaling is responsible for most adaptive changes of the hippocampal-prefrontal synapses associated with extinction training. Not only did in vitro application of BDNF to mPFC slices from animals with Asic1a gene inactivated in vHPC and subjected to extinction training enhance NMDAR/AMPAR ratios at the vHPC→IL/mPFC synapses, but also the infusion of BDNF protein into mPFC, as well as the overexpression of BDNF in vHPC, successfully rescued the impaired fear extinction in these mice. These results are in agreement with the previous finding that activation of BDNF/TrkB receptor signaling enhances the GluN2B subunit–containing NMDAR currents at the synapses of IL/mPFC pyramidal neurons, which serves as a cellular mechanism for consolidation of extinction memory of cocaine-conditioned place preference (39). Our data further suggest a projection-specific regulation of mPFC synapses by vHPC to extinction learning, where ASIC1a regulates BDNF expression in vHPC to signal IL/mPFC neurons through modification of NMDAR activities at the vHPC-IL/mPFC synapses. Moreover, consistent with the NMDAR augmentation at the vHPC→IL/mPFC synapse upon fear extinction learning, the likelihood of burst firing in principal neurons of IL/mPFC was also increased in wild-type (WT) but not Asic1a-deficient animals. The NMDAR-dependent burst firing in vivo in IL/mPFC neurons has been shown to be crucial for consolidation of fear extinction memory (5).
In summary, we propose a working model of vHPC-ASIC1a regulation of fear extinction (Fig. 6F), in which activation of postsynaptic ASIC1a by protons co-released with glutamate from synaptic vesicles during and/or after extinction training augments membrane depolarization and Ca2+ influx induced by glutamate alone, which then enhances BDNF expression in vHPC. This is followed by an antegrade BDNF signal transferred through vHPC projections to mPFC, resulting in modification of NMDAR functions at the vHPC-mPFC synapses, which ultimately leads to suppression of fear behaviors. Our findings reveal a role for vHPC ASIC1a-BDNF signaling in regulating learning-dependent circuit plasticity and fear extinction. This work represents a significant advancement in our understanding of not only neural mechanisms by which ASIC1a is involved in neuronal circuit modification but also how vHPC activity contributes to fear extinction. Our results should inform new strategies in the development of clinical therapies that target extinction abnormality–related symptomatology associated with numerous neuropsychiatric illnesses, including anxiety disorders.
MATERIALS AND METHODS
Animals
WT C57BL/6J mice were purchased from Shanghai Laboratory Animal Center, Chinese Academy of Sciences, Shanghai, China. Asic1a KO (ASIC1a−/−) and floxed (Asic1aflox/flox) mice were prepared as previously described (17, 27). Heterozygous Asic1a (Asic1a+/−) mice were obtained by crossing WT C57BL/6J and Asic1a−/− mice. The littermate Asic1a+/+, Asic1a+/−, and Asic1a−/− mice bred from Asic1a+/− mice were used for behavioral experiments. All mice were bred in the specific pathogen–free class laboratory animal housing of Shanghai Jiao Tong University School of Medicine, where mice were housed on a 12-hour light/12-hour dark cycle with rodent chow and water ad libitum. Adult male mice (8 to 12 weeks old) were used for various experiments. All of the animal procedures were approved by the Animal Ethics Committee of Shanghai Jiao Tong University School of Medicine and approved by the Institutional Animal Care and Use Committee (Department of Laboratory Animal Science, Shanghai Jiao Tong University School of Medicine) (policy number DLAS-MP-ANIM. 01–05).
Virus constructs
Recombinant AAVs (serotype 2/9) were packaged by Shanghai SunBio Biomedical Technology Co. Ltd, China. All viral vectors were stored in aliquots at −80°C until use. The viral titers for injection were more than 1.0 × 1012 virus particles per milliliter (VG/ml), except for AAV-Syn-BDNF-mCherry and AAV-Syn-mCherry, which were diluted to 1.0 × 1011 VG/ml before injection.
