

Abstract
Objective:
Determine the effect of foetal haemoglobin (HbF) and α-thalassaemia on red blood cell (RBC) deformability of patients with sickle cell anaemia (SCA) with and without hydroxyurea (HU).
Methods:
Adult patients were enrolled in the Sickle Cell Program of the Cardeza Foundation (Thomas Jefferson University) and were followed-up prospectively during the period in which the Multicenter Study of Hydroxyurea (MSH) in patients with SCA was conducted. Ninety-one patients did not receive HU, 20 patients were enrolled in MSH and 10 patients were enrolled in an open label study of HU in SCA. Of the 20 patients enrolled in MSH, 11 took HU and 9 took placebo. Control group included 113 normal individuals. Red blood cell deformability index (DI) was measured by ektacytometry.
Results:
Patients with SCA taking HU (n=21) had higher DI than those taking placebo (n=9) or who were not taking this therapy (n=91). In patients without therapy, those with α-thalassaemia (n=31) had higher DI than those without. We showed a significant positive correlation between the level of HbF and DI. SCA patients without α-thalassaemia and HbF less than 10% (n=48) had lower DI than patients with α-thalassaemia and HbF < 10% (n=23) and patients with (n=8) or without α-thalassaemia but with HbF > 10% (n=12). DI measured in patients without α-thalassaemia and HbF > 10% was higher than in the three other subgroups.
Conclusion:
Elevated levels of HbF with or without HU and α-thalassemia improve sickle RBC rheology, which, in turn, improve the clinical picture of SCA.
Keywords: sickle cell anemia, hydroxyurea, red blood cell deformability, α-thalassemia
INTRODUCTION
Therapy of patients with sickle cell anaemia (SCA) with hydroxyurea (HU) improves their clinical and haematological characteristics (1–5). In the randomized, double-blind, placebo-controlled study of hydroxyurea in sickle cell anaemia (MSH), HU had a salutary effect on the clinical course of the disease in adults with three or more painful crises per year (3). Haematological effects of HU included increased Hb F synthesis, Hb levels, mean corpuscular volume (MCV), proportion of F cells, and red blood cell (RBC) hydration status (1–4). White-blood cell (WBC), platelet, reticulocyte and dense-cell counts were reduced in the patients who received HU (3). The purpose of this study was to determine the effects of HU, fetal hemoglobin (Hb F) and α-thalassaemia on the rheological properties of sickle RBC of patients with SCA. Improvement in RBC rheology after treatment with HU was described previously in two patients treated with HU (1) and more recently in an open-label, uncontrolled study from a single institution involving about 30 patients (6, 7). In this study we investigated the modulating role of HbF and α-thalassaemia on RBC deformability in patients with and without HU, with some patients enrolled in MSH.
PATIENTS AND METHODS
Patients.
Adult patients with SCA were enrolled in the Sickle Cell Program of the Cardeza Foundation of Thomas Jefferson University and were followed-up prospectively from 1992 through 1995, the period during which the MSH study was conducted. All patients were adult African Americans. Hematologic and rheological data were performed when the patients were in their usual asymptomatic steady state in the outpatient clinic. Definitions of steady state were as described previously (8). The study was approved by the IRB and written consent was obtained from all patients. The distribution of the patients studied was as follows: 91 patients did not receive HU, 20 patients were enrolled in MSH and 10 patients were enrolled in an open label study of HU in SCA. Of the 20 patients enrolled in MSH, 11 took HU and 9 took placebo; and of the 11 patients who took HU in MSH, 3 were non-responders. A control group mainly composed of non African Americans without acute or chronic disease (n = 113) was used for the measurement of RBC deformability only.
Haematological data.
Haemoglobin (Hb), haematocrit (Hct) white blood cell (WBC) and platelets (PLT) count, RBC indices (mean cell volume, MCV; mean corpuscular haemoglobin concentration, MCHC; red blood cell distribution width, RDW, RET; percentages of reticulocytes) were measured by routine methods. Hb F was determined by alkaline denaturation (9). For the patients enrolled in MSH, blood samples were collected during clinic visits for serial measurements of Hb F and F cells (RBC containing Hb F). For technical reasons, serial Hb F measurements could not be completed and, therefore, F cells were used as a surrogate measure to estimate Hb F, as described by Steinberg et al (10). The definition of non-responders was those patients in whom HU did not decrease the frequency of VOCs as described in MSH (3).
