

ABSTRACT
Breast cancer is a common type of cancer among female cancer patients and the main cause of cancer-related deaths. During the last decades, targeted therapies for breast cancer have been rapidly developing. Among them, MLN4924, a first-in-class NEDD8-activating enzyme (NAE) inhibitor, has performed antitumor activity by inactivating the cullin-RING ligases and causing the accumulation of their substrates to induce apoptosis in a number of studies. In this study, we found that MLN4924 activates the AKT pathway in both HER2-positive and triple-negative breast cancer (TNBC) cell lines. Given that AKT signaling is responsible for tumor progression and drug resistance in some types of cancers, we hypothesized that the AKT inhibitor may synergistically enhance the tumor suppression capability in breast cancer by MLN4924. To demonstrate the sensitizing effect, MK-2206 was chosen as the adjuvant treatment, and cell growth, migration and apoptosis were detected. The results showed that MLN4924 treatment inhibited cell growth and migration and induced apoptosis in both SK-BR3 and MDA-MB231 breast cancer cell lines. More importantly, the combined treatment of MLN4924 and MK-2206 indeed caused stronger cytotoxicity and inhibition of migration and a much higher induction of apoptosis compared with MLN4924 treatment alone. Our study provides the proof-of-concept evidence for strategic drug combination of MLN4924 with an AKT inhibitor for maximal killing of breast cancer cells via the enhancement of apoptosis.
Keywords: Breast cancer, targeted therapy, neddylation, MLN4924, AKT inhibitor, MK-2206
Introduction
Breast cancer is one of the most common cancers and the leading cause of cancer death among female cancer patients [1]. Chemotherapy plays a crucial role in primary treatment for patients with locally advanced or metastatic breast cancer. Although it achieves significant efficacy as a first-line treatment, a vast majority of patients inevitably develop drug resistance in subsequent lines of treatment [2]. Thus, development of novel targeted therapeutic agents that sensitize breast cancer to chemotherapy will be extremely important for overcoming drug resistance to improve therapeutic outcomes [3,4].
The ubiquitin-proteasome system (UPS) controls protein homeostasis by spatiotemporal degradation of numerous proteins with key regulatory roles in a vast array of biological processes, such as cell cycle progression, signal transduction, and cell death. Failure to control protein degradation frequently happens in cancer and other human diseases and contributes to disease progression, cancer metastasis, and drug resistance. Thus, targeting abnormal protein degradation in cancer is considered as an attractive approach for effective cancer treatments and is validated by the FDA approval of Bortezomib, the first class of general proteasome inhibitors. The UPS targets proteins for degradation via a three-step enzymatic process catalyzed by a cascade of enzymes, including ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3). Among the three enzymes, E3 ligase is responsible for the transfer of ubiquitin from E2 to the recognized and recruited target protein, of which cullin-RING ligases (CRLs) are the largest family [5]. As an activated E3 ligase, the cullin protein must be covalently modified by a single molecule of NEDD8, a process known as neddylation, in which NEDD8 is activated by the NEDD8-activating enzyme (NAE) and conjugated to the cullin protein via a three-step enzymatic process like ubiquitination [6,7].
MLN4924, a first-in-class NAE inhibitor, binds to NAE and creates a covalent NEDD8-MLN4924 adduct to inhibit NAE enzymatic activity [8]. Given that cullin neddylation, a process catalyzed by NAE, is required for CRL E3 ligase activity, by blocking cullin neddylation, MLN4924 inactivates CRL E3, causes accumulation of CRL E3 substrates and suppresses tumor cell growth both in vitro and in vivo by inducing apoptosis, senescence and autophagy [5,6,9]. Few studies have demonstrated the antitumor activity of MLN4924 in HER2- and estrogen receptor (ER)-positive breast cancer cell lines [10,11]. With promising preclinical efficacy, MLN4924 has undergone a series of Phase I/II clinical trials against a number of human malignancies [6,9]. However, cancer cells developed MLN4924 resistance either through the development of heterozygous mutations in the NAEβ subunit of NAE or another unknown mechanism [12].
Here, we report that MLN4924 activates the AKT pathway in a feedback manner to promote survival signaling in both HER2-positive and triple-negative breast cancer (TNBC) cell lines. We hypothesized that the AKT inhibitor could enhance the tumor suppressive activity of MLN4924 in breast cancer. MK-2206 is the first allosteric potent inhibitor of all three isoforms of AKT at nanomolar concentrations [13]. It can be given orally and has demonstrated antitumor activity in various cancer cell lines, such as breast cancer, leukemia and multiple myeloma [14–16]. MK-2206 also has been advanced to a series of Phase I/II clinical trials against a number of human malignancies, including HER2 positive or negative breast cancer [17,18]. Therefore, MK-2206 was chosen as the adjuvant therapy of MLN4924 to test our hypothesis. In the present study, HER2-positive and TNBC cell lines, SK-BR3 and MDA-MB231, were treated with MLN4924 alone or in combination with MK-2206. The synergistic effects were investigated by detecting cell growth, migration and apoptosis. Indeed, the combination therapy enhanced the tumor suppressive ability of MLN4924 by inhibiting the AKT pathway, thus providing a promising targeted therapeutic strategy for breast cancer.