Virus injection
Mice at ages of 6 to 7 weeks were anesthetized using 5% chloral hydrate with single intraperitoneal injection (0.01 ml/g body weight) and mounted in a stereotactic frame with nonrupture ear bars (RWD Life Science). After making an incision to the midline of the scalp, small bilateral craniotomies were performed using a microdrill with 0.5-mm burrs. Glass pipettes, with tip diameters of 10 to 20 μm, were made by P-97 Micropipette Puller (Sutter Glass pipettes) for AAV microinjection. The microinjection pipettes were first filled with silicone oil and then connected to a microinjector pump (KDS 310, KD Scientific) with full air exclusion. AAV-containing solutions were loaded into the tips of pipettes and injected at the following coordinates (posterior to bregma, AP; lateral to the midline, ML; below the bregma, DV; in millimeters): BLA: AP −1.60, ML ±3.30, DV −4.80; mPFC: AP +1.70, ML ±0.40, DV −2.60; dHPC: AP −2.00, ML ±1.50, DV −1.25; vHPC: AP −3.16, ML ±3.20, DV −4.75; dHPC: AP −2.00, ML ±1.50, DV −1.25; mPFC: AP +1.70, ML ±0.40, DV −2.60. Virus-containing solutions were injected bilaterally into the BLA (0.3 μl per side), mPFC (0.3 μl per side), vHPC (1 μl per side), or dHPC (0.3 μl per side) at a rate of 0.1 μl/min. After injection, the pipettes were left in place for an additional 10 min to allow the injectant to diffuse adequately. To inactivate Asic1a gene and overexpress BDNF simultaneously in vHPC, a mixture of AAV-Syn-BDNF-mCherry (1.0 × 1011 VG/ml) and AAV-Syn-Cre-GFP (>1.0 × 1012 VG/ml) (ratio, 1:3) were bilaterally injected into vHPC (1 μl per side). To inactivate Asic1a gene and allow optical stimulation of excitatory neurons simultaneously in vHPC, a mixture of AAV-CaMKIIα-ChR2-mCherry (>1.0 × 1012 VG/ml) and AAV-Syn-Cre-GFP (>1.0 × 1012 VG/ml) was bilaterally injected into vHPC (1 μl per side). For controls, AAV-Syn-BDNF-mCherry and AAV-Syn-Cre-GFP were replaced with AAV-Syn-mCherry and AAV-Syn-GFP, respectively. Mice were allowed to recover for at least 4 weeks before behavioral and other tests. The injection sites of virus solutions were examined after experiments by the expression of the fluorescent protein, GFP, YFP, or mCherry.
Cannula implantation and local drug injection
Mice were anesthetized with 5% chloral hydrate and then fixed on a stereotaxic apparatus (RWD Life Science). Stainless steel guide cannulas (RWD Life Science) were bilaterally implanted into the target brain areas, and the tips of cannulas were at following coordinates (in millimeters): BLA: AP −1.40, ML ±3.40, DV −3.50; mPFC: AP +1.70, ML ±1.65, DV −1.70, angled at 30°; dHPC: AP −1.90, ML ±1.90, DV +0.17, angled at 30°; vHPC: AP −3.16, ML ±3.20, DV −3.75. The cannulas were fixed to the skull using acrylic cement and two skull screws. Stainless steel obturators (33 gauges) were inserted into guide cannulas to avoid obstruction until drug infusion. Animals were allowed to recover from surgery for 2 weeks before behavioral tests. Mice were handled and habituated to the infusion procedure several days before drug injection. During drug infusion, mice were briefly head restrained, while the stainless steel obturators were removed and injection cannulas (33 gauges, RWD Life Science) were inserted into guide cannulas. Injection cannulas protrude 1.00 mm from the tips of guide cannulas. Infusion cannula was connected via PE20 tubing to a microsyringe driven by a microinfusion pump (KDS 310, KD Scientific). Drugs were infused bilaterally into the target brain areas at a flow rate of 0.15 μl per min. After finishing drug injection, the injection cannulas were left in place for 2 min to allow the solution to diffuse from the cannula tip. The stainless steel obturators were subsequently reinserted into guide cannulas, and the mice returned to their home cage for 30 min before behavioral tests.
Western blotting
Mice were sacrificed by cervical dislocation. mPFC or hippocampi from both sides of the brain were rapidly isolated. Hippocampi were then divided into dorsal and ventral parts along the longitudinal axis. The tissues were placed into liquid nitrogen followed by long storage at −80°C. For Western blotting analysis, samples were removed from the freezer and homogenized in a lysis buffer containing 20 mM tris-Cl (pH 7.4), 150 mM NaCl, 1% Triton X-100, 5 mM EDTA, 3 mM NaF, 1 mM sodium orthovanadate, 10% glycerol, and complete protease inhibitor set (Sigma-Aldrich, St. Louis, MO). The lysates were vortexed for 20 s, incubated on ice for 40 min, and centrifuged at 13,000 rpm for 15 min. The supernatants were transferred to sterile 1.5-ml Eppendorf tubes for subsequent experiments. Protein concentrations were determined by the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). Twenty micrograms of total protein extract per lane was resolved using denaturing SDS–polyacrylamide gel electrophoresis gels and transferred to polyvinylidene difluoride filters by electroblotting. The filters were incubated overnight at 4°C with appropriate primary antibodies. The following primary antibodies were used: goat anti-ASIC1a (1:1000; Santa Cruz Biotechnology, catalog no. sc-13905) and mouse monoclonal anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (1:1000; KangChen, catalog no. KC-5G4). Secondary antibodies conjugated to horseradish peroxidase were added to the filters and then visualized in enhanced chemiluminescence solution. The visualization was performed via the ImageQuant LAS 4000 Mini Molecular Imaging System (GE Healthcare Life Sciences), and the Quantity One software [National Institutes of Health (NIH)] was used for the analysis of band intensity.
