

ABSTRACT
The heterogeneity in human breast cancer poses a challenge for effective treatment. Better understanding of tumor initiation and development will help to resolve this problem. Current models explaining intratumoral diversity include cancer stem cells, clonal evolution and cancer cell dedifferentiation and reprogramming. Herein, a new model, cancer transmission, is proposed to explain cancer heterogeneity. We found breast cancer cells (MCF10A.NeuT) were capable of transforming normal mammary epithelial cells (MCF10A). The transformed cells exhibited cancerous properties including enhanced proliferation and migration, loss of apical-basal polarity and depolarized acini structure associated with epithelial-mesenchymal transition (EMT). The transformed MCF10A cells displayed distinct EMT characteristics compared to parental cells. We further showed that cancer cell-secreted factors were sufficient to induce cancerous transformation of normal cells. Furthermore, transformed cells were resistant to radiation treatment, providing new insights into mechanisms underlying therapeutic resistance.
KEYWORDS: Breast cancer, ErbB2/Her2, heterogeneity, clone evolution, cancer stem cell, epithelial-mesenchymal transition (EMT)
Introduction
Breast cancer is the most common cancer in women, with an estimated 1.7 million new cases and more than 500 000 deaths annually worldwide (Global Health Estimates, WHO 2013). It is an aggressive disease, with respect to the metastatic potential of the primary tumor and in the time it takes for metastasis to occur. Immune therapy using Trastuzumab has been pivotal in the management of ErbB2-positive breast cancer and has improved patient survival considerably. However, about 20% of early stage breast cancer patients do not respond to Trastuzumab, and 70% of patients with advanced disease develop resistance to Trastuzumab treatment [1,2]. This treatment failure is partly attributed to cancer heterogeneity [3]
Breast cancer is a heterogeneous disease, reflected by intertumor heterogeneity among different patients and intratumor heterogeneity within each individual. Intertumor heterogeneity includes clinical and morphologic differences involving staging and histopathologic classification whilst intratumor heterogeneity occurs at the genomic, transcriptomic, and proteomic levels, which results in tumor cells having different features and sensitivity to cancer therapies [4,5].
Cancer stem cells (CSC) and clonal evolution models have been proposed to explain intratumor heterogeneity [6]. In the clonal evolution model, tumors arise from a single mutated cell and cells accumulate mutations as the tumor develops. The fittest cells with aggressive behaviors will go through natural selection to drive tumor initiation and progression [7]. However, a limitation of this model is it does not take into account the importance of non-genetic variations, minor clones, and potential functional interactions between clones and tumors [8]. The cancer stem cell model asserts that a small pool of cancer cells with self-renewal and differentiation capacity serve as “seeds” to promote tumor initiation, progression and recurrence [9]. A major drawback of this model is that it neglects heterogeneous DNA within the tumor [8]. It has been proposed that clonal evolution and the CSC model are not mutually exclusive and that their combination is also plausible [6].The original CSC model states that tumor progression exists in a unidirectional hierarchy with transformed normal tissue stem cells at the top followed by “tumor cell plasticity”. This opinion has been reconsidered because non-CSCs have been found that have “tumor cell plasticity” enabled by re-programming. It was shown that differentiated human mammary epithelial cells were capable of switching to a CSC state, thus resolving inconsistencies of the current CSC model [10,11].
Understanding the mechanisms of tumor heterogeneity is critical for the development of more effective cancer treatments. Here, we found that human mammary epithelial cells exhibited cancerous properties following co-culture with breast cancer cells. The transformed showed altered cellular proliferation, migration, and EMT phenotypes. Importantly, these cells had intrinsic radiation resistance compared to breast cancer cells. These findings suggest a new model to explain intratumor heterogeneity and have direct implications for cancer therapy, given that heterogeneous events determine therapeutic responsiveness as shown in this study.
Results
Intratumor heterogeneity of ErbB2 in breast cancer
Overexpression of oncogene ErbB2 frequently occurs in human breast cancers and promotes cancer cell growth, migration and invasion [12], which allows tumor spread into surrounding tissues and/or circulation and subsequent metastasis, a central hallmark of poor prognosis. ErbB2 expression heterogeneity has been previously reported [13,14]. Given the disagreement over protein expression evaluation in specimens with +1 and +2 ErbB2 IHC scores, to determine intratumor heterogeneity, we only included specimens with +3 ErbB2 IHC staining. Specimens taken from the primary breast tumor displayed morphological heterogeneity with H&E staining (data not shown), which was further confirmed with IHC of the same areas. Breast cancer characteristics by intratumor heterogeneity of ErbB2 are presented in Figure 1.
Figure 1.
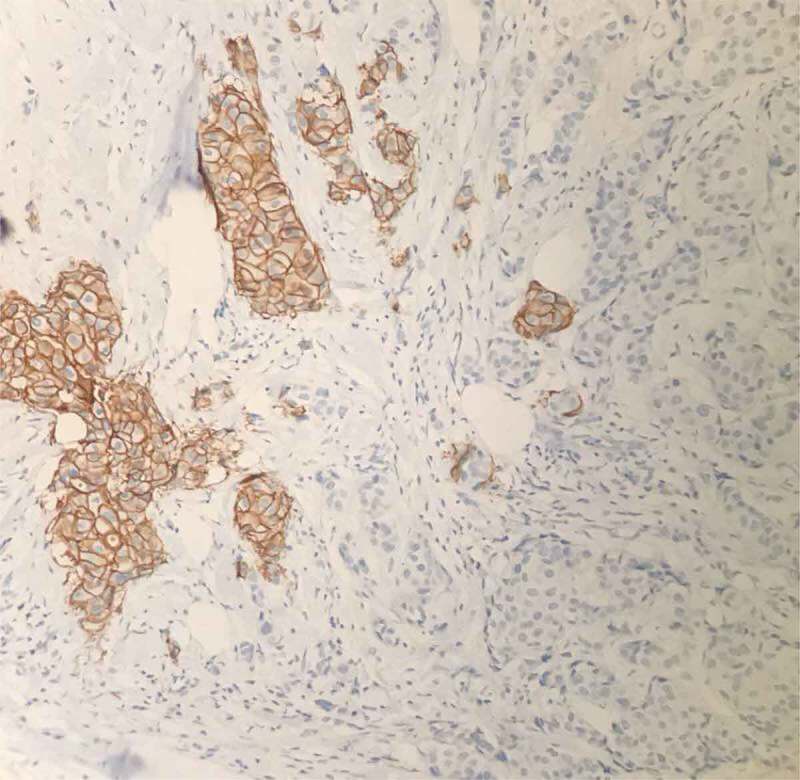
Heterogeneous expression of ErbB2 in breast cancers specimens determined by immunohistochemistry (IHC).
IHC staining of breast cancer tissues for ErbB2 protein expression. Brown staining shows positive staining of ErbB2. In the same observed field, ErbB2-negative breast cancer cells are also present.
We then asked whether the distinctly heterogeneous tumor cells originated from the same initiating cell, which is the basis of the cancer stem cell and evolution theories. However, there was no convincing data to exclude the possibility that ErbB2-positive and ErbB2-negative cells were from different initiating cells. Given ErbB2 is a “driver” oncogene and overexpression of ErbB2 alone is capable of transforming normal breast epithelial cells into cancer [15], we hypothesized that the tumor-initiating cell transformed by ErbB2 can further transform normal epithelial cells through direct or indirect interactions. To test our hypothesis, we established co-culture experiments as outlined in Figure 2(a).
Figure 2.
