

Abstract
Sheep colibacillosis is one of the most common bacterial diseases found at large-scale sheep farms. The aim of this study was to employ RNA-seq to screen differentially expressed (DE) long non-coding RNAs (lncRNAs) that impart antagonistic or sensitive effects on Escherichia coli F17. In this study, individuals who had antagonistic or sensitive responses to E. coli F17 were identified by feeding E. coli F17 strains to Hu lambs. The sensitive group had higher levels of intestinal bacteria than that in the antagonistic group (P < 0.05), the jejunum showed various levels of mucosal tissue damage and had a dark colour, and disintegration of part of the small intestinal villi was observed. Totals of 34 DE lncRNAs and 703 DE mRNAs in two groups were identified. qRT-PCR results for 12 randomly selected DE lncRNAs and DE mRNAs were consistent with the RNA-seq data. Gene Ontology (GO), KEGG Pathway enrichment and lncRNA-mRNA interaction analyses identified 6 co-expressed genes, namely, MYO1G, TIMM29, CARM1, ADGRB1, SEPT4, and DESI2. This is the first study that has performed expression profiling of lncRNAs in the spleen of antagonistic and sensitive lambs. The identification of DE lncRNAs can facilitate investigations into the molecular mechanism underlying resistance to diarrhoea in sheep.
Introduction
Sheep colibacillosis is one of the most common bacterial diseases found at large-scale sheep farms. The traditional method of controlling the bacterial disease is by antibiotic therapy, although this approach also has several disadvantages. Detecting the expression of antagonistic genes in sheep colibacillosis provides information that may facilitate the elucidation of the molecular mechanism underlying disease resistance to Escherichia coli. In 1966, Orskov et al.1 first reported the porcine E. coli adhesion antigen K88, which is an episome-determined antigen2. In addition, the morphology of the pilus situated on the surface of the bacteria has been examined by electron microscopy. To date, numerous animal-derived enterotoxigenic Escherichia coli (ETEC) pili have been identified, including K88, K99, 987P, F17 and F41, which are all vital virulence factors. Long non-coding RNAs (lncRNAs) are a class of non-coding RNAs longer than 200 nucleotides. Several studies have revealed that lncRNAs are closely related to the development of human tumours, cardiovascular diseases, and metabolic diseases. Furthermore, there is growing evidence that lncRNAs play a significant regulatory role in anti-viral and other natural immune responses3–7. However, research on the function of lncRNAs has mainly focused on the regulation of muscle growth, testicular development, hair follicle development, and other traits in sheep, which are less well studied compared to humans8–10. A few studies have recently focused on disease prevention and control in sheep11,12, including disease resistance, yet our understanding of its underlying molecular mechanism is limited. In the present study, the expression levels of lncRNAs in two sheep spleen phenotypes, antagonistic or sensitive to pili of E. coli F17, were assessed by RNA-seq, and key lncRNAs were investigated by target prediction and functional annotation analysis based on the cis mechanism, a transcription activation and expression control method for adjacent mRNAs by non-coding RNAs. The results were further verified by q-PCR. Our results provide a deeper understanding of sheep antagonism to E. coli F17 in terms of lncRNAs, facilitate in the identification of additional functional genes that are antagonistic to E. coli F17, and provide a theoretical basis for solving key issues that are related to breeding indigenous disease-resistant sheep in China.
Results
Histological observation and comparison of the number of intestinal bacteria in the antagonistic and sensitive groups
Based on faecal morphology13, the experimental subjects were divided into the antagonistic group (12p, 13p, 14p) and the sensitive group (15p, 16p, 17p). In the sensitive groups, the bacterial count was arranged from 4.7*108 to 1.9*109, the mean of the bacterial count was 1.22*109, and yet the count dropped 5.1*106 to 9.0*107 in the antagonistic groups, the mean of the bacterial count was 3.37*107 (Table 1). Compared to the antagonistic group, the number of intestinal bacteria in the sensitive group was significantly higher (P < 0.05, Fig. 1). The jejunum mucosal tissues of lambs in the sensitive group were damaged, dull in colour, lysed, and exhibited large lacuna that could be observed at the submucosal layer. The intestinal villi had disintegrated and were highly vascularized (i.e., capillaries), and the intestinal mucosa showed severe damage, thus making it difficult to prepare histological sections for assessment (Fig. 2).
Table 1.
Comparison of the number of intestinal bacteria between the antagonistic and sensitive lambs.
| Group | Intestinal tract | Dilution factor | Bacterial count (CFU·mL−1) | |||
|---|---|---|---|---|---|---|
| 105 | 106 | 107 | 108 | |||
| Antagonistic groups | Duodenum | 70 | 6 | ng | ng | 6.0 × 106 |
| Jejunum | 51 | ng | ng | ng | 5.1 × 106 | |
| Ileum | >500 | 170 | 9 | ng | 9.0 × 107 | |
| Sensitive group | Duodenum | >1,000 | >500 | 128 | 13 | 1.3 × 109 |
| Jejunum | >1,000 | 88 | 47 | ng | 4.7 × 108 | |
| Ileum | >1,000 | >500 | 176 | 19 | 1.9 × 109 | |
Note: “ng” means “no growth”.
