

Abstract
Aim: In-Stent Restenosis (ISR) is the major reason for recurrent ischemia and amputation after endovascular treatment of Peripheral Artery Disease (PAD). Our previous study demonstrated that miR-140-3p is significantly down-regulated in PAD arteries. However, expression and function of miR-140-3p in ISR of human PAD are currently unclear.
The aim of this study is to determine the miR-140-3p expression and its regulative role in ISR of PAD.
Methods: The RNA level was determined by quantitative real-time polymerase chain Reaction (qRT-PCR) and in situ hybridization. Primary cultured ASMCs were isolated from human femoral arterial of the healthy donors or ISR patients. Cell proliferation was determined by Edu incorporation and CCK-8 assay. Apoptosis was determined by Annexin-V/PI Double-Staining assay and TUNEL assay. A rat carotid artery balloon angioplasty model was used to investigate the effect of miR-140-3p on restenosis.
Results: MiR-140-3p was significantly down-regulated in PAD and ISR arteries than normal arteries. Primary cultured ISR ASMCs exhibited elevated proliferation and down-regulated miR-140-3p than normal ASMCs. Transfection of miR-140-3p mimic attenuated PDGF-BB-induced proliferation in cultured ASMCs and induced apoptosis. Luciferase reporter assay indicated that miR-140-3p transfection significantly down-regulated C-Myb and BCL-2 in ISR ASMCs by targeting to their 3′-UTRs. MiR-140-3p transfection induced anti-proliferation and apoptosis in ASMCs, which were ameliorated by over-expression of C-Myb or BCL-2. Moreover, the animal study showed that miR-140-3p can reduce restenosis following angioplasty via targeting C-Myb and BCL-2.
Conclusions: The result suggests that miR-140-3p regulates ASMC function via targeting C-Myb and BCL-2 in the process of ISR in PAD. The novel findings may offer a hopeful therapeutic target for human PAD.
Keywords: In-stent restenosis, Peripheral arterial disease, MiR-140-3p, C-Myb, BCL-2
Introduction
Peripheral artery disease (PAD) of lower extremities affects approximately 12% to 14% of the general population, and 5% to 10% patients with PAD will progress to critical limb ischemia1, 2). Endovascular treatment, including balloon and stent angioplasty, is a common treatment strategy and is becoming the first choice for symptomatic PAD. However, 20%–50% of PAD patients treated with stent angioplasty would develop In-Stent Restenosis (ISR) within 1 year, resulting in recurrent ischemia and limb loss in approximately 14.7% patients2). The vascular injury caused by balloon and stent angioplasty provokes excessive artery smooth muscle cell (ASMC) proliferation, which plays the pivotal step in the process of ISR. The anti-proliferative agents in drug-coated balloon or drug eluting stent, such as paclitaxel, effectively attenuate early ISR by inhibiting ASMC proliferation. However, the non-cell-specific feature of coated agents also damages the endothelial cells, thus leading to thrombosis or atherosclerosis, resulting to late treatment failure3–6).
MicroRNA is a new class of endogenous, single-stranded, short non-coding (∼22 nucleotides) RNAs, which can negatively regulate their target mRNAs at post-transcriptional level by binding the 3′-untranslated region (3′-UTR)7–9). Accumulating evidence has suggested that microRNAs are involved in the pathogenesis of cardiovascular diseases and could be used as the diagnostic markers. For example, Dong et al. have demonstrated that the expression levels of miR-24, miR-33a, miR-103a, and miR-122 in peripheral blood mononuclear cells (PBMCs) are increased in patients with coronary artery disease (CAD), which are the independent risk factors for CAD10). In addition, the panel of the four miRNAs possesses a high diagnostic accuracy of CAD10). Likewise, He et al. have reported that expressions of miR-4284 and miR-4463 were altered in the serum of patients with arteriosclerosis obliterans (ASO) in a time and stage-specific expression manner, which could be used as potential biomarkers for the early diagnosis of ASO11). The microRNA expression profile in rat vessels was first described by Ji et al12), and the microRNA expression profile of human PAD was originally described in our previous study13–15). The cellular tropism of microRNA expression and function demonstrates that it could be an ideal potential therapeutic target for ISR5, 6, 16). However, direct evidence as to whether microRNAs are involved in the process of ISR in human arteries with PAD is still missing16).
Aim
In the current study, we found out that miR-140-3p was down-regulated in ISR artery samples of human PAD and focused on the potential role and underlying mechanisms of miR-140-3p in modulating ASMC function in the process of ISR of human PAD.
Materials and Methods
Sample Acquisition
IRS artery samples were acquired from 10 PAD patients with ISR who accepted lower limb amputation. Normal artery samples were obtained from six donors without PAD of lower extremities. After arterectomy, some specimens were frozen in liquid nitrogen for RNA extraction, and other specimens were embedded in paraffin for further experiments. This study was approved by the Research Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University and conformed to the declaration of Helsinki. Informed consent for the procedure was given by all patients.
Quantitative Real-Time Polymerase Chain Reaction
To detect miRNA or mRNA level, Quantitative Real-time Polymerase Chain Reaction (qRT-PCR) was performed. Total RNA was extracted from cultured ASMCs, human arteries, and rat carotid arteries using TRIzol (Invitrogen, USA) following the manufacturer's protocol. The threshold cycle (Ct) values of miRNA and mRNA were normalized to the reference genes, U6 and GADPH, respectively. The final data were calculated by comparative Ct method (relative gene expression = 2−(ΔCt sample − ΔCt control)) and presented as the fold-change. The primers used in the study are shown: hsa miR-140-3p 5′-TACCACAGGGTAGAACCACGG-3′, rno miR-140-3p 5′-CCACAGGGTAGAACCACGG-3′; has-C-Myb 5′-ATCTCCCGAATCGAACAGATGT-3′ (forward) and 5′-TGCTTGGCAATAACAGACCAAC-3′ (reward); has-BCL-2 5′-GGTGGGGTCATGTGTGTGG-3′ (forward) and 5′-CGGTTCAGGTACTCAGTCATCC-3′ (reward).