Measurement of RBC deformability.
The ektacytometer, a viscodiffractometer, designed and previously described by Bessis and Mohandas (11, 12), was used to measure whole cell deformability in room air as a continuous function of the suspending medium osmolality at a constant applied shear stress of 170 dynes/cm2 (osmotic gradient ektacytometry). For these studies, the deformability index (DI) of RBC was continuously recorded as the suspending medium osmolality was increased from 50 to 500 mosmol/kg as previously described (1). The DI values at 290 mosmol/kg were used for further analyses.
Determination of a-Globin Genotypes.
α-Globin genotypes were determined by Southern blot hybridization (13) of genomic DNA extracted from peripheral blood leukocytes. The restriction endonucleases used were Bam HI and Bgl 11, and the probe was Bam H 1 -linearized α-cDNA JW 10 1 plasmid (14), which was labeled with 32P by nick translation.
Statistical methods.
Unpaired student t test was used for comparisons between two groups. A one-way analysis of variance (ANOVA) for comparisons with more than two groups and post-hoc tests (Newman-Keuls) were used, when appropriate, to locate where differences occurred. Pearson correlation was done between Hb F and RBC deformability. A multivariate analysis was performed to test the independent contribution of HbF level and alpha-thalassaemic status to DI, after excluding patients under HU therapy. Significance level was defined as p<0.05. Analyses were conducted using SPSS (v. 20, IBM SPSS Statistics, Chicago, IL) and data were reported as mean ± SD (minimum-maximum value).
RESULTS
DI and HU.
Hematological values of the patients studied are reported in table 1. HbF and RDW were, respectively, higher and lower in patients taking HU treatment, with or without excluding non-responders, in comparison with patients not receiving HU. Hb, Hct and MCV levels were higher in patients receiving HU treatment, with or without excluding non-responders, compared to patients without HU and patients receiving placebo. The proportion of patients with HbF level above 10% was not different between the different groups: 20/91 in the group without HU, 6/21 in patients treated with HU (6/18 after excluding non-responders) and 2/9 in the placebo group. α-thalassaemia frequency was comparable between the different groups: 31/91 in the group not treated by HU (6 with 2 α-genes deleted), 6/21 in the HU group (6/18 after excluding non responders) and 1/9 in the placebo group. Figure 1 demonstrated that DI at 290 mosmol/kg was higher in SCA patients taking HU compared to SCA patients under placebo or not taking HU. The control group exhibited higher DI compared to the three other groups.
Table 1:
Effects of HU on haematological parameters
| Age (yrs) | HbF (%) | MCHC (g/dL) | MCV (fl) | RDW (%) | Hb (g/dL) | Hct (%) | Retic (%) | WBC (109/L) | PLT (109/L) | |
|---|---|---|---|---|---|---|---|---|---|---|
| SCA without HU (n=91) | 30 ± 11 (16-69) | 6.4 ± 4.3 (1.0-21.0) | 31.5 ± 1.9 (24.8-34.7) | 90.2 ± 9.8 (67.2-109.2) | 22.4 ± 3.1 (16.5-33.1) | 7.9 ± 1.5 (4.1-11.5) | 24.4 ± 4.8 (12.8-36.1) | 14.6 ± 7.9 (3.2-46.4) | 13.2 ± 3.6 (6.9-24.4) | 451 ± 156 (153-793) |