Materials and methods
Cell lines and chemicals
Breast cancer cell lines SK-BR3 and MDA-MB231 were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. In addition, the cells were cultured in a cell incubator at 37°C with 5% CO2. MLN4924 was purchased from Apexbio (B1036) and MK-2206 from Selleck (S1078). For drug treatment, subconfluent cells were incubated with MLN4924 and MK-2206 at various concentrations for various periods of time as indicated.
Western blotting
Cells were cultured in 60-mm dishes and treated with the indicated concentration of MLN4924 and MK-2206. Cells were then harvested with a cell scraper and lysed in NP-40 lysis buffer with proteasome and phosphatase inhibitors on ice for 30 min, followed by centrifugation at 13,600 rpm for 25 min at 4°C. After protein concentration was measured by the BCA Protein Assay Kit (Thermo, 23,225) according to the manufacturer’s instructions, the samples were subjected to SDS-PAGE and transferred to nitrocellulose (NC) membranes (GE, 10,600,001). After blocking with 5% nonfat powdered milk (Sangon, A600669), the membranes were incubated with primary antibodies at 4°C overnight and corresponding secondary antibodies at room temperature for 2 h the following day. At last, bands were visualized using SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo, 34,580) with Medical X-ray Processor (Carestream). Primary antibodies were used as follows: against p-AKT (S473) (4060), t-AKT (4691), p-S6K1(T389) (9234), p-ERK (T202/Y204) (9101), t-ERK (9107), PARP (9542), Caspase 3 (9665), MMP2 (87,809), MMP7 (71,031), MMP9 (13,667), vimentin (5741) (Cell Signaling Technology), NEDD8 (ab81264) (Abcam), t-S6K1 (sc-230) (Santa Cruz) and ACTIN (A5441) (Sigma).
Cell viability and clonogenic survival assays
SK-BR3 and MDA-MB231 cells were plated in triplicate in 96-well plates at 3000 cells per well. Cells were treated with MLN4924 alone at various concentrations for 72 h or in combination with MK-2206 for 48 h. Cell viability was evaluated by the ATP-lite 1step Luminescence Assay System according to the manufacturer’s instructions (PerkinElmer). A total of 300 cells were plated in 0.1% gelatin (Sigma, V900863)-coated 60-mm dishes and treated with the indicated drugs the following day. After 7 days, the cells were stained with the Coomassie brilliant blue solution for 20 min and then flushed with running water. After air drying, the cell colonies were photographed for colony counting.
Migration assays
Cell migration ability was measured by a wound healing assay and a transwell migration assay. After drug treatment, the cells were incubated in serum-free medium for 12–18 h. For the wound healing assay, the cells were photographed at 0 h, 24 h and 48 h after a pipette tip was used to scratch across the plate. For the transwell assay, the cells were harvested and seeded in the upper chamber at 1 × 105 per well with serum-free medium. The bottom well contained medium with 10% FBS. After 24 h, the cells were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet (Aladdin, C110703) for 20 min. Cells in 5 random fields of view were photographed and counted.
Flow cytometry
Cells were cultured in 6-well plates and treated with MLN4924 alone or in combination with MK-2206. After 48 h, the cells were harvested with 0.05% trypsin (Gibco) and centrifuged at 1000 × g for 5 min. Cells were then washed with PBS and stained using the Annexin V-FITC apoptosis detection kit (Beyotime, C1063), followed by flow cytometry. Cells with Annexin V-positive staining were considered as apoptotic cells.
DNA fragmentation assay
Cell were cultured in 100-mm dishes and treated with drugs for 48 h. Then, cells were lysed in 600 μL lysis buffer (5 mM Tris-HCl, pH 8.0, 20 mM EDTA and 0.5% Triton X-100) for 1 h on ice. Genomic DNA was purified by phenol-chloroform extraction. DNA samples were then analyzed by agarose gel electrophoresis at 80 V on a 1.8% agarose (Invitrogen, 75,510–019) gel containing GelRed (Biotium, 41,003). After 1.5 h, the gel was photographed by Gel Doc XR+ System (Bio-Rad).
Statistical analysis
All data are expressed as the mean ± SEM from three independent biological replicates. A student t-test was used with SPSS 20.0 (IBM Armonk). Values of p < 0.05 were considered to be statistically significant.