Fear conditioning and extinction
All auditory fear conditioning and extinction procedures were performed using the Ugo Basile Fear Conditioning System (Ugo Basile Srl). Briefly, mice were first handled and habituated to the conditioning chamber for five successive days. The conditioning chambers (17 cm by 17 cm by 25 cm) equipped with stainless steel shocking grids were connected to a precision feedback current-regulated shocker (Ugo Basile Srl). During fear conditioning, the chamber walls were covered with black and white checkered wallpapers, and the chambers were cleaned with 75% ethanol (context A). On day 1, mice were conditioned individually in context A with five pure tones (CS; 4 kHz, 76 dB, 30 s each) delivered at variable intervals (20 to 180 s), and each tone was co-terminated with a foot shock (US; 0.5 mA, 2 s each). ANY-maze software (Stoelting Co.) was used to automatically control the delivery of tones and foot shocks. Conditioned mice were returned to their home cages 30 s after the end of the last tone, and the floor and walls of the cage were cleaned with 75% ethanol for each mouse. For retrieval, 24 hours after conditioning, animals received two CS-alone presentations in a test chamber, which had gray nonshocking plexiglass floor and dark gray wallpapers and was washed with 4% acetic acid solution between the tests for individual mice (context B). In extinction experiments, animal trained in context A with five CS-US pairings on day 1 was presented with 20 CS without foot shock in context B on day 2. On day 3, mice received eight CS-alone presentations in the extinction context. During the behavioral test, the chamber was placed in a sound-attenuating enclosure with ventilation fan and a single house light (Ugo Basile Srl). The movement of the mouse in the conditioning or test chamber was recorded using a near-infrared camera and analyzed in real time with ANY-maze software. Fear response was operationally defined as measurable behavioral freezing (more than 1-s cessation of movement), which was automatically scored and analyzed by ANY-maze software. The time spent on freezing during the tone (cue) was measured for each tone presentation.
Ex vivo slice electrophysiology
For electrophysiological recordings in mPFC slices, 2 hours after extinction learning (Ext.) or retrieval of fear memory (No Ext.), the mice were deeply anesthetized with 5% chloral hydrate and decapitated. Brains were dissected quickly and chilled in well-oxygenated (95% O2/5% CO2, v/v) ice-cold aCSF containing 125 mM NaCl, 2.5 mM KCl, 12.5 mM d-glucose, 1 mM MgCl2, 2 mM CaCl2, 1.25 mM NaH2PO4, and 25 mM NaHCO3 (pH 7.35 to 7.45). Coronal brain slices (300 μm thick) containing mPFC were cut with a vibratome (Leica VT1000S, Germany). After recovery for 1 hour in oxygenated aCSF at 30° ± 1°C, the slice was transferred to the recording chamber and continuously perfused with oxygenated aCSF at the rate of 1 to 2 ml/min. Some slices from Ext. groups of AAV-Syn-Cre-GFP–injected mice were incubated with BDNF (200 ng/ml diluted in oxygenated aCSF) for 2 hours and then transferred to normal aCSF for recording, with its control staying in normal aCSF at least 2 hours before recording. The superficial layer (layer II/III) pyramidal neurons in IL/mPFC and PL/mPFC were patched under visual guidance using infrared differential interference contrast microscopy (BX51WI, Olympus) and an optiMOS camera (QImaging). The slices were continuously perfused with well-oxygenated aCSF at 35° ± 1°C during all electrophysiological studies. Whole-cell patch clamp recordings were performed using an Axon 200B amplifier (Molecular Devices). Membrane currents were sampled and analyzed using a Digidata 1440 interface and a personal computer running Clampex and Clampfit software (version 10, Axon Instruments). Access resistance was 15 to 30 megohms, and only cells with a change in access resistance of <20% were included in the analysis.