| SCA with HU including non responders (n=21) | 33 ± 9 (22-61) | 11.0 ± 8.3 (1.3-26.9) * | 28.8 ± 2.2 (24.0-33.0) *** †† | 111.1 ± 21.0 (84.5-158.8) *** †† | 20.1 ± 3.4 (12.6-27.2) * | 9.0 ± 1.3 (7.1-11.5) * † | 27.8 ± 4.3 (22.3-35.4) * † | 10.3 ±6.0 (1.9-22.9) | 11.8 ± 5.6 (5.9-23.8) | 396 ± 123 (131-533) |
| SCA with HU excluding non responders (n=18) | 33 ± 9 (22-61) | 14.2 ± 7.5 (4.2-26.9) *** | 29.0 ± 2.4 (24.0-33.0) *** †† | 115.3 ± 21.3 (84.5-158.8) *** ††† | 19.4 ± 3.1 (12.6-25.7) ** | 9.2 ± 1.4 (7.1-11.5) * † | 28.6 ± 4.6 (22.3-35.4) * † | 10.4 ± 6.4 (1.9-22.9) | 9.9 ± 4.1 (5.9-16.9) | 383 ± 152 (131-533) |
| SCA with placebo (n=9) | 28 ± 3 19-37 | 8.1 ± 3.6 (5.5-10.6) | 31.7 ± 2.3 (27.7-33.6) | 91.5 ± 10.9 (76.7-110.8) | 22.3 ± 2.7 (19.7-26.8) | 6.8 ± 1.4 (5.8-7.8) | 20.3 ± 4.1 (17.4-23.2) | 19.0 ± 6.2 (14.6-23.4) | 15.3 ± 5.8 (11.2-19.4) | 308 ± 88 (246-370) |
Different from SCA without HU:
p < 0.05;
p < 0.01;
p < 0.001.
Different from SCA with placebo
p < 0.05;
p < 0.01;
p < 0.001.
Figure 1:
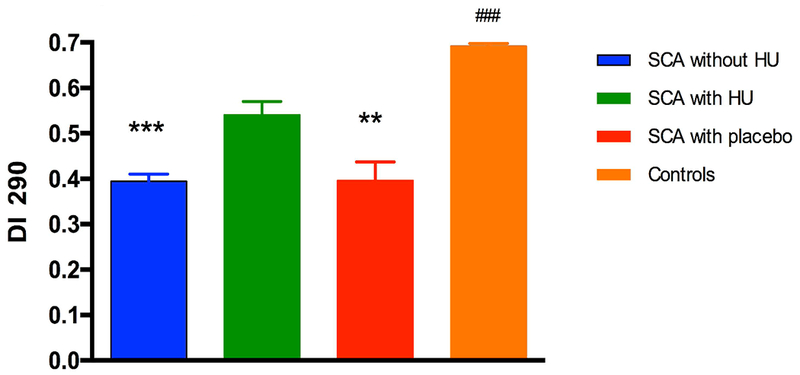
Effects of Hydroyurea (HU) on RBC deformability measured at 290 mmosmol/l (DI 290). SCA without HU group: n = 91; SCA with HU group: n = 21; SCA with placebo group: n = 9; Control group: n = 113. Different from the SCA with HU group: **p < 0.01; ***p < 0.001. Different from the three SCA groups: ###p < 0.001.
DI, Hb F and α-thalassaemia.
Figure 2 showed the significant and positive correlation between DI measured at 290 mosmol/kg and HbF level in all SCA participants, including those with HU. This association was still significant after excluding patients under HU treatment (data not show; r = 0.34, p < 0.01). The comparison between patients with or without α-thalassaemia, after excluding those taking HU, demonstrated higher DI in the former group (Figure 3). Among α-thalassaemic patients, 6 had 2 α-genes deleted and, although the mean value of this subgroup was close to the mean value of patients with only one α-gene deleted, the sample size was considered to be too small to analyze this sub-group separately. In order to determine if the effects of Hb F and α-thalassaemia on RBC deformability were independent of each other or not, we arbitrarily divided the patients into two groups: those with Hb F > 10% and those with Hb F < 10% (Figure 4), as was previously described (15). Our results demonstrated that SCA patients without α-thalassaemia and HbF less than 10% had lower DI than patients with α-thalassaemia and HbF < 10% and patients with or without α-thalassaemia but with HbF > 10%. Finally, DI measured in patients without α-thalassaemia and HbF > 10% was higher than in the three other subgroups. The multivariate analysis including both raw values of HbF and the alpha-thalassaemic status (i.e., presence or not), demonstrated an independent association of these two biological parameters (p < 0.001 and p < 0.05 for HbF and alpha-thalassaemia, respectively) with DI.
