

Summary
The evolutionary history of the wolf-like canids of the genus Canis has been heavily debated, especially regarding the number of distinct species and their relationships at the population and species level [1, 2, 3, 4, 5, 6]. We assembled a dataset of 48 resequenced genomes spanning all members of the genus Canis except the black-backed and side-striped jackals, encompassing the global diversity of seven extant canid lineages. This includes eight new genomes, including the first resequenced Ethiopian wolf (Canis simensis), one dhole (Cuon alpinus), two East African hunting dogs (Lycaon pictus), two Eurasian golden jackals (Canis aureus), and two Middle Eastern gray wolves (Canis lupus). The relationships between the Ethiopian wolf, African golden wolf, and golden jackal were resolved. We highlight the role of interspecific hybridization in the evolution of this charismatic group. Specifically, we find gene flow between the ancestors of the dhole and African hunting dog and admixture between the gray wolf, coyote (Canis latrans), golden jackal, and African golden wolf. Additionally, we report gene flow from gray and Ethiopian wolves to the African golden wolf, suggesting that the African golden wolf originated through hybridization between these species. Finally, we hypothesize that coyotes and gray wolves carry genetic material derived from a “ghost” basal canid lineage.
Keywords: phylogenomics, canid phylogeography, African golden wolf, Ethiopian wolf, canid hybridization, gray wolf, African hunting dog
Graphical Abstract
Highlights
-
•
Extensive gene flow in the genus Canis, especially among the crown group
-
•
Genetic contribution from an unknown canid into the ancestor of the gray wolf and coyote
-
•
The African golden wolf possibly a hybrid species, from the gray wolf and Ethiopian wolf
-
•
Possible ancient admixture between the dhole and African hunting dog
Gopalakrishnan et al. present evidence of pervasive gene flow among species of the genus Canis. In addition to previously known admixture events, they find evidence of gene flow from a “ghost” canid, related to the dhole, into the ancestor of the gray wolf and coyote. Further, they suggest that the African golden wolf is a species of hybrid origin.
Results and Discussion
The genome dataset analyzed in this study contains 12 gray wolves and 14 dogs, chosen from regions overlapping the current ranges of the other basal canids included in this study, five coyotes, one Ethiopian wolf, three golden jackals, six African golden wolves (originally Canis anthus, but recently reclassified as Canis lupaster [1]), two dholes, four African hunting dogs, and one Andean fox (Lycalopex culpaeus) (Figure 1). Short-read sequencing of the samples and subsequent alignment to the recently published wolf genome assembly [7] resulted in genome-wide coverages ranging from 0.6–26.6× (for details, see Data S1). The genome-wide heterozygosity estimates (Figure S1) clearly show reduced levels in the Ethiopian wolf, African hunting dog, and dhole, an observation that is consistent with their small population sizes. The reconstructed phylogenetic relationships within this group of canids (Figure 2B) are of considerable relevance in light of extensive prior debate on the relationships between the Ethiopian wolf, golden jackal, and African golden wolf [2, 3, 4, 5]. Our results corroborate the recent proposition based on both mitochondrial [2, 3] and nuclear [4, 6] data that the African golden wolf is evolutionarily distinct from the golden jackal (Figure 2C, panel labeled 16), but also that the Ethiopian wolf falls basal to both (Figure 2C, panel labeled 12) [5]. For convenience, we henceforth refer to five canid species, viz. the Ethiopian wolf, African golden wolf, golden jackal, gray wolf, and coyote, as “the crown group” in order to distinguish them from the more basal dholes and African hunting dogs. The placement of the Ethiopian wolf as the basal group in this clade is consistent with tree topologies obtained in previous phylogenetic analyses based on concatenated gene sequences [5] and more recent multispecies coalescent analyses [4] of datasets consisting of a subset of exonic and intronic sequences, but differs from the topology based on concatenated analyses in the latter study. We note that this nuclear-DNA-based phylogeny also places dogs as a sister clade to European gray wolves. However, we caution that this placement has only moderate support (0.86 mean local posterior probability); moreover, the gene tree quartet frequencies of alternate resolutions within the dog-gray wolf branches are comparable to that recovered in the main tree (Figure 2B, panel labeled 20–22), and thus no conclusion can be drawn about which wolf population gave rise to dogs. Indeed, our findings are not incompatible with previously suggested hypotheses [9] that either (1) the dog was domesticated from a now-extinct wolf population and/or (2) Eurasian gray wolf population genomic diversity has been reduced since the domestication event.