Figure 1.
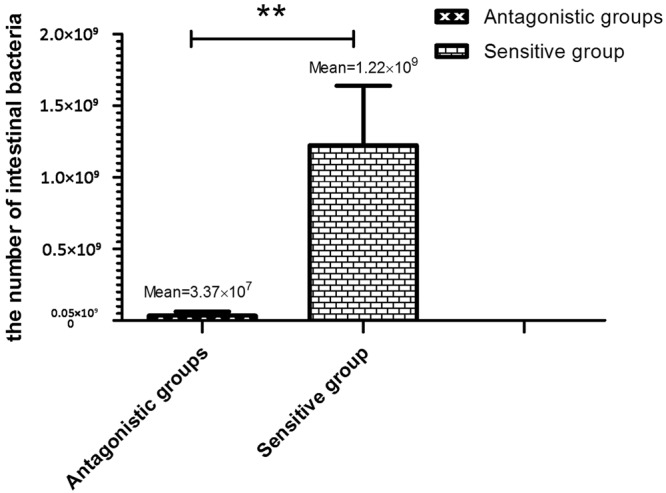
Difference analysis of the intestinal bacteria in antagonistic and sensitive lambs.
Figure 2.

Histological assessment of the jejunum of the antagonistic (A1, A2) and sensitive (B1, B2) groups. (A1 and B1) are 200x microscope observations, (A2 and B2) are 400x microscope observations.
Summary of RNA sequencing of spleens in sheep
cDNA libraries of the lamb spleens from the antagonistic and sensitive groups were constructed. Sequencing was performed using the Illumina HiSeq 2500 platform. The antagonistic and sensitive groups generated a total of 354,943,820 and 370,616,990 raw reads, with GC contents of 48.33% and 49.67%, respectively. The valid reads in the clean reads were mapped to the O. aries v4.0 reference genome, and more than 73.5% of the reads were mapped to the genome. The reads mapped to multiple locations of the reference sequence were less than 4.5% and more than 70% of the reads were uniquely mapped to the reference sequence. Approximately 35% of the reads mapped to the positive and negative chains of the genome. In addition, the number of reads that were mapped to the exonic regions (~60%) was higher than those that were mapped to intergenic and intron regions by annotation analysis. These results indicate that the matching efficiency of our de novo assembly is high, and most reads mapped to the exonic region (Table 2).
Table 2.
Read statistics of the reference genome.
| Sample | Sample_12p | Sample_13p | Sample_14p | Sample_15P | Sample_16p | Sample_17P |
|---|---|---|---|---|---|---|
| Raw reads | 135,067,560 | 100,031,546 | 119,844,714 | 110,990,304 | 122,657,326 | 136,969,360 |
| Total reads | 101,734,214 | 76,072,598 | 82,901,000 | 82,165,530 | 88,819,554 | 93,869,100 |
| Total number of reads mapped | 78,625,843 (77.29%) | 58,935,848 (77.47%) | 62,008,068 (74.80%) | 60,455,546 (73.58%) | 67,337,265 (75.81%) | 69,827,122 (74.39%) |
| Number of reads that mapped to multiple regions | 3,792,793 (3.73%) | 2,606,663 (3.43%) | 3,322,008 (4.01%) | 3,470,863 (4.22%) | 3,937,078 (4.43%) | 3,997,927 (4.26%) |
| Number of reads that were mapped to unique regions | 74,833,050 (73.56%) | 56,329,185 (74.05%) | 58,686,060 (70.79%) | 56,984,683 (69.35%) | 63,400,187 (71.38%) | 65,829,195 (70.13%) |
| Read-1 | 38,037,308 (37.39%) | 286,21,024 (37.62%) | 29,752,817 (35.89%) | 28,436,715 (34.61%) | 32,177,909 (36.23%) | 33,403,729 (35.59%) |
| Read-2 | 36,795,742 (36.17%) | 27,708,161 (36.42%) | 28,933,243 (34.90%) | 28,547,968 (34.74%) | 31,222,278 (35.15%) | 32,425,466 (34.54%) |
| Number of reads that map to ‘+’ | 37,435,639 (36.80%) | 28,177,639 (37.04%) | 29,369,825 (35.43%) | 28,554,241 (34.75%) | 31,731,741 (35.73%) | 32,953,615 (35.11%) |
| Number of reads map to ‘−’ | 37,397,411 (36.76%) | 28,151,546 (37.01%) | 29,16,235 (35.36%) | 28,430,442 (34.60%) | 31,668,446 (35.65%) | 32,875,580 (35.02%) |
| Number of non-splice reads | 65,956,768 (64.83%) | 50,126,819 (65.89%) | 50,492,679 (60.91%) | 48,280,120 (58.76%) | 54,077,425 (60.88%) | 56,555,536 (60.25%) |
| Number of splice reads | 8,876,282 (8.72%) | 6,202,366 (8.15%) | 8,193,381 (9.88%) | 8,704,563 (10.59%) | 9,322,762 (10.50%) | 9,273659 (9.88%) |
| Number of reads mapped in proper pairs | 66,749,602 (65.61%) | 50,161,962 (65.94%) | 51,970,094 (62.69%) | 51,198,774 (62.31%) | 56,791,332 (63.94%) | 58,971,888 (62.82%) |
Identification of transcripts in sheep spleens
After mapping the reference sequence, we identified 1,988 lncRNAs and 38,843 mRNAs from 42,460 compiled transcripts. The length of the lncRNAs was mainly distributed within the range of 200 bp-5,000 bp (Fig. 3a), and the average length was 2,124 bp. Additionally, the lncRNA types mainly include intergenic lncRNAs (character u) (Fig. 3b)and intronic lncRNAs (character i), containing 2 to 3 exons (Fig. 3c).