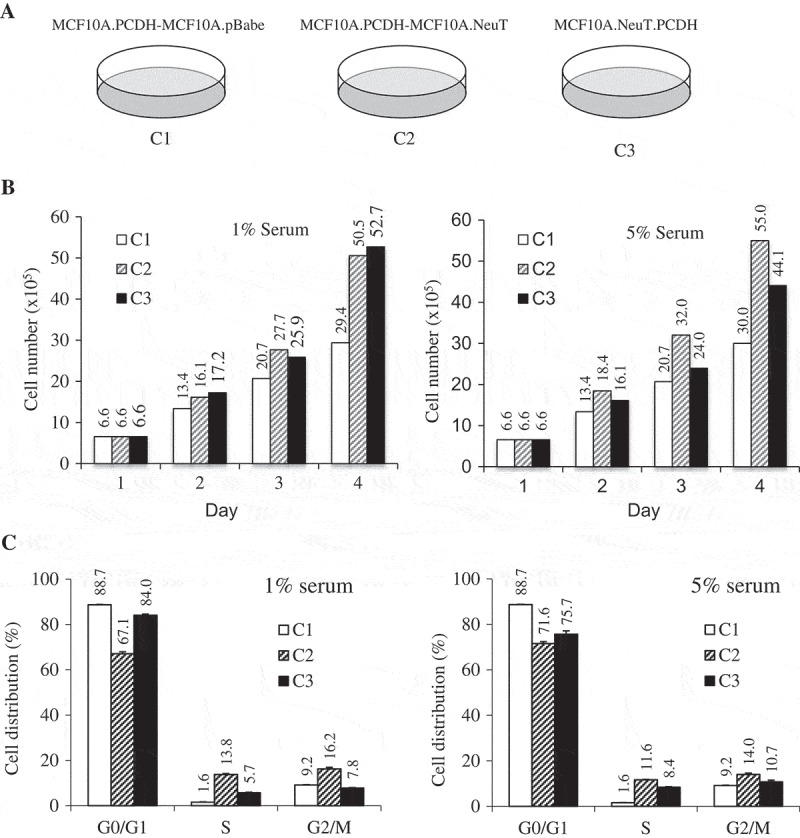
ErbB2-expressing breast tumor cells have a distinct proliferation profile.
(a) MCF10A cells co-cultured with either control (C1) or NeuT (ErbB2) (C2) transformed cells. MCF10A-NeuT cells transduced with pCDH vector (C3) were included as control. (b) 1 × 105 cells were seeded per well in a 6-well cell culture plate. Cellular growth was determined by counting the cells at 24 hr in the presence of either 1% or 5% serum. (c) Cell cycle progression was analyzed by flow cytometry in the presence of either 1% or 5% serum. The co-culture experiments have been done in triplicate.
MCF10A cells gain proliferative advantage after co-culture with MCF10A.NeuT cells
MCF10A.NeuT cells were established by transducing immortalized breast epithelial MCF10A cells with the oncogene NeuT (constitutively active form of ErbB2). This cell model exhibits cancerous properties and clinical characteristics of breast cancer [16,17]. To test our hypothesis, we mixed MCF10A and MCF10A.NeuT or control MCF10A.pBabe cells at a 1:1 ratio. MCF10A cells were stably transduced with pCDH-GFP to allow separation following co-culture. When cells reached confluence, they were kept for an additional 24 hrs before being split into three plates. After three passages of co-culture, the GFP-positive cells were sorted using FACs. MCF10A cells co-cultured with MCF10A.pBabe cells or MCF10A.NeuT cells were designated C1 and C2 respectively. To reduce the potential influences of retroviral transduction and GFP expression, MCF10A.NeuT cells were also transduced with pCDH retrovirus and served as a control (C3) (Figure 2(a)).
Firstly, the proliferation rate of C2 cells was compared to that of C1 and C3 cells. Cells were seeded and cultured in growth medium supplemented with either 1% or 5% serum. The number of cells were counted every day for four days. As shown in Figure 2(b), in both serum conditions, C2 cells showed a 72% (29.4 vs 50.5) and 83% (30 vs 55) increase in cell number compared to C1 parental control cells. The cell cycle distribution of these cells was further analyzed. 24 hrs after cells were seeded, 13.8% cells of C2 cells entered into S-phase compared to 1.6% of C1 cells (Figure 2(c)). These data suggest that normal breast epithelial cells after co-culture with breast cancer cells gain growth advantage.
MCF10A cells co-cultured with MCF10A.NeuT cells show enhanced migration ability
Cancer cells possess a broad spectrum of migration and invasion mechanisms including individual and collective cell migration. Cell motility was determined using a migration assay and following the manufacturer’s protocol. After 12 hrs C2 cells occupied most of the space indicating faster migration, while C1 control cells migrated slower. After 24 hrs, both C1 and C2 cells fully occupied the detected area whilst C3 cells did not (Figure 3(c)). The same results were observed with serum starvation (1% serum) (Figure 3(b)). Thus, following co-culture with cancer cells normal epithelial cells show enhanced migration ability, although cancer cells did not show enhanced motility comparing to normal cells.
Figure 3.
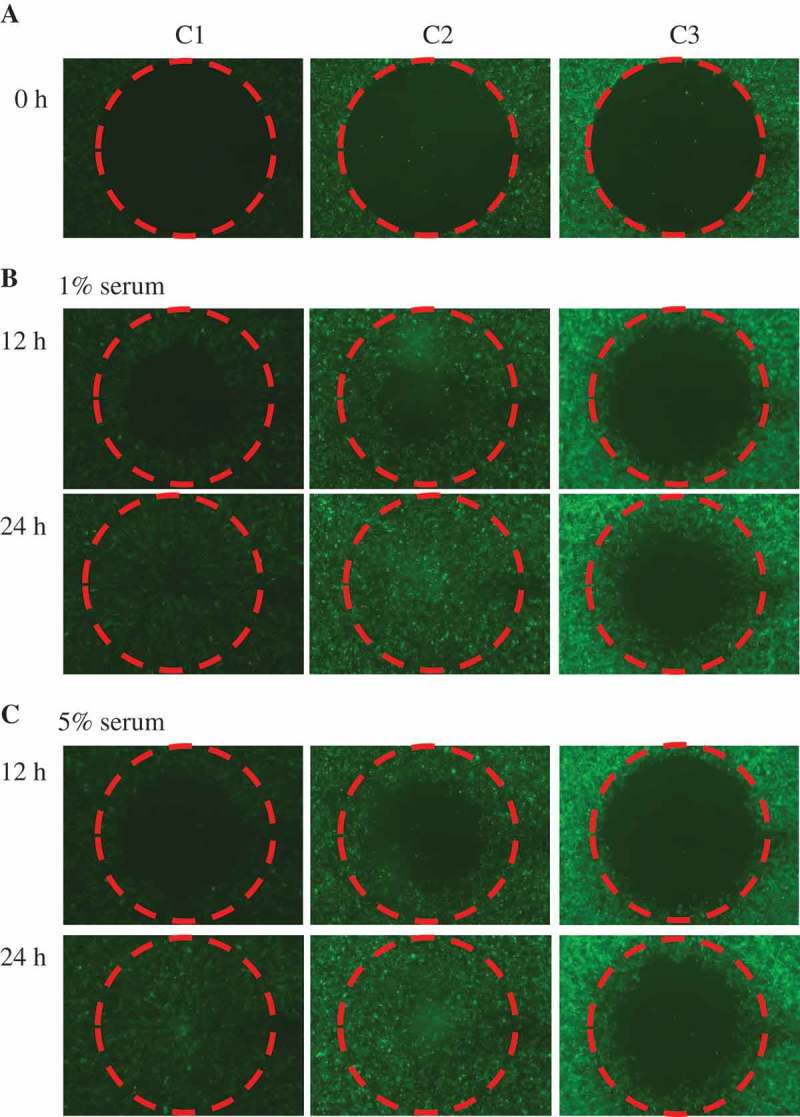
MCF10A.pCDH co-cultured with MCF10A.NeuT cells show enhanced migration ability compared to MCF10A.NeuT cells.