Figure 1.

Map Showing the IUCN Ranges, Range Overlaps, and Sampling Locations of the Canids Included in This Study
The overlaps in ranges are shown in blended colors (orange, dark purple, dark olive green, light teal, etc.). Since IUCN does not have range information for African golden wolf, the IUCN range of golden jackal has been split in two; the Eurasian part is shown as the range of golden jackal, and the African part is shown as the range of African golden wolf. Further details on the samples, including their sampling location and source, can be found in Data S1, and their estimated heterozygosities—which are inversely proportional to their population sizes—are shown in Figure S1.
Figure 2.

Nuclear and Mitochondrial Phylogeny of Basal Canids
(A) The maximum-likelihood estimate of the mitochondrial phylogeny for a subset of the samples, using de novo mitochondrial assemblies obtained with MtArchitect. The node labels show the bootstrap support for the node.
(B) The phylogeny estimated from nuclear DNA by ASTRAL-II, where monophyletic clusters have been collapsed into a single leaf node. The tip labeled “African golden wolf-hybrid” represents a single known hybrid from the Sinai Peninsula—labeled “African golden wolf Egypt” in the mtDNA phylogeny—as described in the main text. The mean local posterior probabilities are shown for branches where this value is less than 1. The full nuclear phylogeny containing the sample relationships, branch supports, branch lengths proportional to divergence times, and estimated split times can be found in Figures S2A and S2B and Table S2.
(C) For a subset of the internal branches in the nuclear phylogeny, the quartet frequencies of the three possible configurations around each branch in the underlying unrooted tree are shown. The red bar represents the configuration shown in the phylogeny, and the two blue bars represent the two alternative configurations. For every quartet, the frequency of the true bipartition has previously been shown to be at least one-third [8], indicated here by a dotted line. Each alternative configuration is labeled by the bipartition it creates, with labels corresponding to those in (A). For example, the second bar of the panel labeled 12 swaps the positions of golden jackal (6) and Ethiopian wolf (5), whereas the third bar puts them as sister to each other. This plot summarizes the gene tree incongruence around examined branches.
Mitochondrial genomes were de novo assembled from all species studied, using MtArchitect [10], which accounts for presence of numts in the reference genome. A maximum-likelihood phylogeny based on these mitochondrial genomes (Figure 2A) is largely consistent with that obtained from the nuclear genome analysis, with one obvious exception—the coyote mitochondrial genomes fall basal to all the other crown canids. This is consistent with Koepfli and colleagues’ [4] results on near-complete mitochondrial genomes and thus contradicts the findings of numerous previous studies that used partial mitochondrial DNA sequences and placed coyotes (1) as sister to gray wolves [11], (2) in an unresolved clade with African golden wolves and Ethiopian wolves [2, 3], (3) as sister to Ethiopian wolves [1, 2, 12, 13], or, finally, (4) as sister to a clade containing Ethiopian wolves and golden jackals [14].
We subsequently explored the degree of interspecific gene flow between the various species. Many publications have reported interspecies gene flow between members of the canid crown group (dog-gray wolf complex, coyotes, Ethiopian wolves, golden jackals, and African golden wolves) [4, 5, 9, 13, 15, 16, 17, 18, 19]—something perhaps unsurprising, given the large geographic overlap of many of the populations. Initial analyses of genetic structure among these canids using NGSadmix [20] (Figure S3A) revealed that the individuals partition according to expected species structure. However, more details became apparent as the number of estimated clusters (K) was increased. For example, at higher values of K, gray wolves form five principal groups (Mexico, Ellesmere-Greenland, East Asia, the Middle East, and the remaining Eurasia), whereas African golden wolves are split into an Eastern and a Northwestern clade, as previously shown [4, 6, 16]. We note that similar east-west population differentiation is observed for several other African mammalian species [21], thus pointing to a general trend that the African golden wolves follow. The NGSadmix analyses also suggest the presence of admixture between the different species. For example, we detected not only dog introgression in the gray wolves from Spain and Israel, but also, perhaps of greater interest, gene flow between African golden wolves, golden jackals, and gray wolves. One example is a highly admixed African golden wolf from the Egyptian Sinai Peninsula, whose genome contains contributions from both Middle Eastern gray wolves and dogs (Figure S3A).