Figure 3.

Summary of the lengths, types, and number of exons of the predicted lncRNAs.
Analysis and validation of DE transcripts
The expression levels of lncRNA and mRNA transcripts were estimated using the FPKM values. We found that the expression level of the lncRNA transcripts was relatively low (Fig. 4). A total of 14 upregulated and 20 downregulated DE lncRNAs and 370 upregulated and 333 downregulated DE mRNAs were screened under conditions of P < 0.05 and |log2 (fold change)| > 1 (Fig. 5). To further verify the reliability of our RNA-seq data, a total of 12 DE lncRNAs and DE mRNAs were randomly selected. Their relative expression levels in the antagonistic and sensitive lambs were confirmed by q-PCR (Fig. 6) and were found to coincide with our RNA-seq results (Fig. 7), thus indicating that our RNA-seq data were reliable. Our analyses also showed that high-throughput sequencing has the advantage of detecting genes that are expressed at relatively very low levels (0 < FPKM < 1).
Figure 4.

Expression patterns of lncRNA and mRNA transcripts. The Box-whisker Plot consists of five statistics: the minimum, the first quartile (25%), the median (50%), the third quartile (75%), and the maximum.
Figure 5.

Differentially expressed lncRNAs and mRNAs between antagonistic and sensitive lambs. Color indicates the amount of expression of the gene. The darker the color, the greater the expression (red is up-regulated, green is down-regulated). Each row represents the expression of each gene in different samples, and each column represents the expression of all genes in each sample. The top tree shows the results of cluster analysis of different samples from different experimental groups, and the left tree shows the results of cluster analysis of different genes from different samples.
Figure 6.
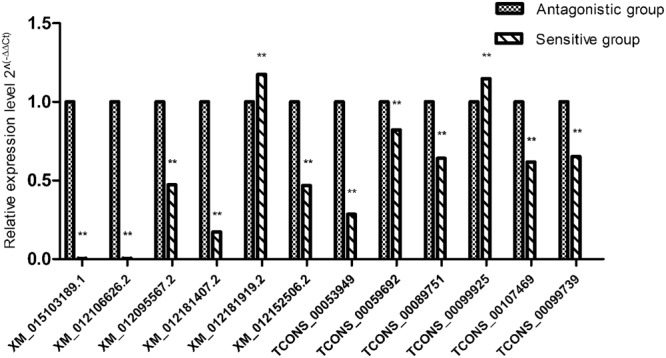
Relative expression levels of DE lncRNAs and mRNAs between antagonistic and sensitive lambs were confirmed by q-PCR. Note: “**” means highly significant correlation; “*” means significant correlation; “ns” or “no SuperiorScript” means no significant correlation. The same as below.
Figure 7.
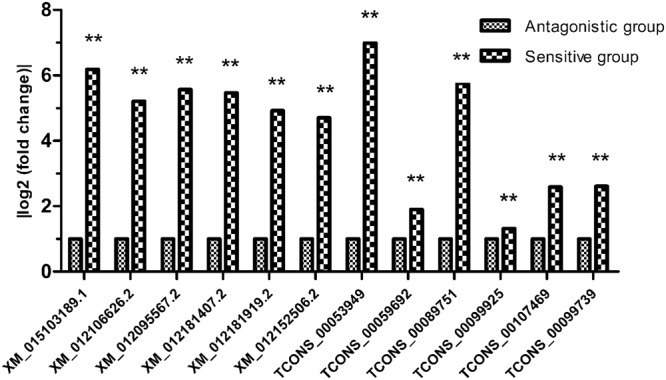
RNA-seq results of DE lncRNAs and mRNAs.
GO and KEGG Pathway enrichment analysis of DE lncRNAs
In the corresponding relationship between “lncRNA name-function prediction Term” (Supplementary 1 and Supplementary 2), we selected the top 500 predictive relationships with the highest predictive reliability (sorted by p-value). The frequency of each function and the number of GO (or pathway) terms with function annotations were analysed to reflect the difference in the distribution of lncRNAs (Fig. 8).
Figure 8.

Gene Ontology and KEGG Pathway enrichment analyses of DE lncRNAs.
Comparisons of the DE lncRNAs and the entries in the GO database identified that a total of 34 lncRNAs could be annotated and classified into 302 functional subclasses. The number of DE lncRNAs in the top 30 functional subclasses is shown Fig. 8. The number of DE lncRNAs in the categories of protein binding (GO:0005515), nucleus (GO:0005634), poly(A) RNA-binding (GO:0044822), cytoplasm (GO:0005737), tissue remodelling (GO:0048771), regulation of endopeptidase activity (GO:0052548, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase complex (GO:0043540), phosphatidylinositol phosphorylation (GO:0046854), fructose-2,6-bisphosphate 2-phosphatase activity (GO:0004331), and calcium-dependent phospholipase C activity (GO:0050429) were higher.