(a) Cells were seeded and grew into 100% confluence. The stoppers were removed at time zero (0). (b) C2 cells migrate similarly to C1 cells but faster than C3 cells 12 hrs and 24 hrs after in the presence of 1% serum. (c) Cells were cultured in medium supplemented with 5% serum.
MCF10A cells co-cultured with MCF10A.NeuT cells show oncogenic transformation potential
Malignant transformation of cells is typically associated with increased proliferation, acquisition of anchorage-independent growth, and enhanced transformation potential. Therefore, we tested whether normal breast epithelial cells co-cultured with cancer cells would acquire transformation phenotypes. Cells were plated for focus formation assay. After two weeks of growth, no foci were observed in the C1 group whereas foci formed in both C2 and C3 groups (Figure 4). This showed that normal epithelial cells after co-culture with cancer cells display oncogenic transformation.
Figure 4.
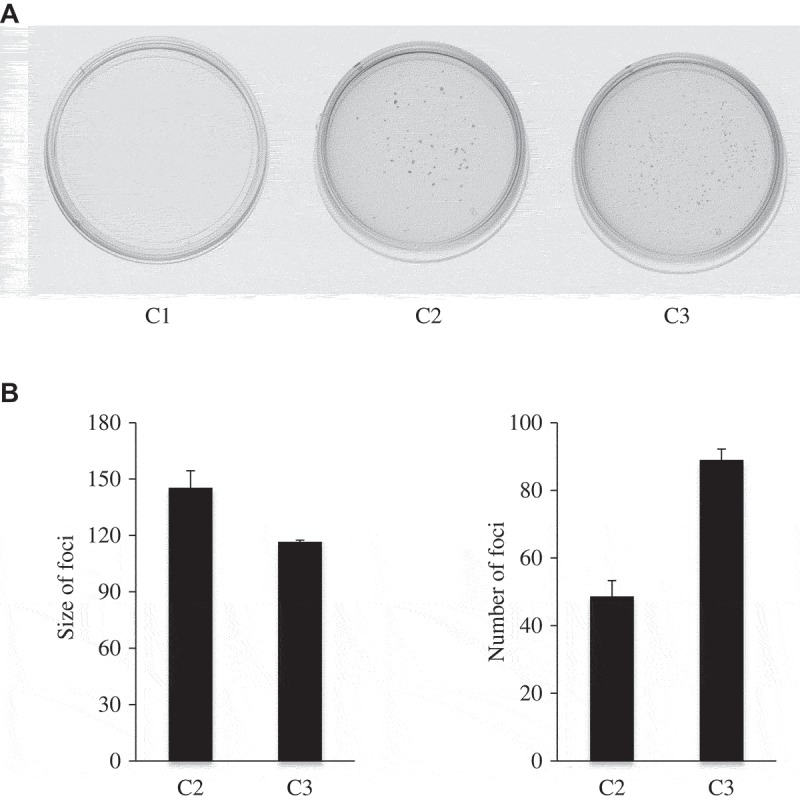
MCF10A cells co-cultured with MCF10A.NeuT cells show enhanced tumorigenic ability.
(a) A total of 1,000 cells were seeded in 6-cm cell culture plate and allowed to grow for 2 weeks. Medium was replaced every three days with fresh medium. Cells were stained with crystal violet. Representative images of the plates are shown. (b) The result was presented as bar graph. The experiments have been repeated three times. Statistical analysis was done by performing Student’s t-test.
MCF10A cells co-cultured with MCF10A.NeuT cells show disrupted apical-basal polarity
Epithelial cells can form acini-like structures with apical–basal polarity that are characterized by a hollow lumen due to centrally-located cells undergoing selective apoptosis [15]. However, activation of ErbB2 in epithelial cells disrupts normal structure of acini morphogenesis by protecting luminal cells from apoptosis and disrupting apical polarity [16], which is a character of early stages of breast cancer (hyperplasia and ductal carcinoma in situ (DCIS)) [18]. Several genes that are required for the development and maintenance of epithelial polarity have been identified including hDlg1 that plays an important role in controlling cell polarization during oriented migration of epithelial cells [19]. Most of the biological functions of hDlg1 rely on its interaction with several regulatory proteins, including ezrin–radixin–moesin (ERM) complex, which participates in cytoskeletal remodeling during cellular migration [20,21]. The normal distribution of hDlg and phosphorylated ERM can be disrupted by ErbB2 overexpression [21].
C1, C2, or C3 cells were grown in 3D matrigel cultures and cellular morphogenesis differences were evaluated. After 12 days, C1 cells displayed a spherical and polarized acinar architecture with a single layer of polarized epithelial cells surrounding a hollow central lumen. C2 cells formed complex multiacinar structured acini ressembling those of C3 cells, with excess cell proliferation and filled luminal spaces [15] (Figure 5(a)). To evaluate apical-basal polarity, acini were immnunostained with anti-hDlg and anti-phospho-ERM antibodies. In C1 cells, hDLg was distributed in the lateral and basal cell membranes. The distribution of phosphorylated ERM proteins was closely subjacent to the plasma membrane. In C2 and C3 cells, hDlg and phospho-ERM distribution was disrupted due to the abnormal acini structures with filled lumens (Figure 5(b)).
Figure 5.
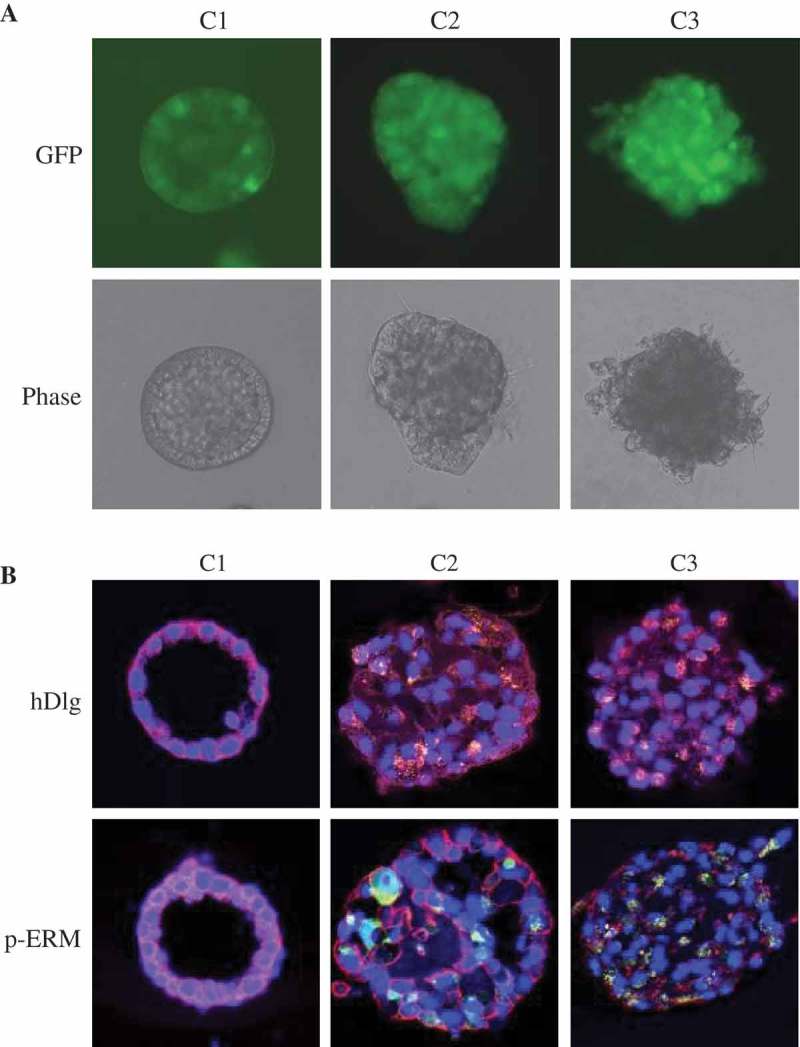
MCF10A cells co-cultured with MCF10A.NeuT cells have similar three-dimensional acini structure with disrupted apical–basal polarity as MCF10A.NeuT cells.