In Situ Hybridization and Immunostaining
To analyze the expression of miR-140-3p, In Situ Hybridization (ISH) was performed in human or rat paraffin-embedded 5-µm-thin sections14). In brief, the slides were deparaffinized using xylene (twice, 5 min) and rehydrated through a gradient ethanol (2 × 100%, 75%, 50%, and 25%, 5 min each). Then, sections were washed three times in phosphate buffer saline (PBS) and digested with 40 µg/mL proteinase K in pre-warmed 50 mM Tris at 37 °C for 5 min, followed by washing in 0.2% glycine/PBS, fixed using 4% paraformaldehyde and acetylated with acetic anhydride/triethanolamine. After prehybridizing in hybridization buffer (50% formamide, 5 × SSC, 0.5 mg/mL yeast tRNA, 1 × Denhardt's solution) at 49.5°C for 2 h, sections were hybridized using probes (1 µmol/L) at 49.5°C overnight. Then, slides were incubated in 1:1000-diluted anti-digoxigenin antibody (Roche, Switzerland) at 4 °C for 16 h. After washing, slides were incubated with 1:100-diluted NBT/BCIP staining buffer for 12 h in the dark. After color development was terminated, slides were washed with PBS and then dehydrated using an ethanol gradient and cover slipped. For determining the co-localization of miR-140-3p and SM α-actin or BCL-2 or C-Myb, the tyramide signal amplification system (Perkin-Elmer) was used to detect the IDH signals according to the manufacturer's protocol. Subsequently, after SM α-actin (ab5694, Abcam, UK) or BCL-2 (NB100-92142, Novus, USA) or C-Myb (NBP1-80306, Novus, USA) and DAPI staining, slides were mounted in H-1500 mounting medium (Vector). Finally, sections were visualized and photographed using a fluorescence microscope, and the IOD values of miR-140-3p on different sections were calculated and analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Inc.).
ASMC and HUVEC Isolation and Cell Culture
ASMCs were isolated from human femoral arteries of healthy donors or ISR patients. ASMCs were maintained in DMEM (Gibco, USA) containing 1% penicillin/streptomycin and 10% heat-inactivated fetal bovine serum in a humidified atmosphere containing 5% CO2 at 37°C. Human umbilical vein endothelial cells (HUVECs) were purchased commercially (Shanghai, China) and cultured in the EC-specific medium EGM-2 (Lonza, Switzerland) supplemented with 10% fetal bovine serum in a humidified atmosphere containing 5% CO2 at 37°C. Cells from passages 4th to 9th were used in this study.
Western Blot Analysis
Western Blot Analysis was performed to detect SM α-actin, SM-MHC, C-Myb, BCL-2, Bax, β-tubulin, and β-actin protein levels. In brief, total proteins were extracted from cultured cells and measured using a BCA Protein Assay kit (Thermo, USA). Equal amounts of protein were subjected to 6% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% BSA for 1 h, followed by incubation with primary antibodies overnight at 4°C. The membranes were incubated with HRP-conjugated secondary antibody (A00098, GenScript, USA). The bands were imaged and analyzed on a GE Image-Quant LAS4000 mini system (GE Healthcare, USA). The primary antibodies used in the study were displayed as follows: anti-SM α-actin (ab5694, Abcam, UK), anti-SM-MHC (ab124679, Abcam, UK), anti-C-Myb (NBP1-80306, Novus, USA), anti-BCL-2 (NB100-92142, Novus, USA), anti-Bax (#2772, Cell Signaling Technology, USA), anti-β-tubulin (#2128, Cell Signaling Technology, USA), and anti-β-actin (#4970, Cell Signaling Technology, USA).
Oligo Transfection
The miR-140-3p mimic, miR-140-3p inhibitor, and negative control oligo were purchased from Ribo-Bio (Guangzhou, China). Oligo transfection was performed using the reagent RNAiMax (Invitrogen, USA) according to the manufacturer's instructions.
Measurement of ASMC Proliferation
ASMC proliferation was determined by CCK-8 assays (Dojindo, Japan) and EdU assays (RiboBio, China). For CCK-8 assays, 10 µL of CCK-8 reagent was added into each well, and absorbance was read at 450 nm after incubation at 37°C for 1 h. For EdU assays, cells were cultured with 10 nM EdU solution at 37°C for 2 h, followed by fixation in 4% formaldehyde for 30 min. Then, the cells were treated with an Apollo cocktail for 30 min and subsequently treated with Hoechst 33342 for nuclear staining according to the manufacturer's instruction. Finally, cell proliferation was detected under an inverted fluorescence microscope (Carl Zeiss Axio Observer Z1, Germany).
Measurement of ASMC Apoptosis
Cell apoptosis analysis was determined by Annexin-VV and PI Double-Staining assays (Annexin-V-FITC Apoptosis Detection kit, Bipec Biopharma, USA) and Terminal Uridine Nick End-Labeling assays (TUNEL, Roche, German). For Annexin-V and PI Double-Staining assays, cells were rinsed with PBS and harvested after 24-h starvation. Flow cytometric analysis (Beckman Coulter, USA) was performed following the double staining of FITC-conjugated Annexin-V and PI according to manufacturer's instruction. For TUNEL assays, cells were grown on coverslips overnight, followed with transfection for 12 h. After starvation for 48 h, TUNEL and DAPI staining were performed following manufacturer's instructions. Images were obtained using fluorescence microscopy, and the apoptosis index of cells was analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Inc.).
Measurement of ASMC Migration
ASMC migration was evaluated by wound closure assays and Transwell assays. For wound closure assays, cells were cultivated on six-well plates overnight and transfected at 80%–90% confluence. Twelve hours after transfection, cells were cultured with serum-free medium to starvation for 24 h. Then, a straight single wound was scratched with a sterilized 200 µL pipette tip. Images were captured at 0 and 12 h after cells were treated with or without PDGF-BB (20 ng/mL) using an inverted microscope (ZEISS Axio Observer, German), and the wound-closure%, which demonstrates the percentage of wound closure, was evaluated using Image-Pro Plus 6.0 software. The initial scratch width was set as 100%. For Transwell assays, after transfection, cells were resuspended at a concentration of 5 × 105 cells/mL in serum-free DMEM, and 200 µL was added to the upper chamber, whereas the lower chamber was filled with 500 µL serum-free DMEM medium with or without PDGF-BB (20 µg/L). After 8 h of incubation at 37°C, cells that had migrated to the lower side of the chamber membrane were fixed with 4% paraformaldehyde and subsequently stained with 0.1% crystal violet, and the upper side of the chamber membrane was cleaned using a cotton swab. Following 3 PBS washes in the chamber, migrating cells were visualized using a microscope and counted.
Immunohistochemical Staining
For detecting C-Myb, BCL-2, CD31, or CD34 protein level in artery walls, IHC was performed in human or rat paraffin-embedded 5-µm-thin sections as previously described13, 14). Sections were incubated with primary antibodies for C-Myb (NBP1-80306, Novus, USA), BCL-2 (NB100-92142, Novus, USA), CD31 (ab28364, Abcam, UK), and CD34 (ab81289, Abcam, UK). After treatment with EnVision™ Detection Systems Kit, Peroxidase/DAB, Rabbit/Mouse, slides were visualized and photographed using Olympus BX53 microscope (Olympus, Japan) and analyzed using Image-Pro Plus 6.0 software (Media Cybernetics, Inc.).
Dual Luciferase Assay
Fragments of the C-Myb or BCL-2 3′-UTR containing the putative wild-type or mutant miR-140-3p binding sites were amplified and cloned into luciferase reporter constructs (Promega Biotech, USA) according to the manufacturer's instruction. The constructs were cotransfected into HEK 293 cells with empty vehicle, miR-140-3p mimic (50 nM), mimic oligocontrol (50 nM), miR-140-3p inhibitor (100 nM), or inhibitor oligo-control (100 nM) using Lipofectamine 2000 (Invitrogen, USA). Forty-eight hours after transfection, the firefly or Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System Kit (Promega Biotech, USA). Renilla luciferase activity was normalized by firefly luciferase activity.