Spontaneous excitatory postsynaptic currents
For recordings of sEPSCs (figs. S2 and S3), the holding potential was −70 mV. Patch pipettes had open tip resistances of 3 to 5 megohms when filled with an intracellular solution that contained 132.5 mM cesium gluconate, 17.5 mM CsCl, 2 mM MgCl2, 0.5 mM EGTA, 10 mM Hepes, 4 mM Mg-ATP (adenosine 5′-triphosphate), and 5 mM QX-314 chloride (280 to 300 mOsm; pH 7.2 adjusted with CsOH). The baseline sEPSCs of IL/mPFC and PL/mPFC neurons were recorded for 5 min and analyzed from 100 to 200 s after the establishment and stabilization of the recording. Data were analyzed using the Mini Analysis Program (Synaptosoft) with an amplitude threshold of 10 pA.
Spike firing
Spiking activity (Fig. 3, E to H, and figs. S5 and S6) and related membrane properties of superficial layer (layer II/III) neurons were measured with an internal solution containing 145 mM potassium gluconate, 5 mM NaCl, 10 mM Hepes, 2 mM Mg-ATP, 0.1 mM Na3GTP, 0.2 mM EGTA, and 1 mM MgCl2 (280 to 300 mOsm; pH 7.2 with KOH). Data were analyzed by Mini Analysis Program (Synaptosoft) with an amplitude threshold of 20 mV. The accommodation ratio (Fig. 3, E to H, and fig. S6) under the step current injection protocol was defined as the action potential number during the last 100-ms current injection to that during the first100-ms current injection.
Light-evoked EPSC
Optical activation of ChR2-expressing axons was performed using a blue collimated light-emitting diode (LED) with a peak wavelength of 470 nm (Lumen Dynamics). The LED was connected to Axon 200B amplifier to trigger photostimulation. The brain slice in the recording chamber was illuminated through a 40× water-immersion objective lens (LUMPLFLN 40XW, Olympus). The intensity of photostimulation was directly controlled by the stimulator, while the duration was set through Digidata 1440 and pCLAMP 10.5 software. To evoke synaptic responses in mPFC by photostimulation of vHPC axons, the slice was illuminated every 20 s with blue light pulses of 5-ms duration (light power density: 2 to 18 mW/mm2 at 470 nm). To prevent polysynaptic activities from being detected in EPSC recordings, we applied appropriate photostimulation intensity that produced 40 to 60% of the maximal synaptic response. For recording the light-evoked EPSCs, the recording pipettes (3 to 5 megohms) were filled with a solution containing 132.5 mM cesium gluconate, 17.5 mM CsCl, 2 mM MgCl2, 0.5 mM EGTA, 10 mM Hepes, 4 mM Mg-ATP, and 5 mM QX-314 chloride (280 to 300 mOsm; pH 7.2 with CsOH). To determine the PPR (Fig. 2), IL/mPFC and PL/mPFC neurons were voltage clamped at −70 mV. The AMPAR oEPSCs were evoked by paired photostimulations (25-, 50-, 100-, 200-, and 500-ms intervals, 5-ms duration) of ChR2-expressing vHPC axons, and PPR was calculated as the peak amplitude ratio of the second to the first oEPSC. To determine the NMDAR/AMPAR ratio (Figs. 3, A to D, and 6, C and D), the AMPAR-mediated oEPSCs were recorded at −70 mV, while the NMDAR-mediated oEPSCs were recorded in the presence of CNQX (20 μM) and picrotoxin (100 μM) at +40 mV. For each mPFC neuron, photostimulations of the same intensity and duration were used to record the AMPAR- and NMDAR-mediated oEPSCs.
RNA sequencing
AAV-injected mice were subjected to extinction learning or retrieval of fear memory 24 hours after conditioning. The vHPC tissues were collected 2 hours after behavioral test and rapidly frozen in liquid nitrogen. Samples from four mice that received the same AAV injection (AAV-Syn-Cre-GFP or AAV-Syn-GFP) and behavioral tests (Ext. or No Ext.) were pulled for RNA sequencing, and for each condition, the RNA sequencing was performed on two independent sets of samples.