(a) Phase-contrast micrographs of acinar structures formed by C1, C2 and C3 cells plated in Matrigel for 12 days. (b) Acini cells were stained for DAPI (blue), hDlg (red, upper panel), and p-Ezrin–Radixin–Moesin (p-ERM) (red, lower panel).
MCF10A cells co-cultured with MCF10A.NeuT cells have different EMT properties compared to MCF10A.NeuT cells
Lumen filling is often followed by EMT, a transdifferentiating process, which facilitates tumor invasion and the metastasis [22]. During EMT, epithelial cells lose the characteristics of epithelial cells (cell junctions, apical-basal polarity and contact inhibition) and gain a mesenchymal phenotype including enhanced invasiveness, increased cell-to-extracellular matrix contact and resistance to apoptosis [23]. In addition, epithelial cells reduce the expression of epithelial markers (epithelial keratins, E-cadherin and β-catenin) and upregulate mesenchymal markers (vimentin and fibronectin) [24,25].
We determined the expression of several proteins known to play a fundamental role in EMT. Epithelial markers (E-cadherin and β-catenin) were detected in MCF10A.pCDH and C1 cells and both cell lines lacked mesenchymal marker expression. C3 cells expressed mesenchymal markers, fibronectin and vimentin and did not express epithelial markers. Co-cultured breast epithelial cells (C2) showed different properties in EMT markers compared to C3 cells. C2 cells lost expression of epithelial markers and upregulated expression of fibronectin and not vimentin compared to C1 normal control cells (Figure 6).
Figure 6.
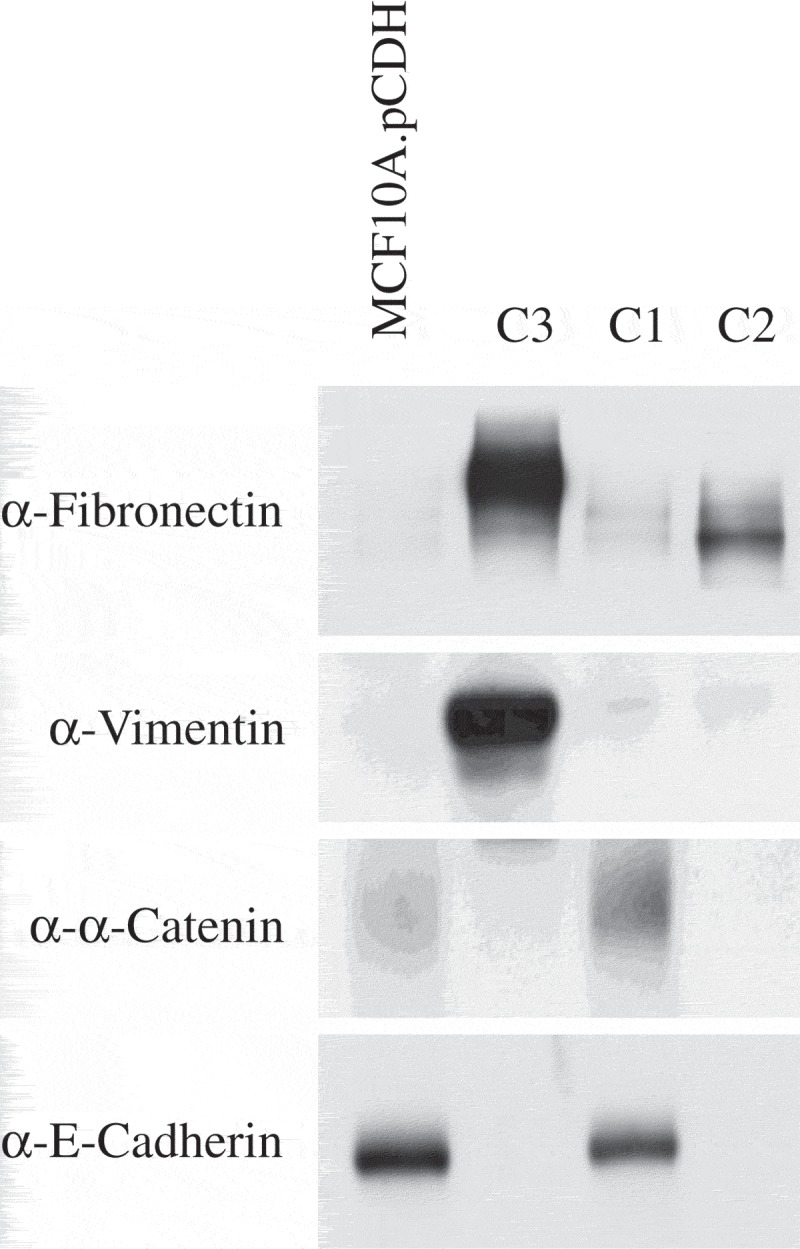
MCF10A cells co-cultured with MCF10A.NeuT cells have different EMT characteristics compared to MCF10A.NeuT cells.
Expression of epithelial (E-cadherin, β-catenin) and mesenchymal (fibronectin, vimentin) protein markers was analyzed by western blot.
MCF10A cells co-cultured with MCF10A.NeuT cells exhibit resistance to γ-irradiation
The focus formation assay was utilized to investigate the proliferative potential upon radiation treatment. C1 normal epithelial cells were unable to form foci (Figure 4). C2 and C3 cells were exposed to γ-irradiation (0, 2 or 4 Gy). Without γ-irradiation exposure, both C2 and C3 cells grew foci similarly (Figure 7). However, reduced foci formation was observed in C3 compared to C2 cells exposed to γ-irradiation. We concluded that normal breast epithelial cells develop resistance to γ-irradiation after co-culture with breast cancer cells.
Figure 7.