Previous studies that have reported admixture between canid species [9] and mitochondrial evidence for overlap of the gray wolf, African golden wolf, and golden jackal in eastern Egypt [4]. This points to the importance of the Sinai Peninsula and the Southwest Levant in canid evolution [4, 9], presumably due to its role as the land bridge between the African and Eurasian continents. We used TreeMix [22], D statistics [23], and admixture graphs [23] to examine signals of admixture between these species. The results confirmed that, in general terms, the level of gene flow between the three species is high, although varying across space in a manner consistent with their natural ranges (Figures 3B and S3A–S3E). For example, gene flow between golden jackals and gray wolves and between African golden wolves and gray wolves is lowest when North American gray wolves are considered, somewhat higher for Asian and European gray wolves, and highest with the gray wolves from the Middle East (e.g., Israel, Syria, and Saudi Arabia) (Figure S3E). Although the latter is not surprising in light of the natural ranges of the species, the evidence of golden jackal ancestry in North American wolves is intriguing. One possible explanation could be that gene flow happened before the divergence of the North American and Eurasian gray wolves. The fact that interspecific gene flow is considerably higher in Middle Eastern than in other gray wolves may also explain the distinctness of this population. The structure between Northwestern and Eastern African golden wolves can be explained using a similar argument—the former have highest levels of golden jackal and gray wolf admixture (Figures 3B, S3A, and S3B), whereas the latter show higher levels of gene flow from Ethiopian wolves. Overall, it is clear that individuals sampled in this land bridge region will be particularly informative for future studies that wish to study canid admixture in greater detail.
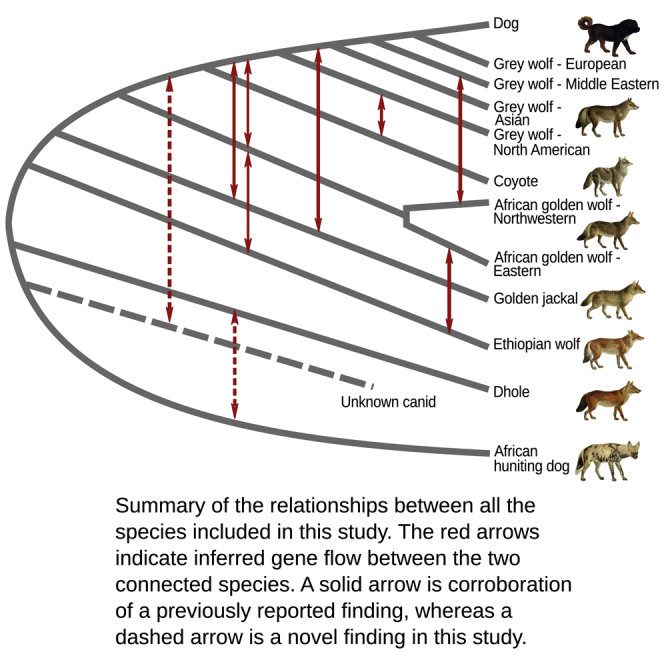
Figure 3.

Gene Flow among the Crown Canid Species
(A) This figure summarizes the relationships among the species (phylogeny) and the various gene flow events inferred from the samples included in this study. Gene flow events are indicated with red arrows, and dotted red arrows show possible gene flow events that have been inferred in this study but have not been previously reported.