Lentiviral Transduction
HEK 293Ta cells were cotransfected with 2.5 µg lentiviral C-Myb (GeneCopoeia, USA) plasmids, 5 µL Lenti-Pac HIV mix, and 15 µL EndoFectinLenti following the manufacturer's protocols (GeneCopoeia, USA). After 48 h of transfection, particles were collected from cell medium by 10 min centrifugation at 500 g and 0.44 µm filtration. To increase the transfection efficiency, virus-containing filtrate, mixed with isometric serum-free medium containing 20 µg/mL polybrene (Sigma, USA), was added to cells overnight and replaced with regular culture medium for subsequent 24 h incubation. Finally, cells used for follow-up experiments were selected by 10 µg/mL puromycin (Sigma, USA) for 48 h.
Rat Carotid Artery Restenosis Model
Male Sprague-Dawley (SD) rats (250 to 300 g) were used to establish the rat carotid artery restenosis model as described previously12, 14, 15). Briefly, rats were anesthetized with ketamine (80 mg/kg)/xylazine (5 mg/kg). Under a dissecting microscope, a midline vertical cervical incision was performed to expose the right carotid artery. After the external carotid arteriotomy, a 2F Fogarty catheter (Edwards Lifesciences, CA) was inserted into the common carotid and generated the carotid artery injury by withdrawing the inflated balloon three times from the edge of the sternum to the carotid bifurcation. After the balloon injury, lentivirus expressing miR-140-3p (LV-miR-140-3p) or controllentivirus expressing green fluorescent protein (LV-GFP) was injected into the right common carotid artery via arteriotomy, and the segment of the common carotid artery was ligated temporarily for 30 min. Subsequently, the right external carotid artery was permanently ligated using 6-0 silk suture, and the blood flow to the right common carotid artery was restored. Twenty one days after balloon injury, the rats were sacrificed with intraperitoneal injection of sodium pentobarbital (200 mg/kg), and bilateral carotid artery samples were collected for further experiments. All of the animal experimental protocols were approved by the Research Ethics Committee of The First Affiliated Hospital, Sun Yatsen University and complied with the International Guidelines for the Care and Use of Laboratory Animals (NIH publication 85-23, revised 1985).
Statistics
All data are expressed as mean ± SD. Student's t-tests were used to test two-groups comparison. Oneway ANOVA was used to analyze the values of multiple independent groups. P value less than 0.05 was considered as statistically significant, and P value less than 0.01 was considered as highly statistically significant. We used SPSS software 17.0 (SPSS Inc, Chicago) and Graph-Pad Prism 5.0 (GraphPad, San Diego, CA) to process the data.
Results
MiR-140-3p Expression Is Decreased in ASMCs of Restenotic Artery Wall
First, using qRT-PCR analysis, we confirmed that miR-140-3p was markedly down-regulated not only in PAD but also in ISR samples compared with normal arteries, which indicated that miR-140-3p might be involved in the process of ISR (Figs. 1A and B). To determine the distribution of miR-140-3p in vascular walls, normal artery samples were dissected into three layers, namely, endothelium, media, and adventitia. Then, qRT-PCR assay was performed to compare miR-140-3p levels in the three layers. As shown in Fig. 1C, miR-140-3p expression level in artery media is significantly higher than that in endothelium and adventitia. Next, immunofluorescence and in situ hybridization assay (ISH) co-staining assay in artery sections demonstrated that miR-140-3p was clearly co-localized with SM α-actin, demonstrating that miR-140-3p is mainly expressed in ASMCs (Fig. 1D). The fluorescence signal was neither observed in two control sections (negative control: no SM α-actin antibody, no miR probe; scramble control: no SM α-actin antibody, but had scramble miR probe) nor observed in endothelium or adventitia of normal artery walls (Fig. 1D). Then, the Integral Optical Density (IOD) values of miR-140-3p staining in both normal and ISR sections were calculated and found to be significantly decreased in ISR sections compared with normal artery sections (Fig. 1E). These data indicated that miR-140-3p was down-regulated in ASMCs of ISR after endovascular treatment.
Fig. 1.
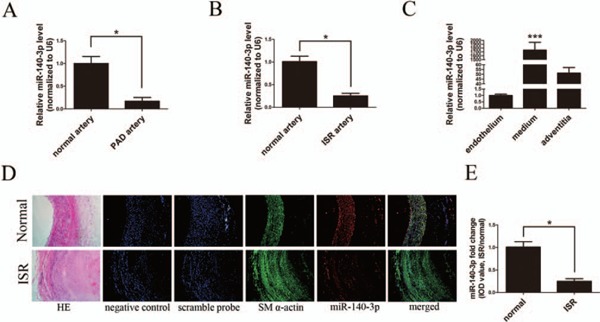
Expression and distribution of miR-140-3p in arterial walls
A and B, Relative expressions of miR-140-3p in normal (n = 6), PAD (n = 10), and ISR (n = 10) arterial walls determined by qRT-PCR. C, Relative expressions of miR-140-3p in endothelium, media, and adventitia of normal arterial walls determined by qRT-PCR. D, Representative graphs of SM α-actin (green) and miR-140-3p (red) co-staining in human normal and ISR sections (scale bars = 100 µm). Hematoxylin–eosin (HE) staining revealed artery structures. Negative control (no SM α-actin antibody and no miRNA probe) and Scrambled probe control (without SM α-actin antibody, but with scrambled miRNA probe) were used to verify no specific staining. Green and red immunofluorescence assays identify the smooth muscle marker SM α-actin and miR-140-3p, respectively. Merged images showed the co-localization of SM α-actin and miR-140-3p. Blue illustrates the cell nuclear staining by DAPI. E, Comparison of the IOD values of miR-140-3p staining in normal and ISR sections; mean miR-140-3p expression levels in normal arteries were defined as 1.0. (n = 10), *P < 0.05, ***P < 0.0001.
MiR-140-3p Expression Is Down-Regulated in ISR ASMCs
ASMC cells isolated from ISR samples were determined by SM α-actin immunofluorescence staining analysis (Fig. 2A). Then, the differences between the normal and ISR ASMCs were studied. As shown in Fig. 2C, the protein levels of SM α-actin and SMMHC were obviously lower in ASMCs from the ISR artery compared with ASMCs from normal arteries. Next, cell proliferation was determined by Edu incorporation and CCK-8 incorporation assay. The proportion of proliferating cells was significantly higher in ISR ASMCs than normal ASMCs (Figs. 2D, E, and F). More importantly, qRT-PCR analysis demonstrated that miR-140-3p expression level was significantly lower in ISR ASMCs compared with ASMCs from normal arteries (Fig. 2G).
Fig. 2.