Results
AKT signaling pathway is activated by MLN4924
Increasing evidence shows that activation of the PI3K/AKT pathway is associated with decreased sensitivity to chemotherapeutic agents and contributes to chemotherapeutic resistance [19]. To determine if the PI3K/AKT pathway is involved in MLN4924 resistance, we first examined AKT activation after treating human breast cancer cells SK-BR3 and MDA-MB231 with MLN4924 at various concentrations. Indeed, the AKT signaling pathway is activated by MLN4924, as reflected by increased AKT phosphorylation on S473 after MLN4924 treatment in a dose-dependent manner in both SK-BR3 and MDA-MB231 cells (Figure 1(a)). We next determined the precise time course of AKT activation upon MLN4924 exposure for up to 48 h in SK-BR3 and MDA-MB231 cells. MLN4924 remarkably activated AKT (increased phosphorylation of AKT), starting as early as 12 h in SK-BR3 cells and reaching the peak at 48 h (Figure 1(b)). Given that the abnormal activation of PI3K/AKT signaling pathway occurs frequently in human breast cancer [20] and is associated with chemotherapeutic resistance [21], we hypothesized that AKT inhibitors may sensitize breast cancer to MLN4924 treatment.
Figure 1.

AKT signaling pathway is activated by MLN4924 treatment.(a) SK-BR3 and MDA-MB231 cells were incubated with various concentrations of MLN4924 (0, 10, 25 and 50 nM) for 24 h, followed by western blotting using antibodies against p-AKT (S473), t-AKT and NEDD8. ACTIN was used as a loading control. The levels of p-AKT (S473) were quantified and expressed as fold change, compared with the control. (b) SK-BR3 and MDA-MB231 cells were treated with MLN4924 (1 μM) for different time periods (0, 12, 24, 36, and 48 h).
AKT inhibitor MK-2206 enhances the suppression of growth in breast cancer cells by MLN4924
Given that MK-2206, the first allosteric inhibitor against all three isoforms of AKT, has been advanced to Phase I/II clinical trials against HER2 positive or negative breast cancer, we chose MK-2206 to verify our hypothesis: the sensitizing effect of AKT inhibitor to MLN4924. First, we treated SK-BR3 and MDA-MB231 cells with various concentrations of MK-2206. The activity of AKT was significantly decreased at 0.15 μM, as reflected by decreased AKT phosphorylation, whereas the activation of S6K1 and ERK1/2 remained unchanged, indicating the selective targeting of AKT by MK-2206 (Figure 2(a)). In addition, there was no significant cytotoxicity at up to 5 μM of MK-2206 (24 h and 48 h), or up to 1.25 μM (72 h) (Figure 2(b)). These data suggest that MK-2206 is an ideal AKT inhibitor with high potency against AKT and minimal cytotoxicity at approximately 0.1 ~ 1.25 μM. Thus, 0.1 ~ 1.25 μM of MK-2206 was chosen for the following drug combination experiments. Cells were exposed to MLN4924 only at various concentrations or in combination with MK-2206 (100 nM) for 48 h. The ATP-lite assay showed that cell viability decreased with the increase of MLN4924 concentration, suggesting its antitumor activity (Figure 2(c)). Compared with the MLN4924-only group, cell viability of the combination group (MLN4924+MK-2206) decreased more. The IC50 values of the combination group were significantly reduced from approximately 300 nM to 150 nM in SK-BR3 cells and from approximately 290 nM to 190 nM in MDA-MB231 cells. These results indicated that MK-2206 can sensitize breast cancer cells to the MLN4924 treatment.
Figure 2.
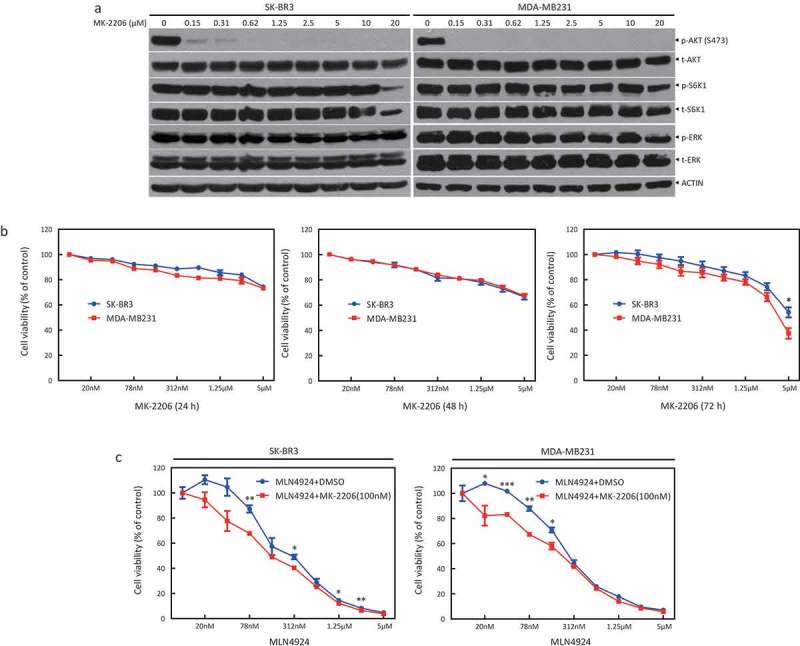
AKT inhibitor MK-2206 enhances the suppression of growth in breast cancer cells by MLN4924.(a) SK-BR3 and MDA-MB231 were treated with indicated concentrations of MK-2206. After 24 h, cells were obtained for western blotting using the indicated antibodies. (b) Cells were treated with the indicated concentrations of MK-2206 for 24 h, 48 h, and 72 h, followed by the ATP-lite assay. (c) Cells were treated with the indicated concentrations of MLN4924 for 72 h and in combination with MK-2206 (100 nM) for 48 h.