Comparison between the DE lncRNAs and the entries in the KEGG Pathway database indicated that a total of 34 lncRNAs could be annotated and classified into 149 KEGG pathways. The number of DE lncRNAs in the top 30 KEGG Pathways is shown in Fig. 8. The number of DE lncRNAs in the thyroid hormone signalling pathway (path:ko04919), spliceosome (path:ko03040), leukocyte transendothelial migration (path:ko04670), neurotrophin signalling pathway (path:ko04722), lysosome (path:ko04142), MAPK signalling pathway-yeast (path:ko04011), sphingolipid signalling pathway (path:ko04071), phagosome (path:ko04145), and oxidative phosphorylation (path:ko00190) categories were relatively higher.
LncRNAs and their adjacent coding genes
We searched for all the coding genes within the 100-kb flanking regions of the lncRNAs, and genes that were significantly co-expressed with the lncRNAs as indicated by Pearson correlation calculations were identified. These co-expressed genes that were adjacent to the lncRNAs were presumed to be regulated by lncRNAs. Therefore, we identified 6 genes may be regulated by their associated lncRNAs (Table 3).
Table 3.
DE lncRNAs and their co-expressed gene.
| lncRNA | Change in expression | mRNA | Gene | Regulation | Chromosome | Pearson p-value | Pearson correlation | Cis distance (bp) |
|---|---|---|---|---|---|---|---|---|
| TCONS_00053949 | Downregulated | XM_012176963.2 | MYO1G | Up | NC_019461.2 | 0.034 | −0.845 | 1,670 |
| TCONS_00059692 | Upregulated | XM_015095635.1 | TIMM29 | Down | NC_019462.2 | 0.01 | −0.916 | 16,037 |
| TCONS_00059692 | Upregulated | XM_015095835.1 | CARM1 | Down | NC_019462.2 | 0.033 | −0.848 | 22,513 |
| TCONS_00089751 | Upregulated | XM_015097644.1 | ADGRB1 | Up | NC_019466.2 | 0.0027 | 0.957 | 8,560 |
| TCONS_00099925 | Downregulated | XM_015098457.1 | SEPT4 | Up | NC_019468.2 | 0.049 | −0.812 | 98,788 |
| TCONS_00107469 | Upregulated | XM_012187185.1 | DESI2 | Up | NC_019469.2 | 0.0026 | 0.958 | 94,714 |
Discussion
Due to the rapid development of transcriptome analysis, lncRNAs have recently been considered as a novel modulator of cell development14. The lncRNAs that we have identified in this study are mainly associated with cancer, such as prostate cancer15, gastric cancer16, lung cancer17 and breast cancer18, as well as reproduction19–22. However, investigations of lamb diarrhoea in relation to lncRNAs are limited. The Hu sheep is a unique breed with high fecundity and strong adaptability to warm-wet climates and can be kept indoors all year. This study has provided the first overview of lncRNAs in relation to diarrhoea in sheep, as well an investigation into their possible roles in disease resistance.
Major economic losses in sheep farms are often due to diarrhoea. In this study, We mainly study the immune status of lambs, and the spleen is the largest immune organ; thus, we selected the spleen as the research object. we found that the expression level of lncRNAs was lower than mRNAs in lamb spleens (Fig. 4), which was in agreement with the results from sheep testicular tissues9, and the average lengths of lncRNAs and mRNAs in sheep were longer than those in pigs (1,713 bp and 1,983 bp, respectively)20. We searched for all the coding genes within the 100-kb flanking regions of the lncRNAs and identified intersecting genes that were significantly co-expressed with the lncRNAs as indicated by Pearson correlation calculations23. We also identified the following 6 genes as being co-expressed with the lncRNAs: myosin IG (MYO1G), translocase of inner mitochondrial membrane 29 (TIMM29), co-activator associated arginine methyltransferase 1 (CARM1), adhesion G protein-coupled receptor B1 (ADGRB1), septin 4 (SEPT4), and desumoylating isopeptidase 2 (DESI2).
MYO1G plays an important role in maintaining cell stiffness in B-cell lymphocytes. The deletion of the myo1g gene results in a reduction in cell stiffness, which in turn affects cell adhesion, proliferation, phagocytosis, and endocytosis in B-cell lymphocytes24. Investigations of TIMM29 are limited. TIMM29 has been identified as the first specific component of the mammalian TIMM22 protein complex and plays an important role in the assembly of the TIMM23 protein25,26. CARM1, a member of the protein arginine methyltransferase (PRMT) family, is an enzyme with a highly conserved domain with methyltransferase activity. CARM1 knockout mice die at birth27, indicating that CARM1 is essential to postnatal survival. It was later discovered that CARM1 inhibition promotes HIV-1 activation28. ADGRB1 is a member of the transmembrane protein-adhesion G protein coupled receptor (aGPCR) family, which is characterized by a conserved GAIN domain that has autologous proteolytic activity that can cleave the receptor near the first transmembrane domain. Studies have shown that the new N-terminal stalk, which is revealed by GAIN domain cleavage, can directly activate aGPCRs as a tethered agonist29. Septins are a highly conserved cytoskeletal family with GTPase activity. The tumour suppressor SEPT4 is a member of the septin family that can induce cancer cell apoptosis30. Mutations in the SEPT4 gene in mice can lead to disorders involving the annuli of spermatozoa31 and adjacent cortical structures, thereby causing low sperm motility, ultimately leading to infertility32. DESI2 is a pro-apoptotic gene; in vitro experiments have shown that its overexpression induces apoptosis in pancreatic cancer and other tumour cells, which can effectively inhibit the proliferation of some cancer cells. Gene therapy using DESI2 and IP10 significantly inhibits tumour growth and effectively prolongs the survival of tumour-bearing mice33,34.