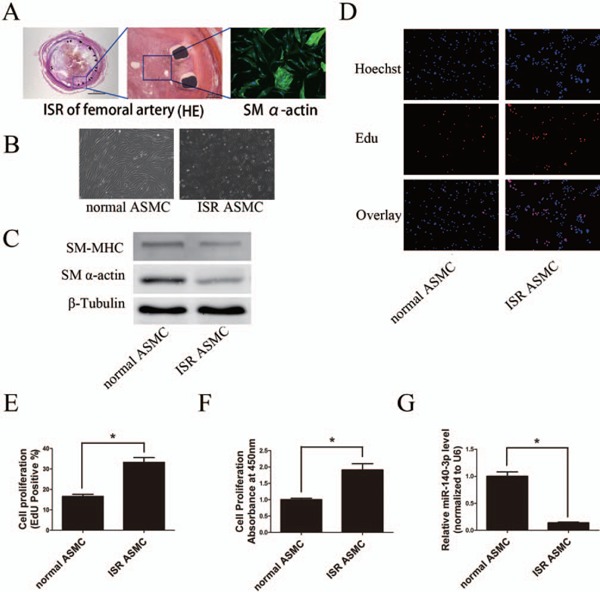
Expression of miR-140-3p in cultured ASMCs in vitro
A, Left (scale bars = 2 mm) and middle pictures (scale bars = 200 µm), hematoxylin–eosin (HE) staining demonstrated artery structures of ISR. Right picture (scale bars = 20 µm), staining of SM α-actin (green) in cells isolated from ISR samples. B, Phase-contrast microphotographs showed the morphology of cultured normal and ISR ASMCs (scale bars = 100 µm). C, Western blot analysis showed that cultured ISR ASMCs expressed lower levels of phenotypic markers (SM-MHC and SM α-actin) compared with normal ASMCs. D to F, ASMCs isolated from ISR arteries were more likely to proliferate than ASMCs isolated from normal arteries, determined by Edu and CCK-8 assays (scale bars = 100 µm). G, The expression level of miR-140-3p was lower in ISR ASMCs than in normal ASMCs, determined by qRT-PCR. (n = 3 to 6), *P < 0.05.
MiR-140-3p Inhibits Cell Proliferation and Increases Apoptosis in Cultured ASMCs
It is well established that excessive ASMC proliferation is a major cause of ISR after endovascular treatment. We have found that miR-140-3p expression is significantly decreased in ASMCs from ISR arteries. To study whether ISR after endovascular treatment is related with miR-140-3p down-regulation, we first investigated the relationship between miR-140-3p level and ASMC proliferation. Primary cultured ASMCs were treated with Platelet-Derived Growth Factor (PDGF)-BB, the strongest stimuli to induce ASMC proliferation as acknowledged. As shown in Fig. 3A, PDGF-BB treatment induced a time-dependent suppression of miR-140-3p expression. Next, miR-140-3p mimic was introduced into cultured ASMCs by oligo transfection. Then, ASMC proliferation was detected by two different methods: EdU assay and CCK-8 assay. As shown in Figs. 3B, C, and D, PDGF-BB (20 ng/mL) induced ASMC proliferation was significantly attenuated by miR-140-3p mimic.
Fig. 3.
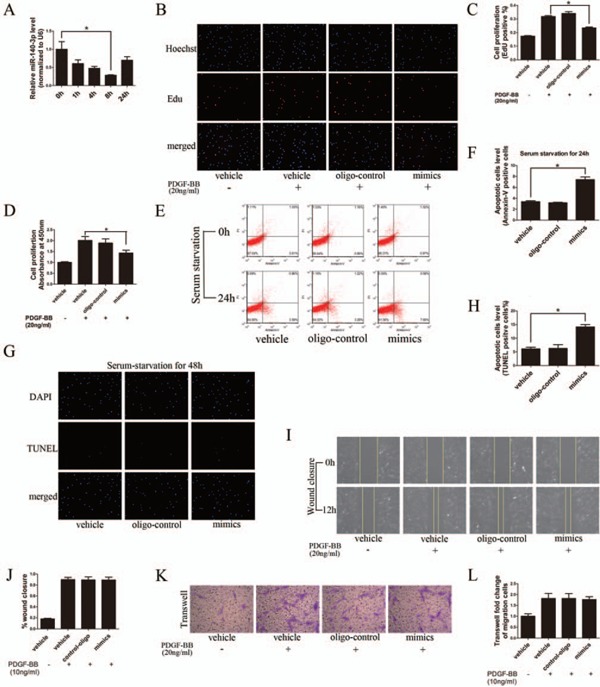
MiR-140-3p inhibits proliferation and increases apoptosis of ASMCs in vitro
A, PDGF-BB (20 ng/mL) induces a time-dependant decrease in miR-140-3p expression in cultured ASMCs, determined by qRT-PCR. B to D, MiR-140-3p (50 nM) mimic transfection into ASMCs could partially inhibit cell proliferation induced by PDGF-BB (20 ng/mL), determined by Edu and CCK-8 assays. E to H, MiR-140-3p (50 nM) mimic transfection could increase apoptosis in cultured ASMCs, demonstrated by Annexin-V/PI dual staining flow cytometry and TUNEL assay. I to L, MiR-140-3p (50 nM) mimic transfection did not affect PDGF-BB (10 ng/mL) induced ASMC migration, determined by wound closure and Transwell assay. (n = 3 to 6), *P < 0.05.
It is well known that the unbalance between ASMC proliferation and apoptosis is another key cellular event of artery restenosis. To determine the role of miR-140-3p in ASMC apoptosis, we applied Annexin-V/PI Double-Staining assay and TUNEL assay. As shown in Figs. 3E and F, miR-140-3p mimic delivery increased early-stage cell apoptosis (Annexin-V positive but PI negative) by over two folds after 24 h of serum-deprived culture. Furthermore, miR-140-3p induced late-stage cell apoptosis after 48 h of serum starvation, determined by TUNEL assay (Figs. 3G and H).
However, we found that miR-140-3p delivery had no effect on ASMC migration, detected by Wound Closure assay and Transwell assay (Figs. 3I, J, K, and L). To sum up, these data suggested that increasing miR-140-3p level inhibited cell proliferation and promoted cell apoptosis in cultured ASMCs.
C-Myb and BCL-2 Are Direct Target Genes of MiR-140-3p
The mechanism by which microRNAs regulate ASMC function depends on their targets. The participation of C-Myb and BCL-2 into ASMC proliferation has been well documented by previous studies17–20). According to the mRNA 3′-UTR complementary sites to miR-140-3p, computational target analysis from microRNA database revealed that C-Myb and BCL-2 harbor conserved binding sites for miR-140-3p21, 22). As shown in Figs. 4A and 4B, immunohistochemistry (IHC) analysis confirmed that C-Myb and BCL-2 staining was significantly stronger in ISR artery sections, compared with those in normal artery sections. Meanwhile, C-Myb and BCL-2were localized in artery media and neointima, which consists of the distribution of miR-140-3p. In addition, both C-Myb and BCL-2 protein expressions were found significantly up-regulated in ISR ASMCs and PDGF-BB-stimulated ASMCs, compared with normal ASMCs (Figs. 4C and D). Next, miR-140-3p modulated C-Myb and BCL-2 expression was studied. As shown in Figs. 4E and 4G, miR-140-3p mimic significantly decreased C-Myb and BCL-2 expressions at both protein level and mRNA level. In contrast, miR-140-3p inhibitor obviously increased C-Myb and BCL-2 expressions at both protein (Fig. 4F) and mRNA levels (Fig. 4H).