(B–D) These figures show the gene flow among the different crown canid species using D statistics. These D statistics show significant gene flow between the gray wolf, African golden wolf, golden jackal, and Ethiopian wolf. One principal new finding is structure within the African golden wolves, splitting into Northwestern and Eastern clades, which show genetic affinity to gray wolves and Ethiopian wolves, respectively. A second principal finding is inferred gene flow from an unknown canid lineage, related to the dhole, into the ancestor of the coyote and the gray wolves. We hypothesize this may explain the unexpected basal placement of the coyote in the mitochondrial tree. Further evidence of gene flow in the crown canids is shown in Figure S3.
Furthermore, D statistics were used to test for gene flow between the dhole and African hunting dog, using members of the crown group as ingroup and the Andean fox as outgroup. Although no gene flow was detected between species of the crown group and the African hunting dogs, the analyses provided strong evidence of gene flow between the African hunting dog and dhole (Figure S3C). This is a surprising finding, since the ranges of the two species do not overlap. However, it is well documented that the dhole existed as far west as Europe during the Pleistocene [24]. Thus, one possible explanation could be the presence of dholes in the Middle East in the past, from where they could have encountered and mixed with African hunting dogs in North Africa. It must, however, be stressed that given that there has never been any reported evidence of dholes in either the Middle East or North Africa, our hypothesis is purely speculative. The timing and location of this admixture event remain unresolved.
Although there have been several reports of hybridization between dogs and Ethiopian wolves [13, 15], the genetic history of the Ethiopian wolf has not previously been investigated using nuclear genomic data. The D-statistics-based analyses provided evidence for gene flow between Ethiopian wolves and not only African golden wolves, but also golden jackals, gray wolves, and coyotes (Figure S3). The finding of considerable gene flow between the Ethiopian and Eastern African golden wolf lineages is not surprising, given their geographical co-occurrence in Africa. We consistently also observed a Northwestern-Eastern split in the African golden wolves and note that this correlates with our finding that the Ethiopian wolf contributes a higher amount to the Eastern African golden wolves. This suggests that admixture from the Ethiopian wolf may be a key factor contributing to African golden wolf population structure.
The presence of gene flow between the Ethiopian wolf and the other crown canid species is more surprising, given their lack of range overlap. However, this might be explained through the previously reported extensive evidence of admixture between African golden wolves and gray wolves, coyotes, and golden jackals [4, 9]. In short, we hypothesize that the signal of Ethiopian wolf admixture into the other crown canid species is mediated by African golden wolves. A summary of all the admixture events inferred in this study is shown in Figure 3A.
The uncertain placement of the African golden wolf (Figure 2C, panel labeled 17), combined with evidence of gene flow from the Ethiopian wolf, led us to investigate whether the African golden wolf is a species of hybrid origin, derived from a mixture between gray and Ethiopian wolves or close relatives. The current distribution ranges of Ethiopian and gray wolves do not overlap, and indeed, the known historical distribution of Ethiopian wolves is restricted to the Ethiopian highlands [15]. However, extensive gene flow with other canids, combined with the two distinct levels of Ethiopian wolf gene flow into the two distinct populations of African golden wolves, suggests that either Ethiopian wolves or a close (now extinct) relative had, in the past, a much larger range within Africa and thus greater opportunity to admix with other canid species. Additionally, mitochondrial analyses of African golden wolves, in this and previous studies, find them to be most closely related to gray wolves [2, 3, 4, 25]. Further, African golden wolves are a sister clade to gray wolves and coyotes in the nuclear phylogeny, whereas they are a sister group to the Middle Eastern gray wolves in the mitochondrial phylogeny. We explored the relationships between the golden jackal, Ethiopian wolf, and African golden wolf using G-PhoCS [26] (Table S1), which supported the finding of gene flow into the Ethiopian wolf from the African golden wolf. To further explore the relationship between these species and the gray wolf, we used TreeMix [22] and admixture graphs [23] to obtain trees, which were used to assess whether the African golden wolf is a hybrid species (Figures 4B and 4C). We initially constructed a graph including the coyote, Ethiopian wolf, gray wolf, and Andean fox and assessed the most likely position for the African golden wolf in this graph. The placement of the two African golden wolf populations in this tree was further investigated by modeling them as sister to all possible nodes and as admixed populations deriving ancestry from two possible nodes. Finally, the model was extended to account for African golden wolf admixture into the Ethiopian wolf. We found that the common ancestor of the African golden wolf populations is best modeled as admixed between a component related to the Ethiopian wolf (∼28%) and another related to the gray wolf (∼72%) (worst-fitting f statistic Z value = −1.086; Figure 4C). Finally, the northwestern African golden wolf population is more closely related to the gray wolf, which is best explained in our model through admixture from gray wolves.