A total of 703 mRNAs and 34 known lncRNAs were differentially expressed between the antagonistic and sensitive groups, including 14 upregulated lncRNAs and 20 downregulated lncRNAs. In addition, the present study identified 1,942 novel lncRNAs. We searched for all the coding genes within the 100-kb flanking regions of the lncRNAs and identified genes shared between the two experimental groups that were significantly co-expressed with lncRNAs as indicated by Pearson correlation calculations. We have determined that 6 genes may be regulated by their associated lncRNAs. To validate our RNA-seq results, q-PCR was performed to verify the expression levels of the 12 known lncRNAs and mRNAs. The final results coincided with our RNA-seq data.
GO is a bioinformatics tool that has been extensively utilized in studying the relationship of various gene functions. GO analysis indicated that 16 out the 34 DE lncRNAs were enriched with the protein binding (GO: 0005515) category. Moreover, KEGG Pathway analysis showed that the sphingolipid signalling (path: ko04071), axon guidance (path: ko04360), and glycosylphosphatidylinositol (GPI)-anchor biosynthesis (path: ko00563) pathways may be important KEGG pathways of genes co-expressed with DE lncRNAs, and the related lncRNAs may be potentially involved in fimbriae adhesion to the intestinal mucosa. However, the role of these pathways in disease resistance remains largely unknown.
In this study, the expression profiles of lncRNAs in the spleens of lambs that were antagonistic or sensitive to diarrhoea were investigated to further understand the regulation of lncRNAs in sheep disease resistance. Several differentially expressed lncRNAs in lamb spleens between the antagonistic and sensitive groups were identified, and we found that 6 genes (MYO1G, TIMM29, CARM1, ADGRB1, SEPT4, and DESI2) may be regulated by their associated lncRNA. Our study may help elucidate the mechanism underlying resistance to diarrhoea in lambs. Further investigations of these sheep lncRNAs in relation to diarrhoea-resistance are warranted.
Methods
Ethics statement
The Institutional Animal Care and Use Committee (IACUC) of the government of Jiangsu Province (Permit Number 45) and the Ministry of Agriculture of China (Permit Number 39) approved the animal study proposal. All experimental procedures were conducted in strict compliance with the recommendations of the Guide for the Care and Use of Laboratory Animals of Jiangsu Province and of the Animal Care and Use Committee of the Chinese Ministry of Agriculture. All efforts were made to minimize animal suffering.
Experimental design and sample collection
The experimental sheep were purchased from Jiangsu Xilaiyuan Ecological Agriculture Co., Ltd. in December 2016. A total of 18 three-day-old lambs showing normal growth and approximately similar weight were randomly selected, and all the sheep were raised with segregation. To ensure their dietary requirements, the sheep were fed with 10% lamb milk powder (Table 4) prior to the experiment. Five-day-old lambs were fed 12.5% lamb milk powder and E. coli F17 bacteria liquid [4.6 × 108 colony-forming units (CFUs)·mL−1], as well as ad libitum access to drinking water. Stool features of the experimental lambs were recorded daily (Table 5). Lambs that exhibited diarrhoea for two days were classified as antagonistic and sensitive and were euthanized. The intestinal tissues were collected in 4% paraformaldehyde. The liver, spleen, duodenum, jejunum, and ileum of each lamb were collected and immediately frozen in liquid nitrogen until RNA extraction.
Table 4.
Lamb Milk Powder Ingredients List (per 100 grams of milk powder).
| Components | Moisture content | Crude protein | Crude fat | Crude fibre | Coarse ash | Calcium | Total phosphorus | Lysine |
|---|---|---|---|---|---|---|---|---|
| Percentage | ≤6 | ≥22 | ≥18 | ≤5 | ≤8 | 0.9~3.0 | ≥0.5 | ≥1.0 |
Table 5.
Bristol Stool Form Scale13.
| Type 1 | Separate hard lumps, similar to nuts |
| Type 2 | Sausage-shaped but lumpy |
| Type 3 | Sausage- or snake-like but with cracks on its surface |
| Type 4 | Sausage- or snake-like, smooth, and soft |
| Type 5 | Soft blobs with distinct edges |
| Type 6 | Fluffy pieces with ragged edges, mushy stool |
| Type 7 | Watery, no solid pieces |
HE staining
The jejunum tissue was washed with 0.9% normal saline and fixed in 4% paraformaldehyde for 48 h at room temperature and then used in histological analysis. Next, 7 μm-thick sections were stained with haematoxylin-eosin and the morphology of the jejunum epithelia was assessed under a microscope.