AKT inhibitor MK-2206 enhances the suppression of survival in breast cancer cells by MLN4924
Given that AKT acts as a critical regulator of cell survival, we next determined whether MK-2206 could enhance the suppression of survival in breast cancer cells by MLN4924. SK-BR3 cells were treated with MLN4924 (0, 100, and 200 nM) alone or in combination with MK-2206 (100 nM) for 7 days, followed by a clonogenic assay to evaluate cell survival. MLN4924 treatment significantly reduced the numbers of forming colonies, indicating the suppression of cell survival by MLN4924 (Figure 3(a)). Furthermore, compared with the MLN4924-only group, colony formation of the combination group (MLN4924+MK-2206) was remarkably reduced (column 4 vs. 3; column 6 vs. 5) (Figure 3(b)). Taken together, MK-2206 synergistically suppresses breast cancer cell survival with MLN4924.
Figure 3.
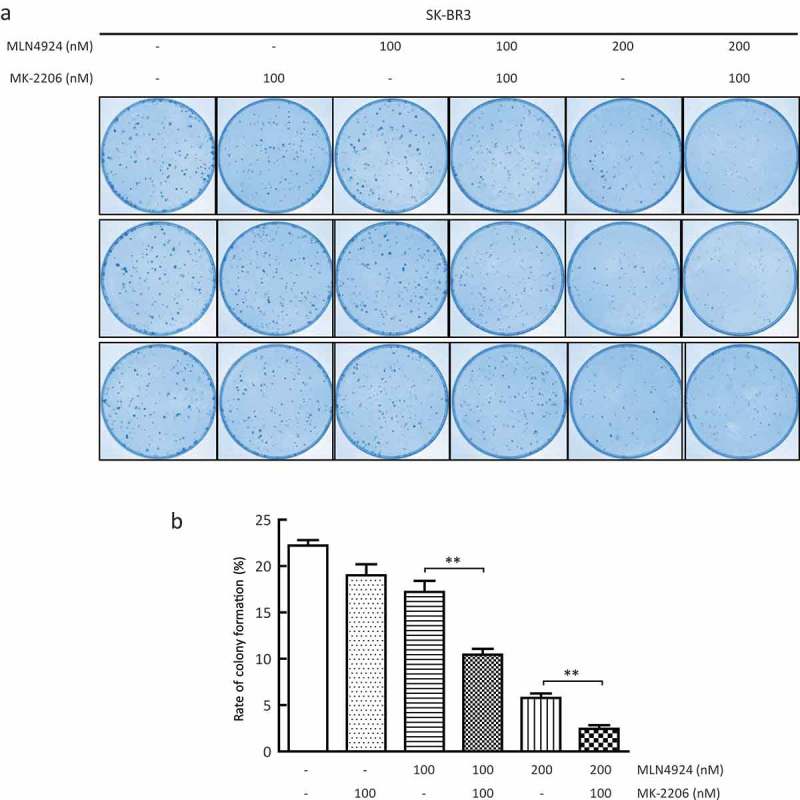
AKT inhibitor MK-2206 enhances the suppression of survival in breast cancer cells by MLN4924.A total of 300 cells were seeded in triplicate in 60-mm dishes and treated with different concentrations of MLN4924 (0, 100, 200 nM) alone or in combination with MK-2206 (100 nM). After 7 days, colonies were stained (a) and counted (> 50 cells in a colony) (b). Rate of colony formation (%) = colony number/300*100% (mean± SEM, n = 3). **p < 0.01.
AKT inhibitor MK-2206 enhances the suppression of migration in breast cancer cells by MLN4924
In addition, the activation of AKT is essential for many events during metastasis including the escape of cancer cells from the tumor environment, in which AKT upregulates matrix metalloproteases (MMPs) to mediate matrix degradation [22]. Next, we explored the synergistic effect of MK-2206 with MLN4924 on cancer cell metastasis. To evaluate the ability of cell migration, a wound healing assay and a transwell migration assay were performed. As shown in Figure 4(a), at 48 h following drug treatment, 1) MK-2206 alone had minimal, if any, effect on cell migration; 2) MLN4924 alone remarkably inhibited cell migration; and 3) compared to MLN4924 alone, the combination group significantly suppressed cell migration. Furthermore, the transwell migration assay also showed that remarkably fewer cells passed through the polycarbonate membrane, considered as the migrated cells, in the combination group than the MLN4924 group (Figure 4(b)). Meanwhile, the levels of MMPs (MMP2, 7, and 9), migration markers, and vimentin, a mesenchymal marker, were further decreased in the combination group, compared with those in the MLN4924-only group (Figure 4(c)). Taken together, MK-2206 could enhance the suppression of cell migration induced by MLN4924.