Figure 2:
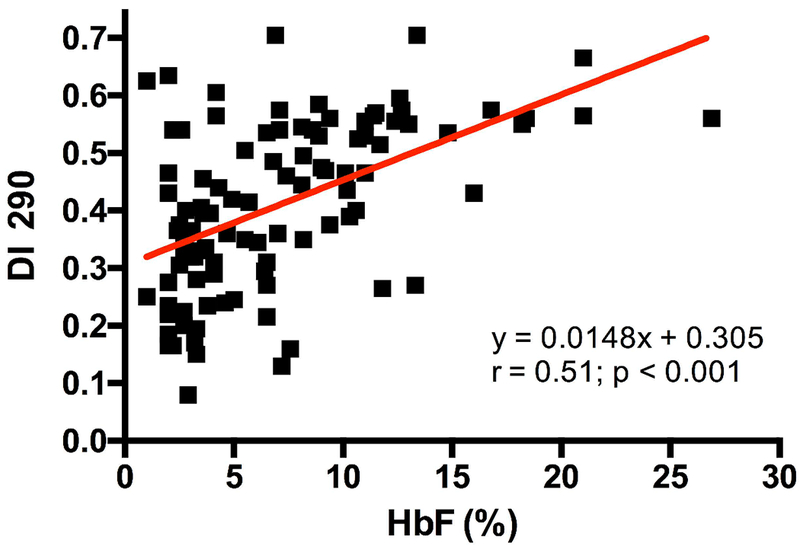
Association between RBC deformability measured at 290 mmosmol/l (DI 290) and HbF level in all SCA patients
Figure 3:
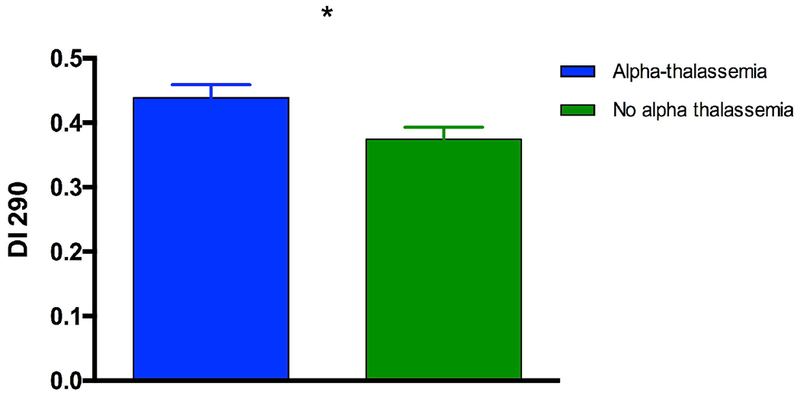
Effects of α-thalassemia on RBC deformability measured at 290 mmosmol/l (DI 290) after excluding those under HU therapy. Alpha-thalassaemic group: n = 31; No alpha-thalassaemia group: n = 60. Difference between groups: *p < 0.05.
Figure 4:
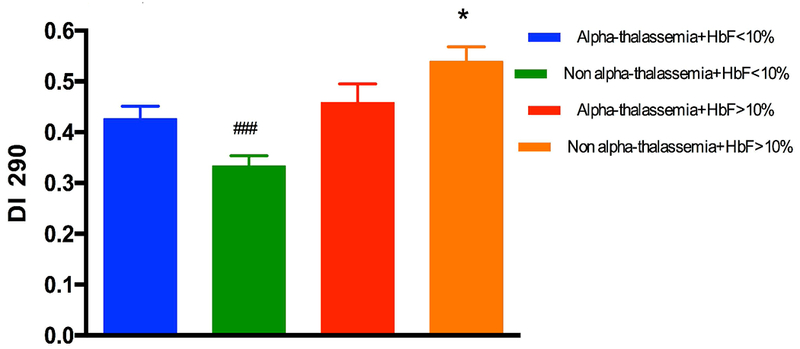
Effects of α-thalassemia and HbF level on RBC deformability measured at 290 mmosmol/l (DI 290) after excluding those under HU therapy. SCA patients with alpha-thalassaemia and HbF < 10%: n = 23; SCA patients without alpha-thalassaemia and HbF < 10%: n = 48; SCA patients with alpha-thalassaemia and HbF > 10%: n = 8; SCA patients without alpha-thalassaemia and HbF > 10%: n = 12. ###Different from the three other groups (p < 0.001); *Different from the three other groups (p < 0.001).