Figure 4.

Modeling the Ancestry of African Golden Wolves
(A) TreeMix tree with all samples, estimated using the pairwise correlation of allele frequencies between all groups of samples. This tree is fit with three migration edges. The first three migration edges all indicate extensive gene flow from the gray and Ethiopian wolves into the African golden wolves, suggesting a hybrid origin for this species.
(B and C) The QP graph is an admixture graph estimated using all pairwise D statistics between samples. Estimated genetic drift is shown along the solid lines in units of f2 distance (parts per thousand), and estimated mixture proportions are given along the dotted lines. Names of specific modern populations are shown in full, whereas hypothetical ancestral individuals are represented by letters.
(B) This tree shows all the possible placements—highlighted in red—for the Northwestern African golden wolf, chosen due to their low levels of gene flow with the Ethiopian wolf. These were modeled as possible internal and external nodes and as an admixed group from all possible node pairs.
(C) The best fitting graph with a Z value closest to 0, modeling the Ethiopian wolf-like and gray wolf-like ancestry of Northwestern and Eastern African golden wolves, as well as gene flow into modern Ethiopian wolves from the Eastern African golden wolves. This admixture graph suggests that the African golden wolves are probably a species of hybrid origin, derived from the gray wolf and Ethiopian wolf as the parental species. Further, Figure S4 shows admixture graphs showing potential gene flow from a “ghost” basal canid lineage into the ancestor of wolves and dogs.
Lastly, our attention was drawn to the curious result of potential gene flow between the lineage representing the ancestor of the coyote and gray wolves and that representing all other canid species, excluding the African hunting dog (Figure S4), in all D statistics analyses computed with the coyote or gray wolf in the ingroup, namely position H2. Notably, these signals disappeared when the sister clade—H3—was replaced with the African hunting dog, leading us to hypothesize that the coyote and gray wolf genomes may contain a basal ancestral component derived from an as-yet-unidentified species that evolved after the divergence of the African hunting dog branch from the other canid species and that the signal of gene flow can be attributed to outgroup attraction of the coyote and gray wolf lineage. Note that such a hypothetical ancient admixture event would also explain the unexpectedly basal position of the coyote mitochondrial genome—the coyote may simply have retained the mitogenome from this unidentified ancestor. We acknowledge that the existence of an unknown ancestral component would be controversial—previous analyses of coyotes and the fossil records from their direct ancestors argue that they have been strictly restricted to North America for over a million years [27, 28]. However, within North America, the coyote has coexisted alongside several now extinct canids, including the American dhole (Cuon sp.) and dire wolf (Canis dirus) [29]. Although the unknown ancestral component to cannot be attributed to any of the known fossil species at this time, future paleogenomic analyses on such materials (if any can be found with surviving DNA) may provide exciting possibilities to test our hypothesis.