Library construction and sequencing
RNA was extracted from the spleen of three individuals from each group. A NanoDrop 2000 Ultra Microscope and an Agilent 2100 Bioanalyzer were utilized in determining the quality control of the extracted total RNAs (Annex 1). Ribosomal RNA was removed using a Ribo-Zero (TM) kit (Epicenter, Madison, WI, USA). Short fragments (approximately 200 bp in length) were obtained and used as templates for first-stand cDNA synthesis. Second-strand cDNA synthesis was performed using a buffer, dNTPs, Rnase H, and DNA polymerase I. After PCR amplification and purification using the Qubit® dsDNA HS Assay Kit, the cDNA library was constructed using an NEBNext® Ultra™ RNA Library Preparation Kit. The cDNA library was sequenced on the Illumina HiSeq 2500 platform at Shanghai OE Biomedical Technology Co. (sequencing read length: 150 bp).
Identification of lncRNAs and mRNAs
The raw data were filtered to eliminate low-quality reads. Clean reads mapped to the reference genome (Ovis aries v4.0) were selected for de novo assembly. Coding and non-coding RNA candidates from the unknown transcripts were categorized using four coding potential analysis methods, CPC35, CNCI36, Pfam37, and PLEK38. The minimum length and the number of exons were set as thresholds, thereby filtering putative encoded RNAs, and transcripts containing two exons and longer than 200 nt were selected as candidate lncRNAs. Different types of lncRNAs were classified by cuffcompare, including intergenic lncRNAs (character u), intronic lncRNAs (character i), anti-sense lncRNAs (character x), and sense-overlapping lncRNAs (character o).
Different expression analysis
Because the fragments per kb per million reads (FPKM) method39 considers the simultaneous effect of sequencing depth and the length of the transcript on the number of fragments, the FPKM value was used to estimate the expression levels of lncRNA and mRNA transcripts. We used DESeq40 to determine the number of DE genes and the FPKM values between the two groups. In cases when RNA-seq data were employed to compare different expression levels in the same transcript in both samples, two criteria were selected: 1) fold change, which is the change in the expression level of the same transcript in both samples; and 2) the p value or false discovery rate (FDR) (adjusted p-value). The FDR error control method was used in correcting the p-value multiple hypothesis test41.
GO and KEGG pathway analyses
After screening for differentially expressed transcripts, functional annotation was performed using GO enrichment analysis. Enrichment analysis employed counting the number of transcripts in each GO term, followed by Fisher’s exact test to assess statistical significance (p < 0.05). KEGG42 is the main public database used in pathway analysis, which was followed by Fisher’s exact test to assess statistical significance (p < 0.05). The analysis of different transcripts was used to identify enriched pathways.
Prediction of the target genes of DE lncRNAs
The target genes of DE lncRNAs were predicted by calculating the Pearson correlation coefficients and P values among multiple genes. The |correlation| ≥ 0.7 and P ≤ 0.05 were used to filter the transcripts43. The DE lncRNAs associated with adhesion were selected, and the target genes of all DE lncRNAs were predicted by cis-acting44.
Verification of the expression level of DE lncRNAs
To verify whether the screened DE lncRNAs play a role in the process of antagonism, q-PCR was used to detect the expression levels of 12 DE lncRNAs and DE mRNAs in the lamb spleens between the antagonistic and sensitive groups. The relative expression of each RNA was normalized to that of GAPDH using the 2−∆∆Ct method45, and the primers used in the amplification of the lncRNAs are shown in Table 6.
Table 6.
The primers for GAPDH, DE lncRNAs, and mRNAs.
| ID | Primer sequence (5′ → 3′) | Product length (bp) | |
|---|---|---|---|
| Primers for the six DE mRNAs | XM_015103189.1 | F: AGCACTTCCTCCTGTCCG | 132 |
| R: CAGCACAGAAGGCAAAGTC | |||
| XM_012106626.2 | F: CCGAGTTTGCAGGTACCCAAC | 121 | |
| R: TTTTGGCGCATGTATACCTG | |||
| XM_012095567.2 | F: TGAGACTCTACTTCGCTGC | 102 | |
| R: TTGCCCATCCTTAATAGCTG | |||
| XM_012181407.2 | F: ACGACGGTGGTTAAATACTC | 125 | |
| R: AGTTGCCCATAGTCACTGGTC | |||
| XM_012181919.2 | F: TCAACCATATGCTGACGGAC | 119 | |
| R: ATGCCGCCTATCAAGGTC | |||
| XM_012152506.2 | F: CCACCTGCGGTTCAAGTTAC | 157 | |
| R: TGCCTGAATCACCTTGTC | |||
| Primers for six DE lncRNA | TCONS_00053949 | F: CCTGGCTATATCCTTACATCAC | 100 |
| R: AAGTTCAAACTCCGCTGCAC | |||
| TCONS_00059692 | F: AATTTCTTCTCGTTCCAAGGC | 131 | |
| R: CCAACAGGGAGCCAACTTC | |||
| TCONS_00089751 | F: AGAAGGCTTTGACCGAAC | 153 | |
| R: TCAATGCCCTCCACGAGAC | |||
| TCONS_00099925 | F: AGTGCCACATGTACCTAGCAG | 146 | |
| R: ACGACAGGCATTTTAACCCATG | |||
| TCONS_00107469 | F: TAGTACAGCCCATATTTATCG | 115 | |
| R: ATTTTCTTTCCACAGGGACG | |||
| TCONS_00099739 | F: CCGACGCTGTCATGATGC | 159 | |
| R: TCCGTCTCCAGAACCAAGGC | |||
| Primers for GAPDH | GAPDH | F: GTTCCACGGCACAGTCAAGG | 127 |
| R: ACTCAGCACCAGCATCACCC |
Statistical analysis
All data were analysed by SPSS (version 20.0), and the relative expression levels of different transcripts were analysed by ANOVA. Tukey’s test was used for multiple comparisons. Statistical significance was determined when p < 0.05. Each group contained three samples, and each experiment was repeated thrice.