Figure 4.
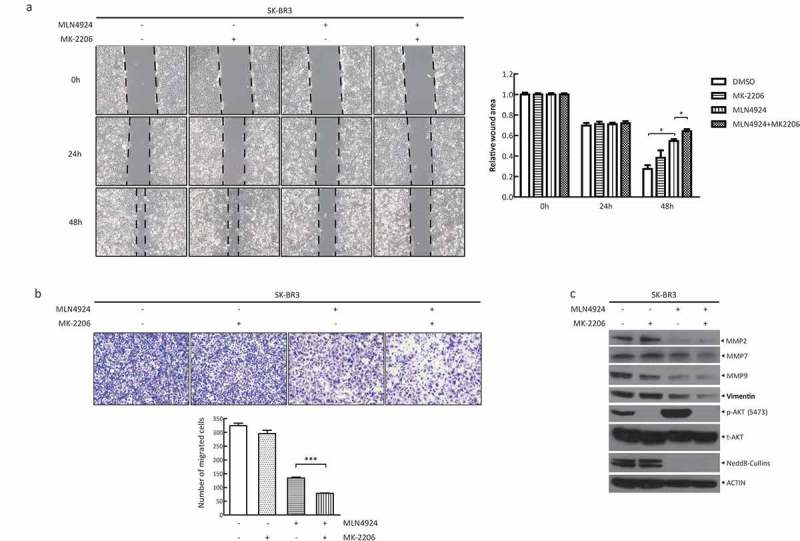
AKT inhibitor MK-2206 enhances the suppression of migration in breast cancer cells by MLN4924.(a) Cells were seeded in 6-well plates and treated with MLN4924 (1 μM) for 24 h, followed by MK-2206 treatment (1 μM) for 24 h. After serum starvation for 12–18 h, the cells were photographed at 0 h, 24 h and 48 h after a pipette tip was used to scratch across the well. Data were shown as the relative wound area normalized to the control (mean± SEM, n = 3). (b) Cells (1x105) were treated with the indicated agents and then seeded into the upper chamber containing serum-free medium. After 24 h, cells were fixed and stained, followed by photography and counting (mean± SEM). (c) Cells were treated with the drugs for 48 h, followed by western blotting using the indicated antibodies. *** p < 0.001.
AKT inhibitor MK-2206 enhances the induction of apoptosis induced by MLN4924
Given that apoptosis is a well-known mechanism for MLN4924-induced suppression of cell growth, finally, we determined whether MK-2206 could synergistically induce cell apoptosis with MLN4924. First, SK-BR3 and MDA-MB231 cells were treated with various concentrations of MLN4924 with or without MK-2206. FACS analysis showed the following: 1) MK-2206 alone had no effect on cell apoptosis, and 2) the combination group induced remarkably more apoptosis than MLN4924 treatment alone, as reflected by a much higher percentage of Annexin V+ population in the combination group (Figures 5(a,b), left panel, a representative FACS profile; and right panel, the percentage of Annexin V+ population, n = 3). Then, DNA fragmentation assay also showed that the induction of apoptotic DNA fragmentation, a key feature of apoptosis, in the combination group is more evident than that in the MLN4924-only group (Figure 5(c)). Finally, increased cleavage of PARP and Caspase 3 in the combination group was also enhanced, compared with the MLN4924-only group (lane 4 vs lane 3, lane 6 vs lane 5, lane 8 vs lane 7), in both SK-BR3 and MDA-MB231 cells (Figure 5(d)). Meanwhile, MK-2206 significantly blocked AKT activation induced by MLN4924 treatment, as evidenced by the change of phosphorylated AKT on S473 (Figure 5(d)). Taken together, these data demonstrate that MK-2206 synergistically enhances the induction of apoptosis induced by MLN4924.
Figure 5.