Total RNA was extracted using a mirVana miRNA Isolation kit (catalog no. AM1561, Ambion) following the manufacturer’s instructions. The quantification and integrity of the RNA samples obtained were determined using Qubit 2.0 Fluorometer (Life Technologies, Waltham, MA) and Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara), respectively. Only samples with RNA integrity numbers of >8 (out of 10) were used for sequencing. Samples were prepared for sequencing by Shanghai Biotechnology Corporation using the VAHTS Stranded mRNA-seq Library Prep Kit for Illumina (Vazyme, Nanjing) and barcoded to allow samples to be multiplexed within a flow cell lane. Barcoded complementary DNA (cDNA) libraries were sequenced on a single lane of the Illumina HiSeq X Ten Sequencing System to obtain 150–base pair (bp) single-end reads at an approximate sequencing depth of 25.6 to 30.3 million reads per sample. Raw reads were trimmed to remove sequencing artifacts (1 bp from 3′ end) and filtered to remove low-quality reads (reads with quality scores of <20 in more than 10% of bases were discarded) before alignment to mouse genome (mm10) using HISAT2 (version 2.0.4). Differential expression analysis was conducted with edgeR, which was used to quantify transcript reads and to obtain z scores and fold change values for individual genes. Genes with corrected P value of ≤0.05 (Benjamini-Hochberg FDR correction) and fold change of ≥1.5 were selected for further analysis. Gene ontology categories were manually curated from results.
Quantitative real-time PCR
For AAV-injected mice, 2 hours after extinction learning (Ext.) or retrieval of fear memory (No Ext.), the vHPC tissues were collected and rapidly frozen in liquid nitrogen. The total RNA was extracted from the vHPC tissues using TRIzol Reagent (Invitrogen). Four micrograms of total RNA was used as template for cDNA synthesis and amplification with the FastQuant RT Kit (Tiangen) according to the manufacturer’s instructions. The cDNA was diluted to an equal concentration of 400 ng/μl, and 80 ng of which was used for further PCR amplification. Real-time PCR was then performed using the cDNA as template in a Power SYBR Green PCR Master Mix (Applied Biosystems). The primers used were as follows: Gapdh, 5′-CCTCGTCCCGTAGACAAAATGGT-3′ (forward) and 5′-TTGAGGTCAATGAAGGGGTCGT-3′ (reverse); Fos, 5′-GAGCTGGAGCCCCTGTGTAC-3′ (forward) and 5′-CAGGGTAGGTGAAGACAAAGGAA-3′ (reverse); Npas4, 5′-CAGATCAACGCCGAGATTCG-3′ (forward) and 5′-CACCCTTGCGAGTGTAGATGC-3′ (reverse); Bdnf, 5′-AGGATCCCCATCACAATCTTACA-3′ (forward) and 5′-GCCACTGACCACACAATTGCT-3′ (reverse). The plate was run in the Applied Biosystems 7500 Fast Real-Time PCR System under the Standard 7500 run mode (one cycle 50.0°C, 20 s; one cycle 95.0°C, 10 min; 35 cycles 95.0°C, 15 s and 60°C, 1 min with fluorescence measured during 60°C step). Data were then analyzed using the 2−ΔΔCt method. All collected data were normalized to the No Ext. group of AAV-Syn-GFP–injected mice.
Quantification of BDNF protein
The vHPC tissues were collected 2 hours after extinction learning (Ext.) or retrieval of fear memory (No Ext.) and were rapidly frozen in liquid nitrogen. Each sample was added to 200 μl of lysis buffer containing 137 mM NaCl, 20 mM tris-HCl (pH 8.0), 1% TERGITOL type NP40, 10% glycerol, complete protease inhibitor set (Sigma-Aldrich, St. Louis, MO), and 0.5 mM sodium vanadate, which was then homogenized and centrifuged at 16,000g for 30 min at 4°C. The supernatants were removed into aseptic Eppendorf tubes and stored at −80°C.
The total protein levels in individual samples were measured by the BCA Protein Assay Kit (Pierce Co., Appleton, WI). The BDNF Emax ImmunoAssay System (Promega Co., Madison, WI) was used to detect the amount of BDNF. Briefly, each well of a 96-well polystyrene plate was incubated overnight at 4°C with 100 μl of anti-BDNF monoclonal antibody (mAb) diluted 1:1000 in carbonate coating buffer [25 mM sodium bicarbonate and 25 mM sodium carbonate (pH 9.7)]. Unabsorbed mAb was removed, and the plates were washed once with tris-buffered saline with Tween 20 (TBST) wash buffer containing 20 mM tris-HCl (pH 7.6), 150 mM NaCl, and 0.05% (v/v) Tween 20. Just before blocking, tissue extracts were removed from the freezer and allowed to cool to room temperature. Plates were blocked using 200 μl of Promega 1× Block and Sample Buffer followed by incubation for 1 hour at room temperature. Plates were then washed using TBST wash buffer. One hundred microliters of each sample or standard (500, 250, 125, 62.5, 31.3, 15.6, 7.8, and 0 pg/ml) was added in duplicates to the plates. The plates were incubated for 2 hours with shaking (600 rpm) at room temperature, which was then followed with five times washing with TBST wash buffer. Anti-human BDNF polyclonal antibody (100 μl of diluted 1:500 in 1× Block and Sample Buffer) was added to each well, and plates were incubated for 2 hours with shaking (600 rpm) at room temperature. Plates were washed again five times using TBST wash buffer. Anti–immunoglobulin G horseradish peroxidase conjugate (100 μl of diluted 1:200 in 1× Block and Sample Buffer) was then added to each well, and plates were incubated for 1 hour with shaking (600 rpm) at room temperature. Plates were emptied again and washed five times using TBST wash buffer. Finally, 100 μl of Promega TMB One Solution was added to the plates. After incubation for 10 min at room temperature, the reaction was stopped using 100 μl of 1 N HCl, which was followed by reading at 450 nm within 30 min. BDNF concentrations were determined on the basis of the absorbance values against the standard curve and reported in pg/mg protein. The values were then normalized to that of the No Ext. group of AAV-Syn-GFP–injected mice in parallel experiments.