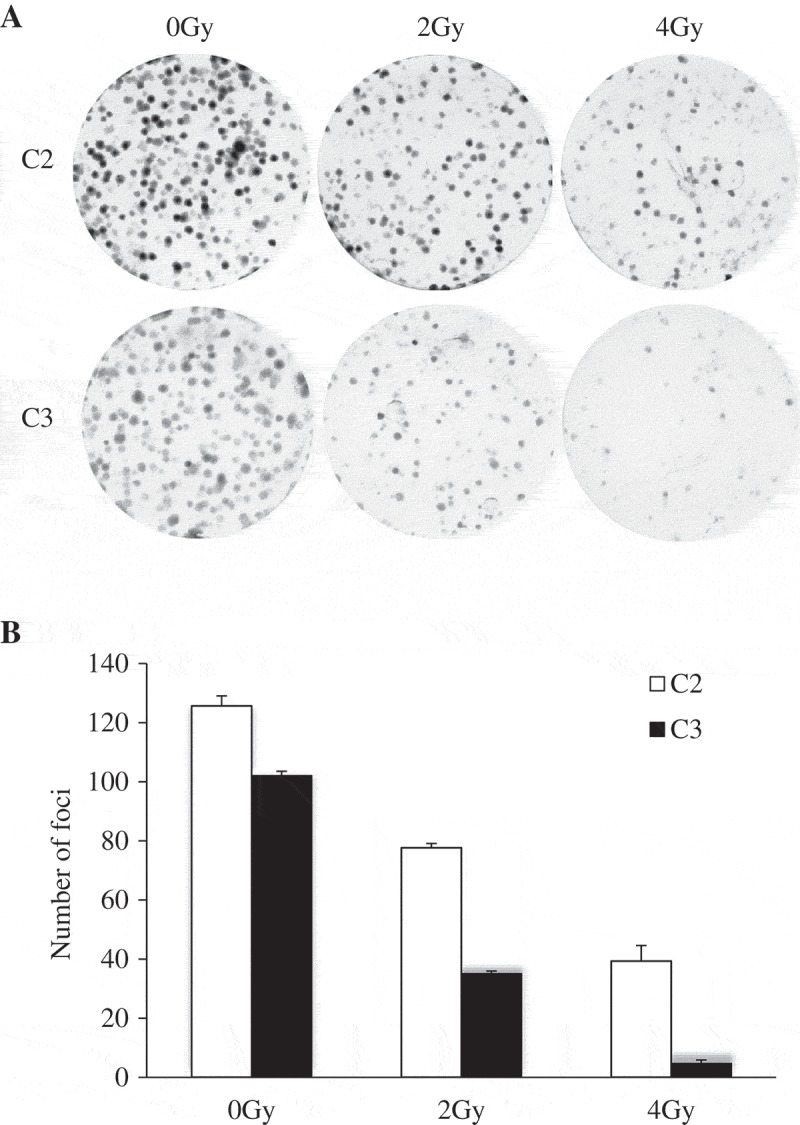
MCF10A cells co-cultured with MCF10A.NeuT cells are more resistant to γ-irradiation compared to MCF10A.NeuT cells.
(a) Represent images of Foci formation assays with C2 and C3 cells with different dose of radiation (0, 2 and 4 Gy). (b) The number of foci was counted showing reduced foci number in a dose-dependent manner. Mean differences were compared by Student’s t test
Factors secreted by cancer cells are sufficient to drive EMT of normal epithelial cells
To determine whether paracrine factors secreted by cancer cells were sufficient to induce to the transformation of normal epithelial cells, we utilized transwell co-culture assays, which only allow the exchange of secreted proteins in the medium shared by C3 and MCF10A.pCDH cells without any direct cell-cell contact. After co-culturing for seven passages, MCF10A.pCDH cells underwent complete EMT which was validated by immunoblotting (data not shown).
We further identified soluble proteins produced by C1 and C3 cells using the Raybio Human Protein Array Kit. Most cytokines and secreted proteins were expressed similarly by C1 and C3 cells (Figure 8(a)), sixteen proteins showed a 2-fold increase and four proteins were down-regulated in C3 vs. C1 cells (Figure 8(b)). C3 cells showed relative abundance of TLR receptor (CD14) and NF-κB (TNFR1, IL-6) pathways. The IGFBP protein family was also upregulated in C3 cells
Figure 8.
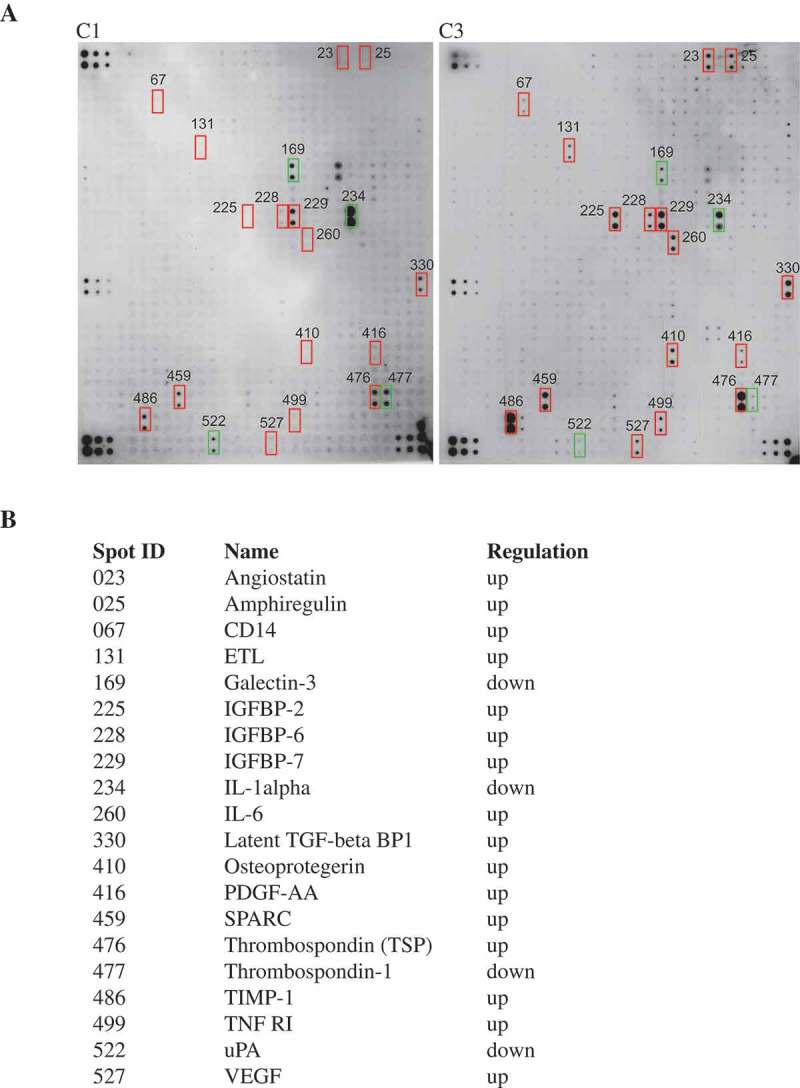
Cytokine profiling of MCF10A.NeuT cells and transformed MCF10A cells.
Experiments were conducted using L-Series Human Antibody Array L-507 Membrane Kit (RayBiotech). (a) Cytokine assay membrane was blotted according to the protocol provided by the manufacturer. (b) Differentially expressed cytokines are listed.
Discussion
Heterogeneity poses a challenge to therapeutic success. Although there are several models that try to explain the underlining mechanism, the driving forces behind tumor initiation and progression have been less understood. The introduction of genetic or epigenetic alterations and the evolutionary selection thereof, are two principle factors driving cancer heterogeneity [26].
This study demonstrated a new perspective to explain cancer heterogeneity and proposed a new mechanism of cancer metastasis (Figure 9). We showed that MCF10A cells became cancerous following co-culture with MCF10A.NeuT cells. It had been established that ErbB2 continuous expression can initiate the early stages of mammary carcinogenesis [16]. C2 and C3 cells displayed common features such as lose of epithelial cell characteristics including loss of apical-basal polarity that resulted in abnormally structured acini. C2 and C3 cells however had some different features, C2 cells had different EMT properties (lacked vimentin expression) compared with C3 cells. We further showed that medium exchange was sufficient to trigger EMT. C2 cells also showed enhanced migration ability and were more resistant to γ-irradiation compared to C3 cells.