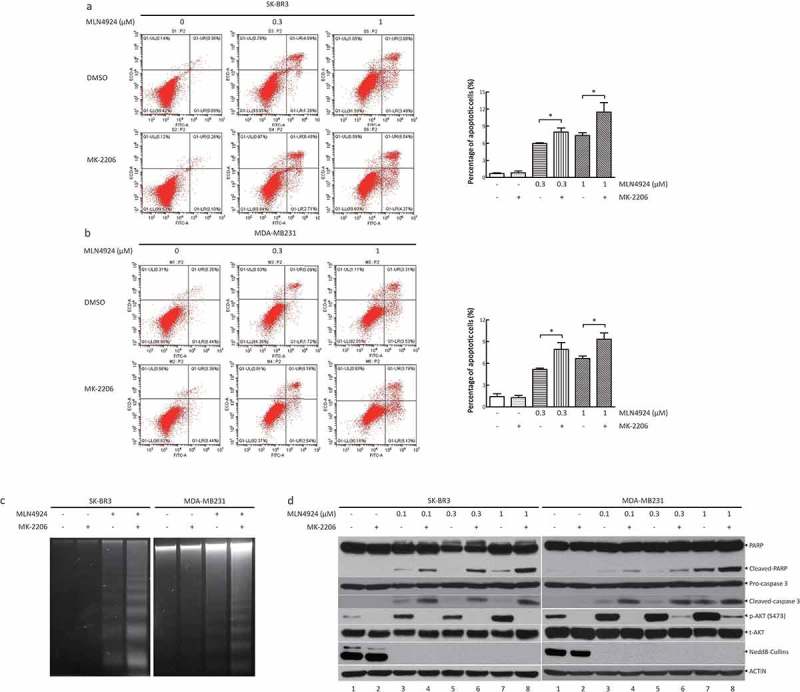
AKT inhibitor MK-2206 enhances the induction of apoptosis induced by MLN4924.(a, b) Cells were treated with MLN4924 (0, 0.3, and 1 μM) alone or in combination with MK-2206 (1 μM). After 48 h, the cells were harvested and stained using the Annexin V-FITC apoptosis detection kit. Cells with Annexin V+ staining located in the right upper and lower quadrants were considered as apoptotic cells (mean± SEM, n = 3). (c) Cells were treated with the drugs for 48 h, followed by DNA extraction. Then, DNA samples were analyzed by agarose gel electrophoresis and were photographed. (d) Cells were incubated with various concentrations of MLN4924 (0, 0.1, 0.3, and 1 μM) for 24 h, followed by MK-2206 (0.5 or 1 μM) or DMSO for 24 h. Cells were harvested for western blotting using indicated antibodies.
Discussion
Breast cancer is characterized by a high prevalence in women and various molecular subtypes that are associated with the reaction to treatment and patient prognosis [23,24]. At present, targeted therapy for breast cancer is mainly based on the amplified RTK pathways, such as HER2 and EGFR [25]. However, therapies targeting RTKs are limited by mutations in the kinases and their downstream targets. Thus, the discovery of novel targeting therapies for breast cancer is receiving much attention.
MLN4924 is a novel potent small molecule inhibitor of NEDD8-Activating Enzyme (NAE) [26] and has proved its antitumor activity in HER2-and estrogen receptor (ER)-positive breast cancer cell lines [10,11,27]. An obvious advantage of MLN4924 over Bortezomib, the first FDA-approved general proteasome inhibitor for the treatment of relapsed/refractory multiple myeloma [28,29], is that MLN4924 selectively inhibits the degradation of a specific set of cellular proteins regulated by CRL E3 ligases (limited cytotoxicity), whereas Bortezomib blocks the degradation of all proteins through 26S proteasome (higher cytotoxicity) [29]. However, cancer cells developed MLN4924 resistance through the development of heterozygous mutations in the NAEβ subunit of NAE [12]. Here, we found that AKT activation may also contribute to MLN4924 resistance.
Previous studies showed that highly increased levels of phosphorylated AKT were discovered in trastuzumab-resistant HER2-positive breast cancer cells and proteasome inhibitors treating HER2-positive and TNBC cell lines [30,31], which partially explains the occurrence of drug resistance and unsatisfactory clinical efficacy. This suggests that AKT inhibitors may overcome trastuzumab resistance or sensitize breast cancer cells to proteasome inhibitors. Indeed, strategic drug combination with agents inhibiting AKT activation significantly enhances the tumor suppression efficacy of the abovementioned targeted therapies [30,31]. In our study, we found that MLN4924 activates the AKT pathway in a feedback manner to promote survival signaling in both HER2-positive and TNBC cell lines, which may contribute to MLN4924 resistance. We further validated that a highly selective AKT inhibitor, MK-2206, synergistically boosts the antitumor activity of MLN4924 via the inhibition of cell growth and migration and the induction of apoptosis.