In conclusion, our results highlight how interspecific gene flow has played an important role in shaping the species and population structure of gray wolves, coyotes, African golden wolves, golden jackals, and Ethiopian wolves and that African golden wolves, coyotes, and gray wolves may have been greatly affected by hybridization events. In particular, we conclude not only that African golden wolves arose through hybridization between a Ethiopian-wolf-like and gray-wolf-like ancestral population, but that subsequently the resulting northwestern and eastern African golden wolf populations underwent continuous admixture with modern gray and Ethiopian wolves, respectively. We furthermore argue that the common ancestor of gray wolves and coyotes differentiated from the lineage leading to golden jackals, in part by admixing with a dhole-like canid. Finally, the robust signal of gene flow observed between African hunting dogs and dholes testifies to an as-yet-undiscovered prehistoric overlap between the two lineages. This underscores how much remains to be discovered about the history of the wolf-like canids and how paleogenomic approaches may be required to advance our understanding of this group. Lastly, our study adds to the growing evidence for the importance of gene flow and hybridization in the evolution of mammalian species in general [23, 30, 31, 32] and that rather than being isolated entities that evolve along tree-like phylogenies, they are interlinked and evolve through interactions in network-like topologies.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| 8 Canid blood or tissue samples | This paper | Data S1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Proteinase K | Sigma-Aldrich | Cat# 3115844001 |
| Phenol | Bionordika | Cat# A0447,0500 |
| Chloroform | Sigma-Aldrich | Cat# 288306-1L |
| Critical Commercial Assays | ||
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69506 |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28006 |
| NEBNext DNA Sample Prep Master Mix Set 2 | New England Biolabs | Cat# E6070 |
| Deposited Data | ||
| 10 Canid genomes | [33] | Data S1 |
| 2 Canid genomes | [34] | Data S1 |
| 3 Canid genomes | [6] | Data S1 |
| 5 Canid genomes | [9] | Data S1 |
| 1 Canid genomes | [4] | Data S1 |
| 4 Canid genomes | [35] | Data S1 |
| 2 Canid genomes | [18] | Data S1 |
| 1 Canid genomes | [36] | Data S1 |
| 5 Canid genomes | [37] | Data S1 |
| 1 African golden wolf | This article | NCBI SRA sample accession number: SAMN10199001 |
| 2 African hunting dogs | This article | NCBI SRA sample accession numbers: SAMN10180432, SAMN10180433 |
| 3 Coyotes | This article | NCBI SRA sample accession numbers: SAMN10180421, SAMN10180422, SAMN10180423 |
| 1 Dhole | This article | NCBI SRA sample accession number: SAMN10180424 |
| 1 Ethiopian wolf | This article | NCBI SRA sample accession number: SAMN10180425 |
| 2 Golden jackals | This article | NCBI SRA sample accession numbers: SAMN10180426, SAMN10180427 |
| 5 Gray wolves | This article | NCBI SRA sample accession numbers: SAMN10180428, SAMN10180429, SAMN10180430, SAMN10180431, SAMN10180511 |
| Gray wolf reference genome | [7] | N/A |
| Oligonucleotides | ||
| Illumina-compatible adapters | [38] | N/A |
| Software and Algorithms | ||
| PALEOMIX | [39] | https://github.com/MikkelSchubert/paleomix; RRID:SCR_015057 |
| AdapterRemoval2 | [40] | https://github.com/MikkelSchubert/adapterremoval; RRID:SCR_011834 |
| bwa v0.7.10 | [41] | http://bio-bwa.sourceforge.net/; RRID:SCR_010910 |
| Picard v1.128 | N/A | https://broadinstitute.github.io/picard; RRID:SCR_006525 |
| GATK v3.3.0 | [42, 43] | https://broadinstitute.github.io/picard; RRID:SCR_001876 |
| ANGSD | [44] | https://github.com/ANGSD/angsd |
| samtools v1.2 | [41] | http://samtools.sourceforge.net/; RRID:SCR_002105 |
| realSFS | [44] | https://github.com/ANGSD/angsd |
| NGSadmix | [20] | http://www.popgen.dk/software/index.php/NgsAdmix; RRID:SCR_003208 |
| ASTRAL-II | [45] | https://github.com/smirarab/ASTRAL |
| RAxML | [46] | https://sco.h-its.org/exelixis/software.html; RRID:SCR_006086 |
| trimal | [47] | http://trimal.cgenomics.org/ |
| FastTree2 | [48] | http://www.microbesonline.org/fasttree/; RRID:SCR_015501 |
| DiscoVista | [49] | https://github.com/esayyari/DiscoVista |
| MtArchitect | [10] | http://biologiaevolutiva.org/tmarques/mtarchitect/ |
| MAFFT | [50] | https://mafft.cbrc.jp/alignment/software/; RRID:SCR_011811 |
| Jalview | [51] | http://www.jalview.org/; RRID:SCR_006459 |
| jmodeltest2 | [52] | https://github.com/ddarriba/jmodeltest2; RRID:SCR_015244 |
| phyML | [53] | http://www.atgc-montpellier.fr/phyml/; RRID:SCR_014628 |
| ADMIXTOOLS | [23] | https://github.com/DReichLab/AdmixTools |
| TreeMix | [22] | https://bitbucket.org/nygcresearch/treemix/wiki/Home |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shyam Gopalakrishnan (shyam@snm.ku.dk).