Fig. 4.
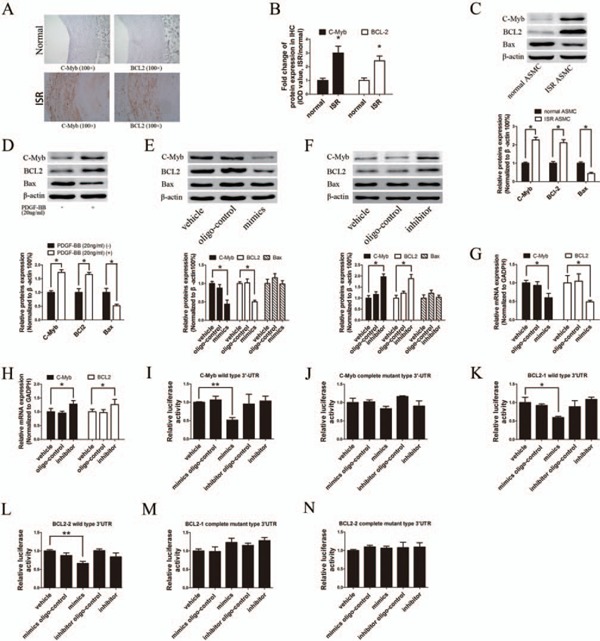
C-Myb and BCL-2 are the direct target genes of miR-140-3p
A and B, Staining of C-Myb (left column, brown) and BCL-2 (right column, brown) were stronger in ISR artery sections (bottom row) than normal artery sections (upper row; scale bars = 100 µm), determined by IHC assay. The relative IOD values of C-Myb and BCL-2 staining were calculated. C, ISR ASMCs expressed higher levels of C-Myb and BCL-2 protein but lower level of Bax protein than normal ASMCs, determined by Western blot. D, PDGF-BB (20 ng/mL) treatment of cultured ASMCs increased C-Myb and BCL-2 protein expression, but decreased Bax protein expression, determined by Western blot. E and F, C-Myb and BCL-2 protein levels were down-regulated by miR-140-3p mimic (50 nM) but up-regulated by miR-140-3p inhibitor (100 nM) transfection, determined by Western blot. G and H, C-Myb and BCL-2 mRNA levels were down-regulated by miR-140-3p mimic (50 nM) but up-regulated by miR-140-3p inhibitor (100 nM) transfection, determined by qRTPCR. I to N, MiR-140-3p mimic (50 nM) or inhibitor (100 nM) was cotransfected with psi-CHECK-2 vectors constructed with C-Myb or BCL-2 3′-UTR wild-type sequences or 3′-UTR mutant sequences, and the luciferase activity was reported. (n = 6), *P < 0.05, **P < 0.01.
To further confirm whether miR-140-3p is able to directly bind to the 3′-UTR seed sequence of C-Myb and BCL-2 mRNA 3′-UTR, we applied dual-luciferase reporter assay. The 3′-UTR fragments of C-Myb or BCL-2 mRNA harboring the predicted binding sites of miR-140-3p were cloned into psiCHECK-2 dualluciferase vector. Since the 3′-UTR of BCL-2 is too long (5279bp) to be constructed into one psi-CHECK2TM vectors, we separated it into two parts (BCL-2-1: 1bp–2600bp; BCL-2-2: 2601bp–5279bp) and cloned it into two individual vectors, with each vector containing three binding sites. Then, the construct was co-transfected with either miR-140-3p mimic or scramble oligo control into HEK 293 cells. In the presence of wild type 3′-UTR sequence, miR-140-3p significantly repressed relative luciferase activity (Figs. 4I, K, and L), and this effect was abrogated by mutation of the miR-140-3p binding sites in the C-Myb or BCL-2 3′-UTR area (Figs. 4J, M, and N). Taken together, these data indicated that miR-140-3p suppresses C-Myb and BCL-2 expressions at post-transcriptional level by directly binding to their mRNA.
C-Myb and BCL-2 Are Involved in MiR-140-3p Mediated Cellular Effects
Provided that C-Myb and BCL-2 were the functional target genes of miR-140-3p, exogenous expression of C-Myb and BCL-2 would compensate the miR-140-3p mediated cellular effects. Thus, vectors containing C-Myb or BCL-2 coding sequences were packaged into lentivirus particles and then were co-transfected with miR-140-3p mimic into ASMCs. A GFP vector was used as negative control. As shown in Figs. 5A and 5B, C-Myb and BCL-2 were successfully overexpressed in ASMCs. Intriguingly, as shown in Figs. 5C to 5E, miR-140-3p mimic induced anti-proliferation effect on ASMCs was significantly attenuated by over expressing C-Myb, but not BCL-2. Moreover, either C-Myb or BCL-2 over expression significantly abrogated the miR-140-3p induced ASMC apoptosis (Figs. 5F and G). Taken together, these data indicated that C-Myb and BCL-2 are the functional target genes which mediated the miR-140-3p cellular effects.
Fig. 5.