Electronic supplementary material
Acknowledgements
This work was performed by National Natural Science Foundation of China(31872333), The Projects of Domesticated Animals Platform of the Ministry of Science, Key Research and Development Plan (modern agriculture) in Jiangsu Province (BE2018354), Major new varieties of agricultural projects in Jiangsu Province (PZCZ201739), Agricultural Science and Technology Independent Innovation Project of Jiangsu Province (CX (18)2003), The Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Major projects of Natural Science Research of colleges and universities in Jiangsu Province, The Project of six peak of talents of Jiangsu Province of China and Postgraduate Innovation Project of Yangzhou University (XKYCX17_060, SJCX18_0804).
Author Contributions
Wei Sun conceived the study and designed the experiment. Jianjun Bao wrote the main manuscript text. Xiaoyang Lv and Wen Gao prepared Figures 1–8. Guoqiang Zhu, Jianjun Bao, Chengyan Jin and Xiaoyang Lv performed the experiments. Buzhong Wang prepared lambs for experiments. Guojun Dai, Yue Wang, Weihao Chen, Tianyi Wu and Lihong Wang helped with sample collection. All authors read and approved the final manuscript.
Data Availability Statement
We guarantee that our data is valid.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-34291-0.
References
- 1.Orskov I, Orskov F. Episome-carried surface antigen K88 of Escherichia coli. I. Transmission of the determinant of the K88 antigen and influence on the transfer of chromosomal markers. J Bacteriol. 1966;91:69–75. doi: 10.1128/jb.91.1.69-75.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stirm S, Orskov I, Orskov F. K88, an episome-determined protein antigen of Escherichia coli. Nature. 1966;209:507–508. doi: 10.1038/209507a0. [DOI] [PubMed] [Google Scholar]
- 3. Kim, T. et al. Role of MYC-regulated long noncoding RNAs in cell cycle regulation and tumorigenesis. J Natl Cancer Inst107, 10.1093/jnci/dju505 (2015). [DOI] [PMC free article] [PubMed]
- 4.Ouyang J, et al. NRAV, a long noncoding RNA, modulates antiviral responses through suppression of interferon-stimulated gene transcription. Cell Host Microbe. 2014;16:616–626. doi: 10.1016/j.chom.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, Rana TM. Decoding the noncoding: prospective of lncRNA-mediated innate immune regulation. RNA Biol. 2014;11:979–985. doi: 10.4161/rna.29937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol. 2014;15:484–491. doi: 10.1038/ni.2887. [DOI] [PubMed] [Google Scholar]
- 7.Heward JA, Lindsay MA. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014;35:408–419. doi: 10.1016/j.it.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Caifang, Deng Mingtian, Fan Yixuan, Yang Hua, Zhang Guomin, Feng Xu, Li Fengzhe, Wang Dan, Wang Feng, Zhang Yanli. Genome-Wide Analysis Reveals Extensive Changes in LncRNAs during Skeletal Muscle Development in Hu Sheep. Genes. 2017;8(8):191. doi: 10.3390/genes8080191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, et al. Long noncoding RNA expression profile changes associated with dietary energy in the sheep testis during sexual maturation. Sci Rep. 2017;7:5180. doi: 10.1038/s41598-017-05443-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yue Y, et al. Integrated Analysis of the Roles of Long Noncoding RNA and Coding RNA Expression in Sheep (Ovis aries) Skin during Initiation of Secondary Hair Follicle. PLoS One. 2016;11:e0156890. doi: 10.1371/journal.pone.0156890.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu XW. Prevention and treatment of sheep coliform disease. The Chinese Livestock and Poultry Breeding. 2017;4:129–130. [Google Scholar]
- 12.Zhang WJ. Prevention and Control of Escherichia coli Disease in Sheep. China Herbivores. 2017;6:76. [Google Scholar]
- 13.Lewis SJ, Heaton KW. Stool form as a useful guide to intestinal transit time. Scand J Gastroenterol. 1997;32:920–924. doi: 10.3109/00365529709011203. [DOI] [PubMed] [Google Scholar]
- 14.Veneziano D, Nigita G, Ferro A. Computational Approaches for the Analysis of ncRNA through Deep Sequencing Techniques. Front Bioeng Biotechnol. 2015;3:77. doi: 10.3389/fbioe.2015.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martignano F, et al. Urinary RNA-based biomarkers for prostate cancer detection. Clin Chim Acta. 2017;473:96–105. doi: 10.1016/j.cca.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Hao Yan-Ping, Qiu Jing-Hui, Zhang Dong-Bo, Yu Cheng-Gong. Long non-coding RNA DANCR, a prognostic indicator, promotes cell growth and tumorigenicity in gastric cancer. Tumor Biology. 2017;39(6):101042831769979. doi: 10.1177/1010428317699798. [DOI] [PubMed] [Google Scholar]