Figure 9.
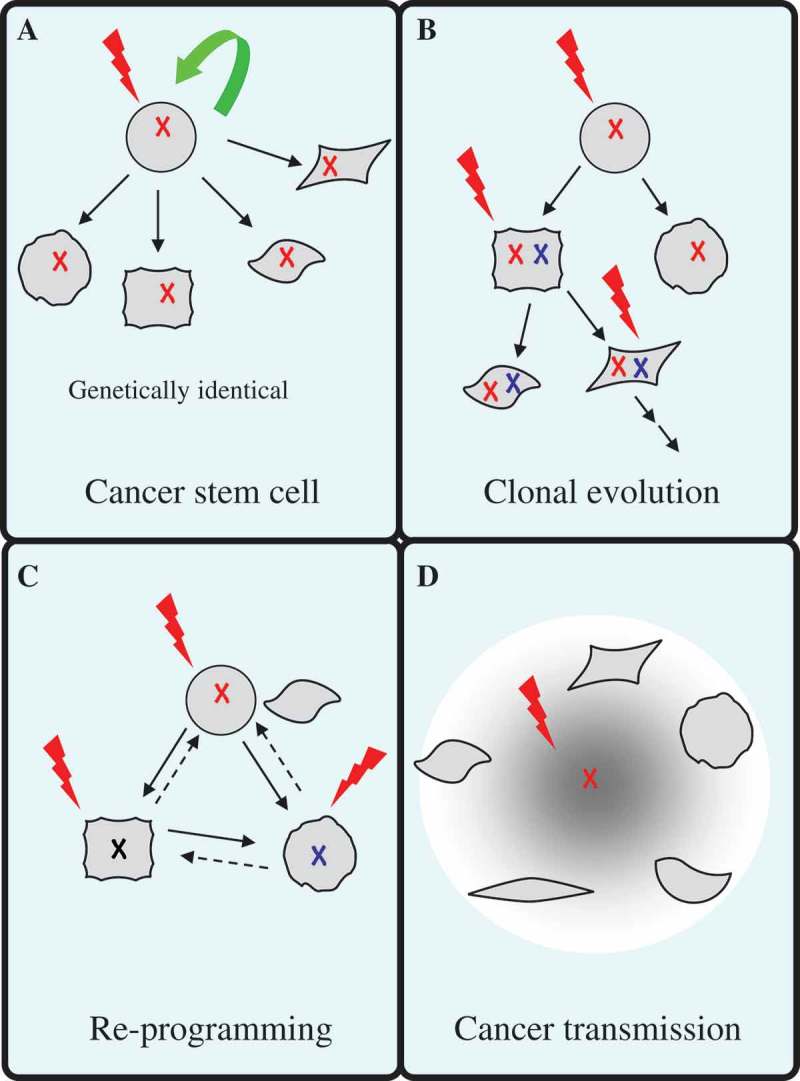
Models depicting cancer heterogeneity.
(a) Cancer stem cell (CSC) model. This model suggests that only a subset of cancer cells possess indefinite self-renewal ability to initiate and maintain tumor growth. CSCs generate tumor heterogeneity by differentiation, which produces a range of distinct cell phenotypes present within the tumor. (b) Clonal evolution model. This model focuses on the process of sequential acquisition of vertically transmittable genetic and/or epigenetic alterations in an individual tumor. Acquired mutations of tumor cells serve as a platform for environmental adaptation and natural selection for the fittest clones to grow. (c) Dedifferentiation and reprogramming model. Differentiated epithelial cells maintain plasticity. Oncogene transformation leads to dedifferentiation of epithelial cells through a process called epithelial-mesenchymal transition (EMT), which generates intratumor heterogeneity. (d) Based on our current study, we propose that normal epithelial cells could acquire cancerous properties through interacting with cancer-initiating cells. Cancer-initiating cells transmit mediators such as cytokines to transform normal cells.
The development and maintenance of epithelial cell functions depends on polarized membrane structures, which are orchestrated by a network of proteins and regulators [27]. Abnormal pathophysiological conditions cause multiple lumen lesions and filling of the luminal space, which is a characteristic of early stages of breast cancer [18]. However, the mechanisms of luminal filling initiation are poorly understood. Recent studies have reported that carcinoma cells directly contact normal epithelial cells through filopodia-like projections in the 3D co-culture system, or they secrete MMP-9 that cleaves E-cadherin disrupting the luminal architecture [28], or alternatively they use exosomes to induce oncogenic transformation of adjacent normal epithelial cells [29]. Our results were consistent with previous reports, and we went a step further and compared the properties of C2 and C3 cancer cells and found that the transmitted cancer cells (C2 cells) were more malignant than the original cancer cells (C3 cells), which could explain how cancer heterogeneity is formed.
The role of the tumor microenvironment including extracellular matrix components and soluble factors in tumor progression [30] and drug resistance [31]has been gradually recognized. Among these factors, abnormal autocrine/paracrine signaling has been associated with breast tumor progression [32]. We therefore identified the microenvironment soluble factors that are exchanged between C1 and C3 cells. Further work needs to be done to determine the key driving factors, which could potentially benefit the treatment of breast cancer. Our results showed that IGFBP-2 was significantly upregulated in MCF10A.NeuT (C3 cells). IGFBP-2 has previously been reported to drive EMT and invasiveness in pancreatic ductal adenocarcinoma through activating the NF-κB pathway [33]. Consistent with this, NF-κB pathway receptors and downstream effectors, i.e. TNF-R1 and IL-6, were upregulated in C3 cells. NF-κB pathway activation has been associated with oncogenesis, tumor progression, and therapy resistance [34].
Radiosensitivity varies between patients and in subclones within the tumor. The response to radiation therapy (RT) and prognosis largely depends on the histological subtype. Luminal subtype breast cancer patients show significant improved overall survival (OS) and lower regional recurrence rate after RT, whereas the HER2-positive subtype is associated with radiation resistance and increased metastatic potential[35]. Intratumor heterogeneity has been characterized in separately evolved tumor subclones, thus their responses to RT were also not identical. Mechanisms of RT resistance can be divided into inherent or acquired resistance. According to the CSC model, CSC density influences radiation treatment response and recurrence, and treatment failure is due to incomplete eradication of CSC [36]. In this setting, RT resistance is an intrinsic characteristic of CSC with increased evidence showing that CSC are more resistant to radiation than non-CSC due to enhanced DNA repair [37] and free radical scavenging ability [36] thus making CSC better able to recover from RT-induced damage. In clonal evolution model, tumors are characterized by stochastic mutation and initial sensitivity to RT. After RT exposure, the fittest subpopulations survive under selective pressure [38], leading to tumor subclone repopulation and gradual development of resistance. RT itself also induces mutations, thus may endow some populations with radiation resistance.
In our tumor transmission model, radiation initially eradicated RT-sensitive tumor cells, while transformed cancer cells from normal epithelial cells were more resistant, explaining how radiation-resistance is formed during tumor progression. We demonstrated that C2 cells form larger colonies than C3 cells and are more resistant to radiation treatment. Radiation induces DNA damage in cells including DNA double-strand breaks and single-strand breaks. In response to DNA damage, sophisticated DNA damage repair pathways are activated to maintain the high-fidelity replication of the genome, which is needed for appropriate cellular proliferation. If DNA lesions are repaired successfully, cells recover from cell cycle arrest and continue to proliferate. If not, the damage will lead to cell death via lethal chromosomal aberrations or direct induction of apoptosis [39]. The different radiation responses observed in C2 and C3 cells may be due to the fact that cancer cells often harbor a reduced repertoire of DNA repair signaling capabilities compared to normal cells, and in some cases cancers also upregulate mutagenic repair pathways that drive oncogenesis [40]. Co-cultured cells transformed from normal cell may still have the normal DNA repair mechanism.