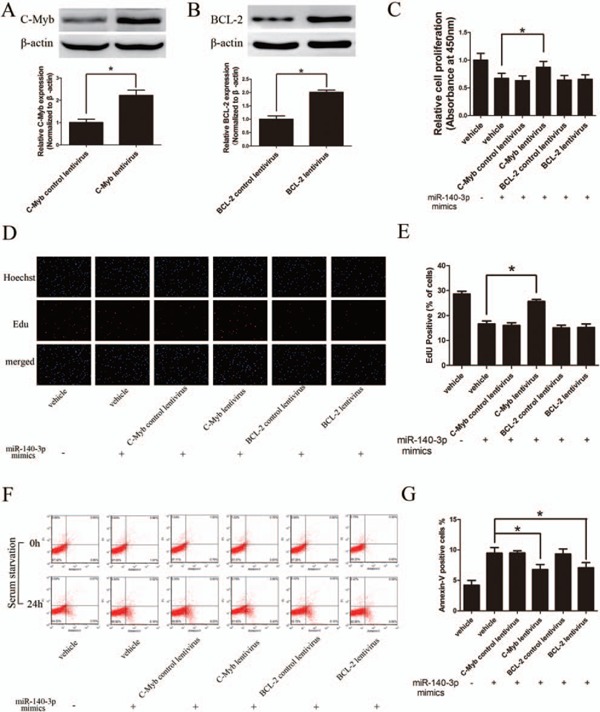
C-Myb and BCL-2 are involved in miR-140-3p mediated cellular effects
A and B, C-Myb and BCL-2 were overexpressed in cultured ASMCs by respective lentivirus vectors, determined by Western blot. C to E, MiR-140-3p mediated anti-proliferation effect on ASMCs was significantly reversed by overexpression of C-Myb but not BCL-2, determined by CCK-8 and Edu assays. F and G, Overexpressing either C-Myb or BCL-2 could partially attenuate the miR-140-3p induced cell apoptosis in ASMCs mediated, determined by Annexin-V/PI dual staining flow cytometry. (n = 3) *P < 0.05
MiR-140-3p Delivery Decreases Artery Restenosis via Targeting C-Myb and BCL-2 in Rat Artery Restenosis Model
As the neointima hyperplasia is the beginning step of artery restenosis, rat carotid artery balloon angioplasty model was used in simulating the artery restenosis after endovascular treatment. As shown in Figs. 6A and 6B, in situ hybridization assay revealed that miR-140-3p expression was significantly down-regulated in restenosis artery sections (LV-GFP group). To further explore the therapeutic role of miR-140-3p in ISR, miR-140-3p coding sequence was constructed into lentivirus vector and then delivered into rat carotid artery after balloon angioplasty. Fourteen days after angioplasty, the carotid arteries were isolated. The in situ hybridization assay demonstrated successful miR-140-3p over expression in rat carotid artery (Figs. 6A, B, and D), compared with the LV-GFP control vector group. As shown in Fig. 6C, miR-140-3p delivery led to an approximately 50% reduction in neointimal hyperplasia after balloon angioplasty, compared with the control group. Intriguingly, in situ hybridization and immunofluorescence dual-staining assay demonstrated that the miR-140-3p target, c-Myb and BCL-2 expression, was down-regulated in the area highly expressing miR-140-3p. On the contrary, their expression was up-regulated in the area lowly expressing miR-140-3p (Figs.6A, B, D, E, and F). Meaningfully, miR-140-3p delivery neither attenuated cell migration nor impaired cell viability in cultured endothelial cells, determined by wound closure assay and CCK-8 incorporation assay (Figs. 6G, H, and I). Furthermore, in vivo miR-140-3p delivery did not affect the endothelial cell marker, CD31 and CD34 expression in rat carotid arteries after angioplasty, demonstrating that the endothelialization was not impeded by exogenous miR-140-3p delivery (Fig. 6J). These data indicated that miR-140-3p could decrease artery restenosis after angioplasty via targeting C-Myb and BCL-2.
Fig. 6.
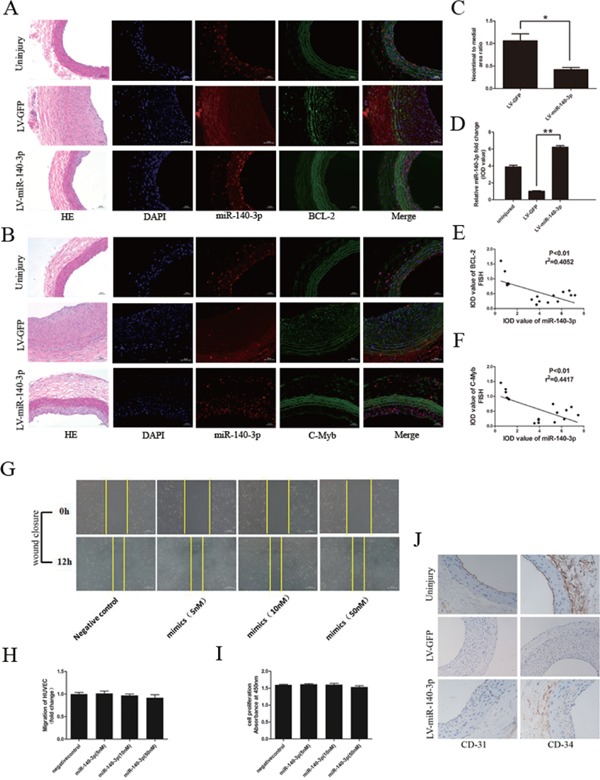
MiR-140-3p delivery decreases artery restenosis via targeting C-Myb and BCL-2 in rat carotid artery after balloon angioplasty
A and B, Representative graphs of miR-140-3p (red) and BCL-2 or C-Myb (green) co-staining in rat carotid artery (scale bars = 400 µm). Uninjury group (upper row) defined as rat carotid artery section without balloon injury. LV-GFP group (middle row) defined as rat carotid artery section with balloon injury transferred with LV-GFP. LV-miR-140-3p group (bottom row) defined as rat carotid artery section with balloon injury transferred with LV-miR-140-3p. HE staining revealed the artery structure. Blue indicated the cell nuclear staining by DAPI. Red immunofluorescence indicated miR-140-3p staining. Green immunofluorescence indicated BCL-2 or C-Myb staining. Merged image showed the co-localization of miR-140-3p and target genes. C, MiR-140-3p delivery into rat carotid artery suppressed neointimal hyperplasia after balloon angioplasty. D, Relative IOD value calculation demonstrated successful miR-140-3p delivery into rat carotid artery. E and F, Relative IOD value calculation demonstrated that BCL-2 and C-Myb staining is decreased in LV-miR-140-3p delivery group, compared with the control group. G to I, MiR-140-3p mimic transfection neither attenuated cell migration nor damaged cell viability in endothelial cells, determined by wound closure assay and CCK-8 incorporation assay. J, CD-31 and CD-34 staining demonstrated that the endothelialization was not attenuated by miR-140-3p (scale bars = 400 µm). (n = 6), *P < 0.05, **P < 0.01.
Discussion
Recent studies demonstrated that miR-21 is upregulated in rat and pig ISR models, and interfering miR-21 releases ISR via inhibiting ASMCs, but not inhibiting endothelial cells23, 24). Such cellular tropism suggests that microRNA could be the ideal target for the intervention of ISR16). Species and temporal specificity are the most important characteristics of micro-RNA expression and function. However, the direct evidence that links microRNA to human artery ISR of PAD is still missing. Our previous study demonstrated that microRNA expression characteristics in human PAD are different from animal models13). Also, miR-21 was found mainly up-regulated in arteries with lumen loss less than 50%, but not the artery sections with severe lumen loss which needed stent angioplasty13). Considering that miR-140-3p was found as the most significantly down-regulated microRNA of PAD samples in our previous study, we focus on the role of miR-140-3p, rather than miR-21, in the process of ISR in human PAD13).