The mechanism of AKT activation by MLN4924, as a potential cause of drug resistance, may be involved in its complex interaction with the mammalian target of rapamycin (mTOR). The mTOR pathway is composed of two complexes, mTORC1 and mTORC2, which are structurally and functionally distinct. By stimulating the phosphorylation of S6K1 and 4E-BP1, mTORC1 mainly promotes cell growth, while mTORC2 promotes cell proliferation and survival by phosphorylating and activating AKT [32]. DEPTOR is a natural inhibitor of both mTORC1 and mTORC2 [33] and a physiological substrate of the CRL1βTrCP E3 ligase [34–36]. MLN4924, serving as an inhibitor of CRL E3 ligases, causes the accumulation of DEPTOR to block the activity of mTORC1 in dose- and time-dependent manners in SK-BR3 and MDA-MB231 cells [27]. It is worth noting that AKT can be activated by relieving the feedback inhibition from S6K1 to PI3K, known as crosstalk between mTORC1 and mTORC2 [37]. Thus, increased DEPTOR induced by MLN4924 treatment may activate AKT in a feedback manner by inhibiting mTORC1, which needs to be further proved by future studies, such as a rescue experiment by depletion of DEPTOR. Interestingly, a previous study showed that, under serum-free conditions, MLN4924 can directly activate AKT at low concentrations through the induction of EGFR dimerization [38]. Taken together, the mechanism of AKT activation by MLN4924 may include 1) feedback activation through mTORC1 inhibition by accumulation of DEPTOR during late-stage MLN4924 treatment and 2) direct activation by promoting EGFR dimerization during early-stage MLN4924 treatment.
In summary, our study demonstrates the following: 1) MLN4924 acts as a potent antitumor therapy in not only HER2-positive but also TNBC cell lines, 2) MLN4924 activates the AKT pathway as a pro-survival signal, and 3) combination with the AKT inhibitor MK-2206 synergistically enhances the tumor suppression capability of MLN4924. Thus, our study provides the proof-of-concept evidence for the strategic drug combination of MLN4924 with an AKT inhibitor for maximal tumor suppression via the inhibition of growth and migration and the enhancement of apoptosis.
Funding Statement
This work was supported by the National Key R&D Program of China [2016YFA0501800 to YZ and XX], the Natural Science Foundation of Zhejiang Province [Grant No. LR16C050001 to YZ], and the National Natural Science Foundation of China [Grant Nos. 31470753 and 81672728 to YZ and 81572708 and 31501129 to XX].
Ethical standards
Ethical approval: This article does not contain any studies with human participants or animals performed by any of the authors.
Author contributions
XC, acquisition and analysis of the data, drafting the manuscript; DC, YB and JS, acquisition of data; XX, analysis and interpretation of the data, revising the manuscript; and YZ, conception and design, analysis and interpretation of the data, drafting and revising the manuscript. All the authors have read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Siegel RL, Miller KD, Jemal A.. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. PMID: 29313949. [DOI] [PubMed] [Google Scholar]
- [2].Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol. 2007;608:1–22. PMID: 17993229. [DOI] [PubMed] [Google Scholar]
- [3].Dear RF, McGeechan K, Jenkins MC, et al. Combination versus sequential single agent chemotherapy for metastatic breast cancer. Cochrane Database Syst Rev. 2013. December 18;(12):CD008792 PMID: 24347031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Robert NJ, Diéras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase iii trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2–negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29(10):1252–1260. PMID: 21383283. [DOI] [PubMed] [Google Scholar]
- [5].Zhao Y, Sun Y. Cullin-RING. Ligases as attractive anti-cancer targets. Curr Pharm Des. 2013;19(18):3215–3225. PMID: 23151137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhao Y, Morgan MA, Sun Y. Targeting neddylation pathways to inactivate cullin-RING ligases for anticancer therapy. Antioxid Redox Signal. 2014. December 10;21(17):2383–2400. PMID: 24410571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16(1):30–44. PMID: 25531226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Brownell JE, Sintchak MD, Gavin JM, et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010. January 15;37(1):102–111. PMID: 20129059. [DOI] [PubMed] [Google Scholar]
- [9].Zhou L, Zhang W, Sun Y, et al. Protein neddylation and its alterations in human cancers for targeted therapy. Cell Signal. 2018. April 1;44:92–102. PMID: 29331584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang D, Tan M, Wang G, et al. The p21-dependent radiosensitization of human breast cancer cells by MLN4924, an investigational inhibitor of NEDD8 activating enzyme. Plos One. 2012;7(3):e34079. PMID: 22457814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Oladghaffari M, Shabestani Monfared A, Farajollahi A, et al. MLN4924 and 2DG combined treatment enhances the efficiency of radiotherapy in breast cancer cells. Int J Radiat Biol. 2017. June;93(6):590–599. PMID: 28291374. [DOI] [PubMed] [Google Scholar]
- [12].Milhollen Michael A, Thomas Michael P, Narayanan U, et al. Treatment-emergent mutations in NAEβ confer resistance to the NEDD8-activating enzyme inhibitor MLN4924. Cancer Cell. 2012;21(3):388–401. PMID: 22439935. [DOI] [PubMed] [Google Scholar]