DISCUSSION
The clinical manifestations of sickle cell disease vary latitudinally among patients and longitudinally in the same patient over time. Some patients with SCA have mild disease, whereas others suffer from a severe form and die at a relatively young age. Age, α-thalassaemia, Hb F level, iron overload, milder anaemia, lower lactate dehydrogenase level, and higher tricuspid regurgitation velocity were associated with greater frequency of severe VOCs (16–18). In addition, the rheology of sickle blood plays an important role in the pathogenesis of vaso-occlusion (19–21). The rheology of blood, in turn, is influenced by several factors including, but not limited, to plasma viscosity, cell hemoglobin concentration, hematocrit, RBC membrane mechanical properties, surface area/volume ratio, Hb F, co-existent genetic factors such as α-thalassaemia, etc. (20, 21). In this study our focus has been on the effect of Hb F, whether natural or induced by hydroxyurea, and α-thalassaemia on the rheological properties of sickle erythrocytes.
It is well known that a high level of Hb F, and its pancellular distribution, is associated with milder clinical picture of SCA. Patients from eastern Saudi Arabia with markedly elevated Hb F (18%–25%) had a relatively mild disease course (22, 23). Even small increments in increased Hb F level have an ameliorative effect on the frequency of VOCs and may ultimately improve survival. High levels of Hb F may decrease the severity of certain sickle cell syndromes because Hb F inhibits polymerization of Hb S during the sickling process. In vitro polymer studies of Hb S and Hb F demonstrated that FS hybrids are excluded from nuclei formation and possibly from the sickling process in vivo (24, 25). Moreover, inhibition of Hb S polymerization improves or normalizes the deformability of sickle RBC as shown in this study.
The salutary effect of Hb F on the clinical picture of SCA and on the rheology of sickle RBCs paved the way for the identification of hydroxyurea (HU) as a pharmacologic agent that increases the synthesis of Hb F in vivo. Hydroxyurea (HU), also known as hydroxycarbamide, has effectively decreased the frequency of VOCs (3) and improved the rheological characteristics of sickle RBCs as shown in this study and others (6, 7). However, there are patients with very low Hb F levels (<2%) and mild disease and patients with high Hb F levels (>20%) and severe disease. In addition, at least 25% of patients do not respond to HU therapy. The incidence of noncompliance and lack of response to hydroxyurea are not well studied and vary among institutions. Moreover, the definition of response depends on HbF level, MCV, leukocyte count, frequency of VOCs, etc (18). The correlation between DI and HbF was moderate (r=0.51 with all subjects and r=0.34 excluding those on hydroxyurea), which indicates that HbF level is not the only determinant of RBC deformability in SCA. Other factors such as, reticulocytes count, mean cell volume, etc., must be involved in determining the final outcome of the clinical picture and of the rheology of sickle RBCs in certain patients with SCA. Alpha thalassaemia is considered a possible such other factor and our results clearly demonstrated that both HbF level and alpha-thalassaemia play an independent role on RBC deformability.