In the present study, the significant miR-140-3p down-regulation in ISR samples was confirmed by qRT-PCR and in situ hybridization assay. The mechanism of microRNA dysregulation is complicated. Micro-RNA genes are initially transcribed as large precursors, called primary microRNAs. This process is mainly regulated by RNA polymerase. Then primary microRNAs are further processed by the RNase-III enzyme Drosha and Dicer to generate mature miRNA products. On the other hand, DNA methylation-mediated epigenetic silencing of microRNAs has been demonstrated in a few reports in atherosclerotic lesions25). Our previous study has found that hypermethylation of the upstream DNA CpG sites accounts for downregulation of miR-1298 in ASO14). At present, the mechanism underlying the down-regulation of miR-140-3p in PAD arteries is still unknown. It should be further investigated if these molecular mechanisms account for down-regulation of miR-140-3p in PAD arteries. To investigate the role of miR-140-3p in ISR, we verified its cellular distribution in artery walls. After dissecting normal arteries into intima, media, and adventitia, miR-140-3p was found mainly expressed in the artery media. This result was further clearly confirmed by in situ hybridization and immunofluorescence costaining assay. Also, the miR-140-3p staining intensity was much weaker in the ASMC layer of ISR artery sections, compared with normal artery sections. Thus, the process of ISR was reasoned with miR-140-3p down-regulation.
It is well established that excessive ASMC proliferation is the main reason for ISR. Interestingly in the present study, compared with ASMCs from normal artery, ASMCs isolated from ISR arteries were more prone to proliferate and expressed lower levels of miR-140-3p as well as lower levels of SMC contractile marker SM-MHC and SM α-actin. In vitro study found that miR-140-3p was critically involved in PDGF-BB-induced ASMC proliferation. In addition, miR-140-3p delivery increased ASMC apoptosis. Meaningfully, exogenous expression of miR-140-3p in rat carotid artery significantly decreased artery restenosis induced by balloon angioplasty injury. More intriguingly in the present study, miR-140-3p neither inhibited endothelial cell proliferation and migration in vitro nor impeded re-endothelialization in vivo. This is most likely because of the low concentration of miR-140-3p in endothelial cells compared with ASMCs. These results indicate that miR-140-3p could be a better therapeutic target for preventing ISR superior to current drug-coated balloon or drug eluting stent.
The functions of microRNA depend on its targets. BCL-2 and C-Myb were predicted as potential miR-140-3p targets according to their seed sequences complementary to miR-140-3p in mRNA 3′-UTR, as well as their opposite expression level with miR-140-3p expression level and their participation in cell proliferation. Whether microRNA induces its target mRNA degradation depends on the number of binding sites and the degree of complementation. The target mRNA is more easily degraded if it harbors extensive complementary sites26, 27). Bioinformatics analysis found that C-Myb harbors four binding sites and BCL-2 harbors six binding sites complementary to miR-140-3p in their mRNA 3′-UTR. Using dual-luciferase assay, we validated that miR-140-3p could directly bind to each seed sequence in C-Myb and BCL-2 mRNA 3′-UTR.
C-Myb is a DNA-binding transcription factor and is involved in the proliferation of ASMCs in vascular pathology17, 18). BCL-2 is localized to the outer membrane of mitochondria, where it plays an important role in promoting cellular survival and inhibiting the actions of pro-apoptotic proteins28). The participation of C-Myb and BCL-2 into ASMC proliferation, apoptosis, and artery pathology has been well documented by previous studies17, 19, 20, 29). In the current study, C-Myb was found directly involved in miR-140-3p mediated anti-proliferation and pro-apoptosis effect in ASMCs, and BCL-2 was only involved in apoptosis. We speculate that there are three possible reasons. First, it has been demonstrated that C-Myb is a positive upstream regulator of BCL-2 and many other proliferative factors29). Thus, miR-140-3p regulates BCL-2 expression not only via binding its mRNA 3′-UTR directly but also via the C-Myb pathway indirectly. Second, Bcl-2 is only one of the multiple target genes of miR-140-3p. Third, the BAX gene is the first identified pro-apoptotic member of the BCL-2protein family. The interaction of BCL-2 and Bax determines cellular death or survival, and the BCL-2/Bax ratio is a vital index for ASMC apoptosis19, 20, 28). Thus, we detected Bax expression even if it was not predicted as a potential target of miR-140-3p. Interestingly, in contrast to BCL-2, Bax expression was found significantly down-regulated in ISR ASMCs and PDGF-BB-treated ASMCs (Figs. 4C and 4D). However, neither miR-140-3p mimic nor inhibitor changed Bax expression (Figs. 4E and 4F). Thus, as excusable, BCL-2 recovery could only partially reverse the miR-140-3p mediated cellular effects. Our results showed that endothelial cells are not affected by the overexpression of miR-140-3p. Cell-type specificity is an important characteristic of biological functions of miRNA. For example, miR-21, miR-221/222, and miR-145 are mainly expressed in ASMCs of arterial walls and have an effect on neointimal growth13, 26, 30). Our results showed that miR-140-3p expression was significantly higher in the ASMCs of the media layer than in the endothelium of the arterial wall, suggesting that miR-140-3p mainly exerts its biological roles in the ASMCs but not in the endothelial cells. As a result, it might be that miR-140-3p has little or no effect on the expressions of C-Myb and BCL-2 in artery endothelial cells.
There are still some limitations of this study. Given that miR-140-3p has many target genes, there should have been some potential sides of miR-140-3p overexpression therapy for PAD. Owing to the cell-type specificity, the potential side effects of miR-140-3p overexpression in PAD should be comprehensively evaluated in an animal model. In addition, although we investigated the effect of miR-140-3p overexpression in ASMC and endothelial cells, we did not know the potential effect of miR-140-3p on other cell types. All of these limitations should be addressed in the following study.
Conclusion
In summary, miR-140-3p is significantly downregulated in ISR samples of PAD patients. Increasing miR-140-3p obviously attenuated ASMC proliferation and increased ASMC apoptosis, via directly targeting C-Myb and BCL-2. More meaningfully, miR-140-3p delivery in animal models dramatically decreased balloon injury that resulted to restenosis, but did not impede re-endothelialization. These novel findings may provide new potential therapeutic targets for treating or preventing ISR in PAD.
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (grant numbers: 81670441 and 81200231), Pearl River S&T Nova Program of Guangzhou (grant number: 201610 010050), and Young Teachers Cultivation Program of Sun Yat-sen University (grant number: 13ykpy21).
Conflicts of Interest
The authors have no financial conflict of interest.