Statistical analyses
Data are presented as means ± SEM unless indicated otherwise. All histograms display individual points, which represent the values and number of individual samples for each condition. Data distributions were tested for normality, and variance equality between groups was assessed using the Levene’s test. Statistical comparisons were performed using unpaired Student’s t tests and one-way ANOVA or two-way repeated-measures ANOVA. For post hoc analysis, we used Bonferroni’s correction for multiple comparisons. Statistical analysis was performed with IBM SPSS Statistics 19 (SPSS), and P < 0.05 was considered statistically significant. Significance is mainly displayed as *P < 0.05, **P < 0.01, and ***P < 0.001, and in some cases is indicated as #P < 0.05, ##P < 0.01, and ###P < 0.001 for multiple comparisons (not significant values are not denoted).
Supplementary Material
Acknowledgments
We thank M. J. Welsh (Howard Hughes Medical Institute, University of Iowa, Iowa City, IA) and C.-C. Lien (National Yang-Ming University, Taiwan) for providing Asic1a KO and floxed mice. Funding: This study was supported by grants from the National Natural Science Foundation of China (91632304, 81730095, and 81771214), the National Basic Research Program of China (2014CB910300), the Shanghai Committee of Science and Technology (18QA1402500), and the NIH (NS102452). Author contributions: Qin Wang, W.-G.L., and T.-L.X. conceived the project, designed the experiments, and interpreted the results. Qin Wang performed the majority of behavioral experiments and data analysis. X.-L.S., Q.J., and Qin Wang performed Western blotting, immunohistochemistry, qPCR, and ELISA experiments. Qi Wang, Y.-J.W., and W.-G.L. performed slice recording and data analysis. T.-F.Y., S.Z., N.-J.X., and M.X.Z. contributed to data interpretation and experimental design. Qin Wang, M.X.Z., W.-G.L., and T.-L.X. wrote the manuscript with contributions from all authors. All authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the supplementary materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau3075/DC1
Fig. S1. Validation of knockdown of ASIC1a and cannula implantation in different brain regions.
Fig. S2. ASIC1a in vHPC is involved in extinction learning–driven changes of sEPSC (as global synaptic inputs) in mPFC neurons.
Fig. S3. US only does not alter sEPSC in mPFC neurons.
Fig. S4. Current-voltage relationships of NMDAR-mediated synaptic currents of different vHPC→mPFC projections.
Fig. S5. ASIC1a-dependent effects of fear extinction training on the action potential firing of pyramidal neurons in mPFC.
Fig. S6. Effects of NMDAR antagonism on action potential firing of pyramidal neurons in mPFC.
Fig. S7. Fear acquisition and extinction/retrieval of Asic1aflox/flox mice that received AAV-Syn-Cre-GFP and AAV-Syn-GFP injection at vHPC.
Fig. S8. Validation of AAV-mediated overexpression of BDNF or ASIC1a at vHPC.
Fig. S9. Effects of Asic1a gene overexpression in dHPC on cued fear extinction.