Overall, our model shows an acquired phenotype heterogeneity that mediates tumor initiation and progression and enables us to better understand the driving forces of tumor heterogeneity. The current study raises a new possibility of how heterogeneous radiation sensitivity develops during tumor progression. The clinical impact of these findings is that targeting the interaction between cancer cells and the microenvironment through targeting cytokine signaling networks could provide effective cancer treatment strategies.
Materials & methods
Breast cancer specimens and immunohistochemistry (IHC) staining
Formalin-fixed and paraffin-embedded tumor specimens used in this study were from the tissue bank of the Tianjin Tumor Hospital, China. All tumors were primary and untreated before surgery with complete clinicopathological information. All the specimens were anonymous and tissues were collected in compliance with institutional review board regulations. IHC staining for ErbB2 expression was conducted as described previously [41]. Hematoxylin and eosin (H&E) staining were reviewed to confirm cancer tissue and normal epithelia. All staining was assessed by pathologists blinded to the origin of the samples using a semi-quantitative method. Tissue was scored (H-score) based on the total percentage of positive cells and the intensity of the staining (1+, 2+or 3+), where H = (% “1+” x 1) + (% “2+” x 2) + (% “3+” x 3). A minimum of 100 cells was evaluated in calculating the H-score.
Cell lines, cell culture and 3-D culture of mammary epithelial cells
MCF10A, MCF10A.pBabe and MCF10A-NeuT were generously provided by Dr. Pestell (Kimmel Cancer Center, Philadelphia, PA). MCF10A cells were transduced with retroviral vector pCDH-CMV-MCS-EF1-copGFP as control. All cells were cultured in DMEM/F12 supplemented with 5% horse serum, 10 µg/ml insulin, 20 ng/ml EGF, 0.5µg/ml hydrocortisone, and 100 ng/ml cholera toxin. MCF10A.pCDH was co-cultured with MCF10A.NeuT or MCF10A.pBabe respectively at a ratio of 1:1. After three passages, GFP-positive cells were separated from GFP-negative cells by fluorescence-activated cell sorting (FACS) (FACSVantage SE; BD Biosciences, San Jose, CA). The co-culture experiments have been done in triplicate.
For three-dimensional culture, MCF10A cells were mixed with 1:1-diluted Matrigel (BD) and seeded into four-well chambers according to a previously described protocol [15]. Confocal and immunofluorescence microscopy were conducted.
Cell migration assay
Collagen-I coated 96-well plates and Cell Seeding Stoppers, made from a medical-grade silicone, were used to monitor cell migration according to the manufacturer’s protocol. Cells were seeded in the center of each well to restrict cell seeding to the outer annular regions of the wells. Following cell attachment (4 hr at 37°C), the stoppers were removed leaving a 2mm diameter unseeded region in the center of each well into which the seeded cells could migrate. Cell migration was visualized and captured by light microscopy.
Cytokine analysis
The RayBio™ Human Antibody Array Kit(AAH-BLM-1) was used according to the recommended protocol. This kit detected 507 soluble proteins in cell culture supernatants including cytokines, chemokines, growth factors, angiogenic factors, and other soluble factors and receptors. Medium from C1 and C3 cells were collected by culturing cells in serum free medium for 48 hr.
Western blot analysis
Western blotting was performed to detect EMT markers as previously described [42]. All antibodies were used at 1:1,000 dilutions. Western blot analysis was performed using antibodies raised against Fibronectin (Santa Cruz, clone EP5), E-cadherin (BD, clone 36), β-catenin (Santa Cruz, clone E-5), and Vimentin (Santa Cruz, clone H-84)
Cell viability assay and proliferation assay
1 × 105 cells were plated in 6-well plates. Cell growth was determined by cell counting. The total number of cells per well was counted every day for 4 days. Each condition was replicated in triplicate (mean ± SEM). Statistic analysis was performed by standard two-tailed Student’s t-test.
Foci formation assay
C1, C2 and C3 cells were seeded in a 6-well plate at a density of 1,000 cells per well. After 24h of incubation and attachment, the cells were irradiated with a 137Cesium γ-ray irradiator (Atomic Energy of Canada, Chalk River, ON, Canada) at a dose of 0, 2 and 4 Gy at a rate of 1 Gy/min. After 7 days, cell formed clumps that were visible to the naked eye (50 cells per clone). The cells were fixed in methanol for 15 min and then stained with Giemsa for 10 min. The number of foci was counted.
Funding Statement
This study was supported by the National Natural Science Foundation of China [31670859, 81572744]; CAMS Innovation Fund for Medical Science [2017-I2M-1-016]; Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences [2016ZX310068, 2016ZX310198, 2016RC310019, 2016RC310017]; PUMC Youth Fund and the Fundamental Research Funds for the Central Universities [3332016100,10023201601602]; Research Funds for the Innovation Team of IRM-CAMS [1650] (Q. L.). Work conducted at the Institute of Radiation Medicine was supported by the startup grant from the Chinese Academy of Medical Sciences (C.W.). This project is funded in part by the National Natural Science Foundation of China [81102006 (Y.T.)] and the Shandong Natural Science Foundation [Z2008C13].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Harris LN, You F, Schnitt SJ, et al. Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin Cancer Res. 2007. February 15;13(4):1198–1207. PubMed PMID: 17317830; eng. [DOI] [PubMed] [Google Scholar]
- [2].Wolff AC, Hammond ME, Schwartz JN, et al. American society of clinical oncology/college of American pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J clin oncol. 2007. January 1;25(1):118–145. PubMed PMID: 17159189; eng. [DOI] [PubMed] [Google Scholar]
- [3].Lee HJ, Seo AN, Kim EJ, et al. HER2 heterogeneity affects trastuzumab responses and survival in patients with HER2-positive metastatic breast cancer. Am J Clin Pathol. 2014;142(6):755–766. [DOI] [PubMed] [Google Scholar]
- [4].Axelson H, Fredlund E, Ovenberger M, et al. Hypoxia-induced dedifferentiation of tumor cells–a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol. 2005. Aug-Oct;16(4–5):554–563. PubMed PMID: 16144692; eng. [DOI] [PubMed] [Google Scholar]
- [5].Heppner GH. Tumor heterogeneity. Cancer Res. 1984. June;44(6):2259–2265. PubMed PMID: 6372991; eng. [PubMed] [Google Scholar]
- [6].Polyak K. Breast cancer: origins and evolution. J Clin Invest. 2007. November;117(11):3155–3163. PubMed PMID: 17975657; PubMed Central PMCID: PMCPMC2045618. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72(19):4875–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Janiszewska M, Polyak K. Clonal evolution in cancer: a tale of twisted twines. Cell Stem Cell. 2015. January 8;16(1):11–12. PubMed PMID: 25575078; eng. [DOI] [PubMed] [Google Scholar]
- [9].Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014. March 6;14(3):275–291. PubMed PMID: 24607403; eng. [DOI] [PubMed] [Google Scholar]