- [13].Hirai H, Sootome H, Nakatsuru Y, et al. MK-2206, an allosteric AKt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9(7):1956–1967. PMID: 20571069. [DOI] [PubMed] [Google Scholar]
- [14].Blackburn Jessica S, Liu S, Wilder Jayme L, et al. Clonal evolution enhances leukemia-propagating cell frequency in T cell acute lymphoblastic leukemia through Akt/mTORC1 pathway activation. Cancer Cell. 2014;25(3):366–378. PMID: 24613413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Will M, Qin ACR, Toy W, et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS–ERK signaling. Cancer Discov. 2014;4(3):334–347. PMID: 24436048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xiang R-F, Wang Y, Zhang N, et al. MK2206 enhances the cytocidal effects of bufalin in multiple myeloma by inhibiting the AKT/mTOR pathway. Cell Death Dis. 2017. May 11 online;8:e2776. PMID: 28492559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ma CX, Suman V, Goetz MP, et al. A phase II trial of neoadjuvant MK-2206, an AKT inhibitor, with anastrozole in clinical stage II or III PIK3CA-mutant ER-positive and HER2-negative breast cancer. Clin Cancer Res. 2017;23(22):6823–6832. PMID: 28874413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wisinski KB, Tevaarwerk AJ, Burkard ME, et al. Phase I study of an AKT inhibitor (MK-2206) combined with lapatinib in adult solid tumors followed by dose expansion in advanced HER2+ breast cancer. Clin Cancer Res. 2016;22(11):2659–2667. PMID: 27026198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].West KA, Sianna Castillo S, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resistance Updat. 2002. December 1;5(6):234–248. PMID: 12531180. [DOI] [PubMed] [Google Scholar]
- [20].Koboldt DC, Fulton RS McLellan MD, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012. October 4;490(7418):61-70. PMID: 23000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Knuefermann C, Lu Y, Liu B, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003. May 22 online;22:3205. PMID: 12761490. [DOI] [PubMed] [Google Scholar]
- [22].Qiao M, Sheng S, Pardee AB. Metastasis and AKT activation. Cell Cycle. 2008. October 1;7(19):2991–2996. PMID: 18818526. [DOI] [PubMed] [Google Scholar]
- [23].Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000. August 17;406(6797):747–752. PMID: 10963602. [DOI] [PubMed] [Google Scholar]
- [24].Sorlie T. The impact of gene expression patterns in breast cancer. Clin Chem. 2016. August;62(8):1150–1151. PMID: 27022070. [DOI] [PubMed] [Google Scholar]
- [25].Luo M, Fu LW. Redundant kinase activation and resistance of EGFR-tyrosine kinase inhibitors. Am J Cancer Res. 2014;4(6):608–628. PMID: 25520855. [PMC free article] [PubMed] [Google Scholar]
- [26].Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009. April 9;458(7239):732–U67. PMID: 19360080. [DOI] [PubMed] [Google Scholar]
- [27].Zhao Y, Xiong X, Jia L, et al. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012;3:e386. PMID: 22951983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kane RC, Bross PF, Farrell AT, et al. Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003;8(6):508–513. PMID: 14657528. [DOI] [PubMed] [Google Scholar]
- [29].Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008. March 15 2008;14(6):1649–1657. PMID: 18347166. [DOI] [PubMed] [Google Scholar]
- [30].Fujita T, Doihara H, Washio K, et al. Proteasome inhibitor bortezomib increases PTEN expression and enhances trastuzumab-induced growth inhibition in trastuzumab-resistant cells. Anticancer Drugs. 2006. April;17(4):455–462. PMID: 16550004. [DOI] [PubMed] [Google Scholar]
- [31].Yy S, Gw S, Rz O. Proteasome inhibitors induce a p38 Mitogen-activated Protein Kinase (MAPK)-dependent anti-apoptotic program involving MAPK phosphatase-1 and Akt in models of breast cancer. Breast Cancer Res Treat. 2006. November 1;100(1):33–47. PMID: 16807678. [DOI] [PubMed] [Google Scholar]
- [32].Laplante M, Sabatini David M. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. PMID: 22500797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009. May 29;137(5):873–886. PMID: 19446321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCFβTrCP E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell. 2011;44(2):304–316. PMID: 22017876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gao D, Inuzuka H, Tan M-K, et al. mTOR drives its own activation via SCFβTrCP-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell. 2011;44(2):290–303. PMID: 22017875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Duan S, Skaar Jeffrey R, Kuchay S, et al. mTOR generates an auto-amplification loop by triggering the βTrCP- and CK1α-dependent degradation of DEPTOR. Mol Cell. 2011;44(2):317–324. PMID: 22017877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhao Y, Sun Y. Targeting the mTOR-DEPTOR pathway by CRL E3 ubiquitin ligases: therapeutic application. Neoplasia. 2012;14(5):360–367. PMID: 22745582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhou X, Tan M, Nyati MK, et al. Blockage of neddylation modification stimulates tumor sphere formation in vitro and stem cell differentiation and wound healing in vivo. Proc Natl Acad Sci U S A. 2016. May 24;113(21):E2935– E2944. PMID: 27162365. [DOI] [PMC free article] [PubMed] [Google Scholar]