REFERENCES AND NOTES
- 1.Milad M. R., Quirk G. J., Fear extinction as a model for translational neuroscience: Ten years of progress. Annu. Rev. Psychol. 63, 129–151 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunsmoor J. E., Niv Y., Daw N., Phelps E. A., Rethinking extinction. Neuron 88, 47–63 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milad M. R., Quirk G. J., Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420, 70–74 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Morgan M. A., Romanski L. M., LeDoux J. E., Extinction of emotional learning: Contribution of medial prefrontal cortex. Neurosci. Lett. 163, 109–113 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Burgos-Robles A., Vidal-Gonzalez I., Santini E., Quirk G. J., Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron 53, 871–880 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Burgos-Robles A., Vidal-Gonzalez I., Quirk G. J., Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J. Neurosci. 29, 8474–8482 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sotres-Bayon F., Sierra-Mercado D., Pardilla-Delgado E., Quirk G. J., Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron 76, 804–812 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho J. H., Deisseroth K., Bolshakov V. Y., Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron 80, 1491–1507 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu C., Krabbe S., Gründemann J., Botta P., Fadok J. P., Osakada F., Saur D., Grewe B. F., Schnitzer M. J., Callaway E. M., Lüthi A., Distinct hippocampal pathways mediate dissociable roles of context in memory retrieval. Cell 167, 961–972.e16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marek R., Jin J., Goode T. D., Giustino T. F., Wang Q., Acca G. M., Holehonnur R., Ploski J. E., Fitzgerald P. J., Lynagh T., Lynch J. W., Maren S., Sah P., Hippocampus-driven feed-forward inhibition of the prefrontal cortex mediates relapse of extinguished fear. Nat. Neurosci. 21, 384–392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peters J., Dieppa-Perea L. M., Melendez L. M., Quirk G. J., Induction of fear extinction with hippocampal-infralimbic BDNF. Science 328, 1288–1290 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellenberger S., Schild L., International Union of Basic and Clinical Pharmacology. XCI. Structure, function, and pharmacology of acid-sensing ion channels and the epithelial Na+ channel. Pharmacol. Rev. 67, 1–35 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Waldmann R., Champigny G., Bassilana F., Heurteaux C., Lazdunski M., A proton-gated cation channel involved in acid-sensing. Nature 386, 173–177 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Xiong Z. G., Zhu X. M., Chu X. P., Minami M., Hey J., Wei W. L., MacDonald J. F., Wemmie J. A., Price M. P., Welsh M. J., Simon R. P., Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell 118, 687–698 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Mazzuca M., Heurteaux C., Alloui A., Diochot S., Baron A., Voilley N., Blondeau N., Escoubas P., Gélot A., Cupo A., Zimmer A., Zimmer A. M., Eschalier A., Lazdunski M., A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat. Neurosci. 10, 943–945 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Ziemann A. E., Schnizler M. K., Albert G. W., Severson M. A., Howard M. A. III, Welsh M. J., Wemmie J. A., Seizure termination by acidosis depends on ASIC1a. Nat. Neurosci. 11, 816–822 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wemmie J. A., Chen J., Askwith C. C., Hruska-Hageman A. M., Price M. P., Nolan B. C., Yoder P. G., Lamani E., Hoshi T., Freeman J. H. Jr, Welsh M. J., The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34, 463–477 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Wemmie J. A., Askwith C. C., Lamani E., Cassell M. D., Freeman J. H. Jr, Welsh M. J., Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J. Neurosci. 23, 5496–5502 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du J., Reznikov L. R., Price M. P., Zha X.-m., Lu Y., Moninger T. O., Wemmie J. A., Welsh M. J., Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc. Natl. Acad. Sci. U.S.A. 111, 8961–8966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreple C. J., Lu Y., Taugher R. J., Schwager-Gutman A. L., Du J., Stump M., Wang Y., Ghobbeh A., Fan R., Cosme C. V., Sowers L. P., Welsh M. J., Radley J. J., LaLumiere R. T., Wemmie J. A., Acid-sensing ion channels contribute to synaptic transmission and inhibit cocaine-evoked plasticity. Nat. Neurosci. 17, 1083–1091 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W.-G., Liu M.-G., Deng S., Liu Y.-M., Shang L., Ding J., Hsu T.-T., Jiang Q., Li Y., Li F., Zhu M. X., Xu T.-L., ASIC1a regulates insular long-term depression and is required for the extinction of conditioned taste aversion. Nat. Commun. 7, 13770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smoller J. W., Acierno J. S., Rosenbaum J. F., Biederman J., Pollack M. H., Meminger S., Pava J. A., Chadwick L. H., White C., Bulzacchelli M., Slaugenhaupt S. A., Targeted genome screen of panic disorder and anxiety disorder proneness using homology to murine QTL regions. Am. J. Med. Genet. 