Experimental Model and Subject Details
The current study uses short read sequencing data from the full genomes of 47 canids spanning 8 different species (when the domestic dog is considered a different species from the gray wolf) from Africa, Eurasia and North America, to address questions about the genetic affinities of these species to each other, and the role of interspecific gene flow in shaping the evolution of the genus Canis. All known information on the context and sequencing coverage of the samples is provided in Data S1.
Method Details
Whole-genome sequencing
DNA was extracted from 10 modern samples of fresh blood or tissue using the DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany) following the manufacturer’s protocol. Three samples (‘African hunting dog Kenya 1’, ‘African hunting dog Somalia’ and ‘Golden jackal Calcutta’) are from historical museum hides and were digested in a proteinase K-containing buffer following [54]; these digests were subsequently treated in a phenol chloroform step following [55]. The supernatant was then mixed 1:10 with a binding buffer following [56] in a binding apparatus following [57], including a Minelute column (QIAGEN, Hilden, Germany) that was then washed and DNA was eluted according to the manufacturer’s guidelines. All extracts were incorporated into double-stranded DNA libraries build using the NEBNext DNA Sample Prep Master Mix Set 2 (E6070 - New England Biolabs, Beverly, MA, USA) following the manufacturer’s protocol and Illumina-compatible adapters [38]. Libraries were sequenced using 50 base pair single (Golden jackal Calcutta, Hunting dog Kenya 1 and Hunting dog Somalia) or 100 base pair paired end (remaining samples) read chemistry on Illumina HiSeq 2000 and 2500 (Illumina, San Diego, CA, USA) platforms.
Read mapping
The short-read data from each sample, including samples from previous publications, was processed using the PALEOMIX pipeline [39]. As the first step of the pipeline, low quality and missing bases were trimmed from the reads, followed by removal of adapters using AdapterRemoval2 [40]. Additionally, all paired end reads where the two reads overlapped by more than 10 base pairs were merged into a single read. Subsequently, the reads from each sample were mapped to the wolf reference genome [7] using bwa (v0.7.10; aln algorithm) [41]. The mapped reads were filtered for PCR and optical duplicates using Picard (v1.128, https://broadinstitute.github.io/picard), and reads that mapped to multiple locations in the genome were excluded. GATK (v3.3.0) [42, 43] was used to perform an indel realignment step to adjust for increased error rates at the end of short reads in the presence of indels. In the absence of a curated dataset of indels in wolves, this step relied on a set of indels identified in the specific sample being processed. After the initial mapping and quality control, the coverages of the samples ranged from 0.6 to 26.6x (for details see Data S1).
Genotype calling
The samples in this study span a wide range of genomic coverages. To avoid introducing biases in various analyses resulting from genotype calling in low coverage samples [58], the uncertainty in genotypes was instead propagated through to downstream analyses using genotype likelihoods. The genotype likelihoods at variant sites were computed in ANGSD [44] using the mapped reads, with the model for reads used by samtools (v1.2) [41]. Bases with base qualities lower than 20 and reads with mapping quality lower than 20 were discarded. Only sites with data present in at least 46 out of the 48 samples were retained. All sites with minor allele frequencies below 0.1 were excluded.
Quantification and Statistical Analysis
Heterozygosity
The heterozygosity for each sample was calculated using ANGSD, by estimating the per-sample folded site frequency spectrum (SFS) and using the fraction of singletons in the sample as a measure of heterozygosity. The variance of the estimate was obtained by bootstrapping the sites 100 times to obtain 100 bootstrapped estimates of the SFS. Briefly, for each sample, the site allele frequency for every site was estimated (“-doSaf 1 -fold 1”) using the reference genome as ancestral, while keeping all other parameters as above. Afterward, the SFS and their corresponding bootstraps was estimated for each sample using realSFS and, for each case, the fraction of singletons was calculated. The sample heterozygosities are shown in Figure S1.