- [10].Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008. May 16;133(4):704–715. PubMed PMID: 18485877; PubMed Central PMCID: PMCPMC2728032. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chaffer CL, Brueckmann I, Scheel C, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA. 2011. May 10;108(19):7950–7955. PubMed PMID: 21498687; PubMed Central PMCID: PMCPMC3093533. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Verbeek BS, Adriaansen-Slot SS, Vroom TM, et al. Overexpression of EGFR and c-erbB2 causes enhanced cell migration in human breast cancer cells and NIH3T3 fibroblasts. FEBS Lett. 1998. March 20;425(1):145–150. PubMed PMID: 9541025; eng. [DOI] [PubMed] [Google Scholar]
- [13].Buckley NE, Forde C, McArt DG, et al. Quantification of HER2 heterogeneity in breast cancer-implications for identification of sub-dominant clones for personalised treatment. Sci Rep. 2016. March;21(6):23383 PubMed PMID: 26996207; PubMed Central PMCID: PMCPMC4800308. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Potts SJ, Krueger JS, Landis ND, et al. Evaluating tumor heterogeneity in immunohistochemistry-stained breast cancer tissue. Lab Invest. 2012. September;92(9):1342–1357. PubMed PMID: 22801299; eng. [DOI] [PubMed] [Google Scholar]
- [15].Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods (San Diego, Calif). 2003. July;30(3):256–268. PubMed PMID: 12798140; eng. [DOI] [PubMed] [Google Scholar]
- [16].Muthuswamy SK, Li D, Lelievre S, et al. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001. September;3(9):785–792. PubMed PMID: 11533657; PubMed Central PMCID: PMCPMC2952547. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kadota M, Yang HH, Gomez B, et al. Delineating genetic alterations for tumor progression in the MCF10A series of breast cancer cell lines. PloS one. 2010. February 15;5(2):e9201 PubMed PMID: 20169162; PubMed Central PMCID: PMCPMC2821407. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rew DA. Diseases of the breast, J.R. Harris et al. (Eds.); Lippincott Williams and Wilkins, Baltimore, 1999 - Surgical oncology. Surg Oncol. 1999;8(3):170–171. [Google Scholar]
- [19].Mimori-Kiyosue Y, Matsui C, Sasaki H, et al. Adenomatous polyposis coli (APC) protein regulates epithelial cell migration and morphogenesis via PDZ domain-based interactions with plasma membranes. Genes to Cells. 2007. February;12(2):219–233. PubMed PMID: 17295841; eng. [DOI] [PubMed] [Google Scholar]
- [20].Lue RA, Marfatia SM, Branton D, et al. Cloning and characterization of hdlg: the human homologue of the Drosophila discs large tumor suppressor binds to protein 4.1. Proc Natl Acad Sci USA. 1994. October 11;91(21):9818–9822. PubMed PMID: 7937897; PubMed Central PMCID: PMCPMC44908. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ju X, Katiyar S, Wang C, et al. Akt1 governs breast cancer progression in vivo. Proc Natl Acad Sci USA. 2007. May 1;104(18):7438–7443. PubMed PMID: 17460049; PubMed Central PMCID: PMCPMC1863437. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tiwari N, Gheldof A, Tatari M, et al. EMT as the ultimate survival mechanism of cancer cells. Semin Cancer Biol. 2012. June;22(3):194–207. PubMed PMID: 22406545; eng. [DOI] [PubMed] [Google Scholar]
- [23].Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006. September 1;66(17):8319–8326. PubMed PMID: 16951136; eng. [DOI] [PubMed] [Google Scholar]
- [24].Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer. 2016. February 24;15:18 PubMed PMID: PMC4765192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ioachim E, Charchanti A, Briasoulis E, et al. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. European J Cancer (Oxford, England: 1990). 2002. December;38(18):2362–2370. PubMed PMID: 12460779; eng. [DOI] [PubMed] [Google Scholar]
- [26].Alizadeh AA, Aranda V, Bardelli A, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21(8):846–853. PubMed PMID: PMC4785013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tepass U, Tanentzapf G, Ward R, et al. Epithelial cell polarity and cell junctions in Drosophila. Annu Rev Genet. 2001;35:747–784. PubMed PMID: 11700298; eng. [DOI] [PubMed] [Google Scholar]
- [28].Patil PU, D’Ambrosio J, Inge LJ, et al. Carcinoma cells induce lumen filling and EMT in epithelial cells through soluble E-cadherin-mediated activation of EGFR. J Cell Sci. 2015. December 1;128(23):4366–4379. PubMed PMID: 26483386; PubMed Central PMCID: PMCPMC4712814. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Melo SA, Sugimoto H, O’Connell JT, et al. Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell. 2014. November 10;26(5):707–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tryfonidis K, Zardavas D, Katzenellenbogen BS, et al. Endocrine treatment in breast cancer: cure, resistance and beyond. Cancer Treat Rev. 2016. November;50:68–81. PubMed PMID: 27643748; eng. [DOI] [PubMed] [Google Scholar]
- [31].Almendro V, Marusyk A, Fau - Polyak K, et al. Cellular heterogeneity and molecular evolution in cancer. (1553-4014 (Electronic)). eng Annu Rev Pathol Mech Dis. 2013;8: 277–302. [DOI] [PubMed] [Google Scholar]
- [32].Allinen M, Beroukhim R, Cai L, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004. July;6(1):17–32. PubMed PMID: 15261139; eng. [DOI] [PubMed] [Google Scholar]
- [33].Gao S, Sun Y, Zhang X, et al. IGFBP2 activates the NF-kappaB pathway to drive epithelial-mesenchymal transition and invasive character in pancreatic ductal adenocarcinoma. Cancer Res. 2016. November 15;76(22):6543–6554. PubMed PMID: 27659045; PubMed Central PMCID: PMCPMC5315491. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J Clin Investig. 2001;107(3):241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kyndi M, Sorensen FB, Knudsen H, et al. Estrogen receptor, progesterone receptor, HER-2, and response to postmastectomy radiotherapy in high-risk breast cancer: the Danish breast cancer cooperative group. J clin oncol. 2008. March 20;26(9):1419–1426. PubMed PMID: 18285604; eng. [DOI] [PubMed] [Google Scholar]
- [36].Krause M, Dubrovska A, Linge A, et al. Cancer stem cells: radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Deliv Rev. 2017. January;15(109):63–73. PubMed PMID: 26877102; eng. [DOI] [PubMed] [Google Scholar]
- [37].Duru N, Candas D, Jiang G, et al. Breast cancer adaptive resistance: HER2 and cancer stem cell repopulation in a heterogeneous tumor society. J Cancer Res Clin Oncol. 2014. August 30;140(1):1–14. PubMed PMID: PMC3889683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–313. PubMed PMID: 22258609; PubMed Central PMCID: PMCPMC3367003. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Willers H, Dahmdaphi J, Powell SN. Repair of radiation damage to DNA. Br J Cancer. 2004;90(7):1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jeggo PA, Lobrich M. How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem J. 2015. October 1;471(1):1–11. PubMed PMID: 26392571; eng. [DOI] [PubMed] [Google Scholar]
- [41].Yang YL, Fan Y, Lang RG, et al. Genetic heterogeneity of HER2 in breast cancer: impact on HER2 testing and its clinicopathologic significance. Breast Cancer Res Treat. 2012. August;134(3):1095–1102. PubMed PMID: 22476857; eng. [DOI] [PubMed] [Google Scholar]
- [42].Qie S, Chu C, Li W, et al. ErbB2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J Cell Biochem. 2014. March;115(3):498–509. PubMed PMID: 24122876; PubMed Central PMCID: PMCPMC4518873. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]