The effects of α-thalassaemia on the rheologic properties of sickle RBCs are most apparent in patients homozygous for α-thalassemia (−α/−α) and intermediate for heterozygotes (−α/αα). This combination results in better cell deformability, less cellular dehydration, less dense cells, less irreversibly sickled cells (ISCs), increased whole-blood viscosity, and possibly less propensity to adhere to the endothelium than in patients with SCA and four α-genes. Although it was not possible to differentiate the effects of homozygous and heterozygous alpha-thalassaemia on DI in this study (the number of patients with 2 alpha genes deletes was too small), deformability of sickle RBCs from subjects with coexistent α-thalassemia is better than that from non-thalassemic patients with SCA as shown in this and other studies (20, 21, 26–28). Subjects with SCA and α-thalassaemia have less (ISCs) and dense cells in their peripheral blood than non-thalassemic patients. Moreover, the effect of α-thalassaemia in response to HU therapy was recently described in the Multicenter Study of Hydroxyurea (MSH) cohort (29). Briefly, this study found that HU decreases the frequency of VOCs in SCA patients with and without α-thalassaemia, and the degree of VOC reduction was more pronounced in the patients with alpha-thalassaemia. Thus, changes in standard laboratory parameters such as MCV and HbF percent remain useful in monitoring HU therapy in the presence of α-thalassaemia despite lower baseline values.
Sickle RBCs are mechanically fragile, and dense sickle RBCs are particularly susceptible to mechanical damage, probably because of decreased membrane stability (30). Because patients with SS and α-thalassaemia have decreased numbers of dense cells, it is likely that their RBCs are less mechanically fragile. This, in turn, may be a contributing factor to the decreased hemolytic rate observed in these individuals.
CONCLUSION
This study shows that elevated levels of Hb F with or without HU and α-thalassemia improve the rheology of sickle RBCs which, in turn, improve the clinical picture of SCA. The limitation of the study is the small number of patients with α-thalassemia.
ACKNOWLEDGMENTS
This study was supported in part by the NIH Multicenter Study of Hydroxyurea in Sickle Cell Anemia Grant HL 45692 and in part by the NIH Comprehensive Sickle Cell Center Grant HL38632 and in part by the Sickle Cell Program of the Commonwealth of Pennsylvania.
APPENDIX
The following institutions and investigators participated in the Multicenter Study of Hydroxyurea in Sickle Cell Anemia:
Clinical Centers (the numbers in parentheses are the numbers of patients enrolled at each center) — University of North Carolina, Chapel Hill (19): E. Orringer, S. Jones, and D. Strayhorn; Duke University, Durham, N.C. (16): W. Rosse, G. Phillips,* D. Peace, and A. Johnson-Telfair; Medical College of Georgia, Augusta (15): P. Milner, A. Kutlar, and A. Tracy; Thomas Jefferson University, Philadelphia (21): S.K. Ballas, G.E. Allen, J. Moshang, and B. Scott; University of Mississippi, Jackson (19): M. Steinberg, A. Anderson, and V. Sabahi; University of Miami, Miami (12): C. Pegelow, D. Temple, E. Case, R. Harrell, and S. Childerie; San Francisco General Hospital, San Francisco (6): S. Embury, B. Schmidt, and D. Davies; University of Illinois, Chicago (57): M. Koshy, N. Talischy-Zahed, L. Dorn, G. Pendarvis, and M. McGee; Michael Reese Hospital, Chicago (11): M. Telfer and A. Davis; Howard University, Washington, D.C. (20): O. Castro, H. Finke, E. Perlin, and J. Siteman; University of Medicine and Dentistry of New Jersey, Newark (10): P. Gascon, P. di Paolo, and S. Gargiulo; Emory University, Atlanta (14): J. Eckman, J.H. Bailey, A. Platt, and L. Waller; St. Luke’s– Roosevelt Medical Center, New York (18): G. Ramirez, V. Knors, S. Hernandez, E.M. Rodriguez, and E. Wilkes; Children’s Hospital of Oakland, Oakland, Calif. (5): E. Vichinsky, S. Claster, A. Earles, K. Kleman, and K. McLaughlin; Medical College of Virginia, Richmond (19): P. Swerdlow, W. Smith, B. Maddox, L. Usry, A. Brenner, K. Williams, R. O’Brien, and K. Genther; Case Western Reserve University, Cleveland (5): S. Shurin, B. Berman, K. Chiarucci, and L. Keverline; Hospital for Sick Children, Toronto (6): N. Olivieri, D. Shaw, and N. Lewis; Brigham and Women’s Hospital, Boston (5): K. Bridges, B. Tynan, and C. Winograd; Interfaith Medical Center, Brooklyn, N.Y. (8): R. Bellevue, H. Dosik, M. Sheikhai, P. Ryans, and H. Souffrant; University of Alabama, Birmingham (8): J. Prchal, J. Braddock, and T. McArdle; and University of Pittsburgh, Pittsburgh (5): T. Carlos, A. Schmotzer, and D. Gardner.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Ballas SK, Dover GJ, Charache S. Effect of hydroxyurea on the rheological properties of sickle erythrocytes in vivo. Am J Hematol. 1989; 32(2): 104–11. [DOI] [PubMed] [Google Scholar]
- 2.Charache S, Dover GJ, Moore RD, Eckert S, Ballas SK, Koshy M, Milner PF, Orringer EP, Phillips G Jr., Platt OS, et al. Hydroxyurea: effects on hemoglobin F production in patients with sickle cell anemia. Blood. 1992; 79(10): 2555–65. [PubMed] [Google Scholar]
- 3.Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995; 332(20): 1317–22. [DOI] [PubMed] [Google Scholar]
- 4.Orringer EP, Blythe DS, Johnson AE, Phillips G Jr., Dover GJ, Parker JC. Effects of hydroxyurea on hemoglobin F and water content in the red blood cells of dogs and of patients with sickle cell anemia. Blood. 1991; 78(1): 212–6. [PubMed] [Google Scholar]
- 5.Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest. 1984; 74(2): 652–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemonne N, Charlot K, Waltz X, Ballas SK, Lamarre Y, Lee K, Hierso R, Connes C, Etienne-Julan M, Romana M, Connes P. Hydroxyurea treatment does not increase blood viscosity and improves red blood cell rheology in sickle cell anemia. Haematologica. 2015; 100(10): e383–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemonne N, Mockesch B, Charlot K, Garnier Y, Waltz X, Lamarre Y, Antoine-Jonville S, Etienne-Julan M, Hardy-Dessources MD, Romana M, Connes P. Effects of hydroxyurea on blood rheology in sickle cell anemia: A two-years follow-up study. Clin Hemorheol Microcirc. 2017; 67(2): 141–8. [DOI] [PubMed] [Google Scholar]
- 8.Ballas SK. More definitions in sickle cell disease: steady state v base line data. Am J Hematol. 2012; 87(3): 338. [DOI] [PubMed] [Google Scholar]
- 9.Betke K, Marti HR, Schlicht I. Estimation of small percentages of foetal haemoglobin. Nature. 1959; 184(Suppl 24): 1877–8. [DOI] [PubMed] [Google Scholar]
- 10.Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S, Dover GJ. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter Study of Hydroxyurea. Blood. 1997; 89(3): 1078–88. [PubMed] [Google Scholar]
- 11.Bessis M, Mohandas N, Feo C. Automated ektacytometry: a new method of measuring red cell deformability and red cell indices. Blood Cells. 1980; 6(3): 315–27. [PubMed] [Google Scholar]
- 12.Groner W, Mohandas N, Bessis M. New optical technique for measuring erythrocyte deformability with the ektacytometer. Clin Chem. 1980; 26(10): 1435–42. [PubMed] [Google Scholar]
- 13.Poncz M, Solowiejczyk D, Harpel B, Mory Y, Schwartz E, Surrey S. Construction of human gene libraries from small amounts of peripheral blood: analysis of beta-like globin genes. Hemoglobin. 1982; 6(1): 27–36. [DOI] [PubMed] [Google Scholar]