- 17.Huang Q, et al. Fine particulate matter 2.5 exerted its toxicological effect by regulating a new layer, long non-coding RNA. Sci Rep. 2017;7:9392. doi: 10.1038/s41598-017-09818-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, et al. Induction of a novel isoform of the lncRNA HOTAIR in Claudin-low breast cancer cells attached to extracellular matrix. Mol Oncol. 2017;11:1698–1710. doi: 10.1002/1878-0261.12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoneda R, et al. A genomic region transcribed into a long noncoding RNA interacts with the Prss42/Tessp-2 promoter in spermatocytes during mouse spermatogenesis, and its flanking sequences can function as enhancers. Mol Reprod Dev. 2016;83:541–557. doi: 10.1002/mrd.22650. [DOI] [PubMed] [Google Scholar]
- 20.Ran M, et al. Systematic Identification of Long Noncoding RNAs in Immature and Mature Porcine Testes. Biol Reprod. 2016;94:77. doi: 10.1095/biolreprod.115.136911. [DOI] [PubMed] [Google Scholar]
- 21.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearson K. Determination of the coefficient of correlation. Science. 1909;30:23–25. doi: 10.1126/science.30.757.23. [DOI] [PubMed] [Google Scholar]
- 24.López-Ortega O, et al. Myo1g is an active player in maintaining cell stiffness in B-lymphocytes. Cytoskeleton (Hoboken) 2016;73:258–68. doi: 10.1002/cm.21299. [DOI] [PubMed] [Google Scholar]
- 25.Callegari S, et al. TIM29 is a subunit of the human carrier translocase required for protein transport. FEBS Lett. 2016;590:4147–4158. doi: 10.1002/1873-3468.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kang, Y. et al. Tim29 is a novel subunit of the human TIM22 translocase and is involved in complex assembly and stability. eLife5, 10.7554/eLife.17463 (2016). [DOI] [PMC free article] [PubMed]
- 27.Yadav N, et al. Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc Natl Acad Sci USA. 2003;100:6464–6468. doi: 10.1073/pnas.1232272100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Z, et al. Crosstalk between histone modifications indicates that inhibition of arginine methyltransferase CARM1 activity reverses HIV latency. Nucleic Acids Res. 2017;45:9348–9360. doi: 10.1093/nar/gkx550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kishore A, Purcell RH, Nassiri-Toosi Z, Hall RA. Stalk-dependent and Stalk-independent Signaling by the Adhesion G Protein-coupled Receptors GPR56 (ADGRG1) and BAI1 (ADGRB1) J Biol Chem. 2016;291:3385–3394. doi: 10.1074/jbc.M115.689349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeon TW, et al. Electro-hyperthermia up-regulates tumour suppressor Septin 4 to induce apoptotic cell death in hepatocellular carcinoma. Int J Hyperthermia. 2016;32:648–656. doi: 10.1080/02656736.2016.1186290. [DOI] [PubMed] [Google Scholar]
- 31.Ihara M, et al. Cortical organization by the septin cytoskeleton is essential for structural and mechanical integrity of mammalian spermatozoa. Dev Cell. 2005;8:343–352. doi: 10.1016/j.devcel.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 32.Kissel H, et al. The Sept4 septin locus is required for sperm terminal differentiation in mice. Dev Cell. 2005;8:353–364. doi: 10.1016/j.devcel.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 33.Shen CC, et al. High Phosphorylation Status of AKT/mTOR Signal in DESI2-Reduced Pancreatic Ductal Adenocarcinoma. Pathol Oncol Res. 2015;21:267–272. doi: 10.1007/s12253-014-9817-3. [DOI] [PubMed] [Google Scholar]
- 34.Lin C, et al. Combination of DESI2 and IP10 gene therapy significantly improves therapeutic efficacy against murine carcinoma. Oncotarget. 2017;8:56281–56295. doi: 10.18632/oncotarget.17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong L, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35:W345–349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun L, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013;41:e166. doi: 10.1093/nar/gkt646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li A, Zhang J, Zhou Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014;15:311. doi: 10.1186/1471-2105-15-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/S0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- 42.Kanehisa M, et al. KEGG for linking genomes to life and the environment. Nucleic Acids research. 2008;36:480–484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ping X, Yasong T, Binbin L, Xu L. Hydrological Trend Variation Classification Method Based on Correlation Coefficient. Journal of basic science and engineering. 2014;22:1089–1097. [Google Scholar]
- 44.Lam MT, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. 2014;39:170–182. doi: 10.1016/j.tibs.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We guarantee that our data is valid.