References
- 1). Shammas NW. Epidemiology, classification, and modifiable risk factors of peripheral arterial disease. Vasc Health Risk Manag, 2007; 3: 229-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2). Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg, 2007; 45 Suppl S: S5-S67 [DOI] [PubMed] [Google Scholar]
- 3). Nakazawa G. Stent thrombosis of drug eluting stent: pathological perspective. J Cardiol, 2011; 58: 84-91 [DOI] [PubMed] [Google Scholar]
- 4). Otsuka F, Finn AV, Yazdani SK, Nakano M, Kolodgie FD, Virmani R. The importance of the endothelium in atherothrombosis and coronary stenting. Nat Rev Cardiol, 2012; 9: 439-453 [DOI] [PubMed] [Google Scholar]
- 5). Choe N, Kwon DH, Shin S, Kim YS, Kim YK, Kim J, Ahn Y, Eom GH, Kook H. The microRNA miR-124 inhibits vascular smooth muscle cell proliferation by targeting S100 calcium-binding protein A4 (S100A4). FEBS Lett, 2017; 591: 1041-1052 [DOI] [PubMed] [Google Scholar]
- 6). Jin C, Zhao Y, Yu L, Xu S, Fu G. MicroRNA-21 mediates the rapamycin-induced suppression of endothelial proliferation and migration. FEBS Lett, 2013; 587: 378-385 [DOI] [PubMed] [Google Scholar]
- 7). Jamaluddin MS, Weakley SM, Zhang L, Kougias P, Lin PH, Yao Q, Chen C. miRNAs: roles and clinical applications in vascular disease. Expert Rev Mol Diagn, 2011; 11: 79-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Quintavalle M, Condorelli G, Elia L. Arterial remodeling and atherosclerosis: miRNAs involvement. Vascul Pharmacol, 2011; 55: 106-110 [DOI] [PubMed] [Google Scholar]
- 9). Ambros V. The functions of animal microRNAs. Nature, 2004; 431: 350-355 [DOI] [PubMed] [Google Scholar]
- 10). Dong J, Liang YZ, Zhang J, Wu LJ, Wang S, Hua Q, Yan YX. Potential Role of Lipometabolism-Related Micro-RNAs in Peripheral Blood Mononuclear Cells as Biomarkers for Coronary Artery Disease. J Atheroscler Thromb, 2017; 24: 430-441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11). He XM, Zheng YQ, Liu SZ, Liu Y, He YZ, Zhou XY. Altered Plasma MicroRNAs as Novel Biomarkers for Arteriosclerosis Obliterans. J Atheroscler Thromb, 2016; 23: 196-206 [DOI] [PubMed] [Google Scholar]
- 12). Ji R, Cheng Y, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of Micro-RNA in vascular neointimal lesion formation. Circ Res, 2007; 100: 1579-1588 [DOI] [PubMed] [Google Scholar]
- 13). Wang M, Li W, Chang GQ, Ye CS, Ou JS, Li XX, Liu Y, Cheang TY, Huang XL, Wang SM. MicroRNA-21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler Thromb Vasc Biol, 2011; 31: 2044-2053 [DOI] [PubMed] [Google Scholar]
- 14). Hu W, Wang M, Yin H, Yao C, He Q, Yin L, Zhang C, Li W, Chang G, Wang S. MicroRNA-1298 is regulated by DNA methylation and affects vascular smooth muscle cell function by targeting connexin 43. Cardiovasc Res, 2015; 107: 534-545 [DOI] [PubMed] [Google Scholar]
- 15). Huang SC, Wang M, Wu WB, Wang R, Cui J, Li W, Li ZL, Li W, Wang SM. Mir-22-3p Inhibits Arterial Smooth Muscle Cell Proliferation and Migration and Neointimal Hyperplasia by Targeting HMGB1 in Arteriosclerosis Obliterans. Cell Physiol Biochem, 2017; 42: 2492-2506 [DOI] [PubMed] [Google Scholar]
- 16). Schaer GL, Zhang C. Implementation of miRNAs to Reduce In-Stent Restenosis in the Future. J Am Coll Cardiol, 2015; 65: 2328-2330 [DOI] [PubMed] [Google Scholar]
- 17). Simons M, Edelman ER, DeKeyser JL, Langer R, Rosenberg RD. Antisense c-myb oligonucleotides inhibit intimal arterial smooth muscle cell accumulation in vivo. Nature, 1992; 359: 67-70 [DOI] [PubMed] [Google Scholar]
- 18). Lambert DL, Malik N, Shepherd L, Gunn J, Francis SE, King A, Crossman DC, Cumberland DC, Holt CM. Localization of c-Myb and induction of apoptosis by antisense oligonucleotide c-Myb after angioplasty of porcine coronary arteries. Arterioscler Thromb Vasc Biol, 2001; 21: 1727-1732 [DOI] [PubMed] [Google Scholar]
- 19). Kutuk O, Basaga H. Bcl-2 protein family: implications in vascular apoptosis and atherosclerosis. Apoptosis, 2006; 11: 1661-1675 [DOI] [PubMed] [Google Scholar]
- 20). Saxena A, McMeekin JD, Thomson DJ. Expression of Bcl-x, Bcl-2, Bax, and Bak in endarterectomy and atherectomy specimens. J Pathol, 2002; 196: 335-342 [DOI] [PubMed] [Google Scholar]
- 21). Saito T, Saetrom P. MicroRNAs--targeting and target prediction. N Biotechnol, 2010; 27: 243-249 [DOI] [PubMed] [Google Scholar]
- 22). Witkos TM, Koscianska E, Krzyzosiak WJ. Practical Aspects of microRNA Target Prediction. Curr Mol Med, 2011; 11: 93-109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). McDonald RA, Halliday CA, Miller AM, Diver LA, Dakin RS, Montgomery J, McBride MW, Kennedy S, McClure JD, Robertson KE, Douglas G, Channon KM, Oldroyd KG, Baker AH. Reducing In-Stent Restenosis: Therapeutic Manipulation of miRNA in Vascular Remodeling and Inflammation. J Am Coll Cardiol, 2015; 65: 2314-2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24). Wang D, Deuse T, Stubbendorff M, Chernogubova E, Erben RG, Eken SM, Jin H, Li Y, Busch A, Heeger CH, Behnisch B, Reichenspurner H, Robbins RC, Spin JM, Tsao PS, Schrepfer S, Maegdefessel L. Local Micro-RNA Modulation Using a Novel Anti-miR-21-Eluting Stent Effectively Prevents Experimental In-Stent Restenosis. Arterioscler Thromb Vasc Biol, 2015; 35: 1945-1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25). Borghini A, Cervelli T, Galli A, Andreassi MG. DNA modifications in atherosclerosis: from the past to the future. Atherosclerosis, 2013; 230: 202-209 [DOI] [PubMed] [Google Scholar]
- 26). Liu X, Cheng Y, Zhang S, Lin Y, Yang J, Zhang C. A necessary role of miR-221 and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ Res, 2009; 104; 476-487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27). Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004; 304: 594-596 [DOI] [PubMed] [Google Scholar]
- 28). Geng YJ. Molecular signal transduction in vascular cell apoptosis. Cell Res, 2001; 11: 253-264 [DOI] [PubMed] [Google Scholar]
- 29). Farrell KA, Withers SB, Holt CM. C-Myb function in the vessel wall. Front Biosci (Elite Ed), 2011; 3: 968-977 [DOI] [PubMed] [Google Scholar]
- 30). Cheng Y, Liu X, Yang J, Lin Y, Xu DZ, Lu Q, Deitch EA, Huo Y, Delphin ES, Zhang C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res 2009; 105: 158-166 [DOI] [PMC free article] [PubMed] [Google Scholar]