105, 195–206 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Smoller J. W., Gallagher P. J., Duncan L. E., McGrath L. M., Haddad S. A., Holmes A. J., Wolf A. B., Hilker S., Block S. R., Weill S., Young S., Choi E. Y., Rosenbaum J. F., Biederman J., Faraone S. V., Roffman J. L., Manfro G. G., Blaya C., Hirshfeld-Becker D. R., Stein M. B., Van Ameringen M., Tolin D. F., Otto M. W., Pollack M. H., Simon N. M., Buckner R. L., Ongür D., Cohen B. M., The human ortholog of acid-sensing ion channel gene ASIC1a is associated with panic disorder and amygdala structure and function. Biol. Psychiatry 76, 902–910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gugliandolo A., Gangemi C., Caccamo D., Currò M., Pandolfo G., Quattrone D., Crucitti M., Zoccali R. A., Bruno A., Muscatello M. R., The RS685012 polymorphism of ACCN2, the human ortholog of murine acid-sensing ion channel (ASIC1) gene, is highly represented in patients with panic disorder. Neuromol. Med. 18, 91–98 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Ziemann A. E., Allen J. E., Dahdaleh N. S., Drebot I. I., Coryell M. W., Wunsch A. M., Lynch C. M., Faraci F. M., Howard M. A. III, Welsh M. J., Wemmie J. A., The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell 139, 1012–1021 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coryell M. W., Wunsch A. M., Haenfler J. M., Allen J. E., McBride J. L., Davidson B. L., Wemmie J. A., Restoring Acid-sensing ion channel-1a in the amygdala of knock-out mice rescues fear memory but not unconditioned fear responses. J. Neurosci. 28, 13738–13741 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu P.-Y., Huang Y.-Y., Chen C.-C., Hsu T.-T., Lin Y.-C., Weng J.-Y., Chien T.-C., Cheng I. H., Lien C.-C., Acid-sensing ion channel-1a is not required for normal hippocampal LTP and spatial memory. J. Neurosci. 33, 1828–1832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strange B. A., Witter M. P., Lein E. S., Moser E. I., Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 15, 655–669 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Parent M. A., Wang L., Su J., Netoff T., Yuan L.-L., Identification of the hippocampal input to medial prefrontal cortex in vitro. Cereb. Cortex 20, 393–403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zucker R. S., Regehr W. G., Short-term synaptic plasticity. Annu. Rev. Physiol. 64, 355–405 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Senn V., Wolff S. B., Herry C., Grenier F., Ehrlich I., Gründemann J., Fadok J. P., Müller C., Letzkus J. J., Lüthi A., Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 81, 428–437 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Lisman J. E., Bursts as a unit of neural information: Making unreliable synapses reliable. Trends Neurosci. 20, 38–43 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Sun X., Lin Y., Npas4: Linking neuronal activity to memory. Trends Neurosci. 39, 264–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park H., Poo M.-m., Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Duvarci S., Pare D., Amygdala microcircuits controlling learned fear. Neuron 82, 966–980 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weng J.-Y., Lin Y.-C., Lien C.-C., Cell type-specific expression of acid-sensing ion channels in hippocampal interneurons. J. Neurosci. 30, 6548–6558 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mango D., Braksator E., Battaglia G., Marcelli S., Mercuri N. B., Feligioni M., Nicoletti F., Bashir Z. I., Nisticò R., Acid-sensing ion channel 1a is required for mGlu receptor dependent long-term depression in the hippocampus. Pharmacol. Res. 119, 12–19 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Yermolaieva O., Leonard A. S., Schnizler M. K., Abboud F. M., Welsh M. J., Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc. Natl. Acad. Sci. U.S.A. 101, 6752–6757 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otis J. M., Fitzgerald M. K., Mueller D., Infralimbic BDNF/TrkB enhancement of GluN2B currents facilitates extinction of a cocaine-conditioned place preference. J. Neurosci. 34, 6057–6064 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maren S., Phan K. L., Liberzon I., The contextual brain: Implications for fear conditioning, extinction and psychopathology. Nat. Rev. Neurosci. 14, 417–428 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau3075/DC1
Fig. S1. Validation of knockdown of ASIC1a and cannula implantation in different brain regions.
Fig. S2. ASIC1a in vHPC is involved in extinction learning–driven changes of sEPSC (as global synaptic inputs) in mPFC neurons.
Fig. S3. US only does not alter sEPSC in mPFC neurons.
Fig. S4. Current-voltage relationships of NMDAR-mediated synaptic currents of different vHPC→mPFC projections.
Fig. S5. ASIC1a-dependent effects of fear extinction training on the action potential firing of pyramidal neurons in mPFC.
Fig. S6. Effects of NMDAR antagonism on action potential firing of pyramidal neurons in mPFC.
Fig. S7. Fear acquisition and extinction/retrieval of Asic1aflox/flox mice that received AAV-Syn-Cre-GFP and AAV-Syn-GFP injection at vHPC.
Fig. S8. Validation of AAV-mediated overexpression of BDNF or ASIC1a at vHPC.
Fig. S9. Effects of Asic1a gene overexpression in dHPC on cued fear extinction.