- 14.Ballas SK, Larner J, Smith ED, Surrey S, Schwartz E, Rappaport EF. The xerocytosis of Hb SC disease. Blood. 1987; 69(1): 124–30. [PubMed] [Google Scholar]
- 15.Ballas SK, Marcolina MJ, Dover GJ, Barton FB. Erythropoietic activity in patients with sickle cell anaemia before and after treatment with hydroxyurea. Br J Haematol. 1999; 105(2): 491–6. [PubMed] [Google Scholar]
- 16.Darbari DS, Onyekwere O, Nouraie M, Minniti CP, Luchtman-Jones L, Rana S, Sable C, Ensing G, Dham N, Campbell A, Arteta M, Gladwin MT, Castro O, Taylor JGt, Kato GJ, Gordeuk V. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia. J Pediatr. 2012; 160(2): 286–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Machado RF, Mack AK, Martyr S, Barnett C, Macarthur P, Sachdev V, Ernst I, Hunter LA, Coles WA, Nichols JP, Kato GJ, Gladwin MT. Severity of pulmonary hypertension during vaso-occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol. 2007; 136(2): 319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014; 312(10): 1033–48. [DOI] [PubMed] [Google Scholar]
- 19.Connes P Editorial: Special issue on sickle cell disease: old and new concepts. Clin Hemorheol Microcirc. In press. [DOI] [PubMed] [Google Scholar]
- 20.Connes P, Alexy T, Detterich J, Romana M, Hardy-Dessources MD, Ballas SK. The role of blood rheology in sickle cell disease. Blood Rev. 2016; 30(2): 111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Connes P, Renoux C, Romana M, Abkarian M, Joly P, Martin C, Hardy-Dessources MD, Ballas SK. Blood rheological abnormalities in sickle cell anemia. Clin Hemorheol Microcirc. In press. [DOI] [PubMed] [Google Scholar]
- 22.Brown MJ, Weatherall DJ, Clegg JB, Perrine RP. Benign sickle-cell anaemia. Br J Haematol. 1972; 22(5): 635. [PubMed] [Google Scholar]
- 23.Perrine RP, Pembrey ME, John P, Perrine S, Shoup F. Natural history of sickle cell anemia in Saudi Arabs. A study of 270 subjects. Ann Intern Med. 1978; 88(1): 1–6. [DOI] [PubMed] [Google Scholar]
- 24.Nagel RL, Bookchin RM, Johnson J, Labie D, Wajcman H, Isaac-Sodeye WA, Honig GR, Schiliro G, Crookston JH, Matsutomo K. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A. 1979; 76(2): 670–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nibu K, Adachi K. Effect of FS (alpha 2 gamma beta s) hybrid hemoglobin on Hb S nucleation and aggregation. Biochim Biophys Acta. 1985; 829(1): 97–102. [DOI] [PubMed] [Google Scholar]
- 26.Ballas SK, Larner J, Smith ED, Surrey S, Schwartz E, Rappaport EF. Rheologic predictors of the severity of the painful sickle cell crisis. Blood. 1988; 72(4): 1216–23. [PubMed] [Google Scholar]
- 27.Embury SH, Clark MR, Monroy G, Mohandas N. Concurrent sickle cell anemia and alpha-thalassemia. Effect on pathological properties of sickle erythrocytes. J Clin Invest. 1984; 73(1): 116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Renoux C, Connes P, Nader E, Skinner S, Faes C, Petras M, Bertrand Y, Garnier N, Cuzzubbo D, Divialle-Doumdo L, Kebaili K, Renard C, Gauthier A, Etienne-Julan M, Cannas G, Martin C, Hardy-Dessources MD, Pialoux V, Romana M, Joly P. Alpha-thalassaemia promotes frequent vaso-occlusive crises in children with sickle cell anaemia through haemorheological changes. Pediatr Blood Cancer. 2017; 64(8). [DOI] [PubMed] [Google Scholar]
- 29.Darbari DS, Nouraie M, Taylor JG, Brugnara C, Castro O, Ballas SK. Alpha-thalassaemia and response to hydroxyurea in sickle cell anaemia. Eur J Haematol. 2014; 92(4): 341–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messmann R, Gannon S, Sarnaik S, Johnson RM. Mechanical properties of sickle cell membranes. Blood. 1990; 75(8): 1711–7. [PubMed] [Google Scholar]