Admixture
Using the genotype likelihoods obtained from the ANGSD pipeline, the ancestry clusters and admixture proportions for 48 samples representing all species (for details see Data S1) were estimated using NGSadmix [20] based on 5.7 million SNPs. Admixture analyses were performed using only markers with minor allele frequency greater than 0.1. We used a range of values for the number of clusters (2-15), to explore the structure in the dataset. To avoid convergence to local optima, the admixture analysis was repeated at least 200 times with different random initial parameter values, and the replicate with the highest likelihood was chosen.
Nuclear genome phylogeny
Using 28 individuals representing all species in this study (for details see Data S1), nuclear genome phylogenetic reconstruction based on coalescence of gene trees was performed using 100 ASTRAL-II trees [45], and an extended majority rule consensus tree was made with RAxML [46] using default parameters. Each tree was based on gene trees inferred from 5000 regions, each roughly 10 kb long sampled from a consensus genome sequence per individuals generated in ANGSD [44] using the “-doFasta 1” option. Regions with missing data were excluded using trimal [47] under the parameters “-gappyout -resoverlap 0.60 -seqoverlap 60.” Each gene tree was generated in FastTree2 [48] using a generalized time-reversible model for sequence evolution. A cut-off at a minimum of four samples per tree was selected, before generation of individual ASTRAL-II trees. Local posterior probabilities and quartet frequencies for the three possible unrooted resolutions around each internal branch were computed using ASTRAL [59] and visualized using DiscoVista [49]. Two support values are computed on the consensus ASTRAL tree: i) frequency of each branch in the 100 replicates and ii) means of local posterior probability across the 100 replicates. The local posterior probability is computed as the probability that the proportion of gene trees consistent with the bipartition shown in the full phylogeny is greater than 0.33, under a multinomial model with three possible outcomes, each representing a bipartition at the interior branch.
Since the branch lengths in the ASTRAL-II analysis are in terms of coalescent time units, another phylogeny was generated to get branch lengths proportional to evolutionary distances, from 1000 randomly sampled 1 kb regions across the genome using a concatenated analysis in RaxML [46], using a GTR-GAMMA model of sequence evolution.
Species split times
The divergence times between the different species were computed using the two plus two (TT) method [60], which uses a pair of samples, and the distribution of derived alleles at all sites, to compute the split time for a focal population from a contrast population. Specifically, the method uses the counts of sites in the genome where the samples fit into one of 9 configurations, i.e., both samples carry 0 derived alleles, one sample carries 1 derived allele and the other carries 0, and so on, to get an estimate of the time of either sample from the most recent common ancestor of the pair of samples. The method provides two estimates of split times for each pair of samples, with one sample treated as the focal population and the other as the contrast population. One of the main advantages of this method is that it is not affected by the population size dynamics of the two populations after the split, but it does assume no migration and constant population size in the ancestor of the two populations (before the split).
In order to reduce the number of comparisons in this model, we chose one representative of each population for this analysis, viz., dhole – Beijing Zoo, African hunting dog – Kenya 1, golden jackal – Syria, African golden wolf Northwestern – Morocco, African golden wolf Eastern – Kenya, Ethiopian wolf – Ethiopia, coyote – California, gray wolf European – Spain, gray wolf Asian – Altai, gray wolf American – Greenland and Mexico 1, dog – India 1 and Qatar 2. The TT statistic was computed for each pair of samples, using only scaffolds longer than 1 Mb (705 in all), excluding sites with less than 5x coverage in either sample. The bootstrap estimate of the statistic and its variance was obtained treating each scaffold as a single block [61].
Mitochondrial reconstruction using de novo assembly
We used MtArchitect [10] to reconstruct de novo the mitochondrial genomes for 17 canids representing all species (for details see Data S1). The genomes were aligned using MAFFT [50] and curated with Jalview [51]. MtArchitect is designed to deal with the presence of numts, by aligning the reads to the mitochondrial and nuclear genome seperately, and including only read pairs (or single end reads), where both reads of the pair map unambiguously and with high mapping quality to the mitochondria. We tested a total of 56 phylogenetic models with jmodeltest2 [52] and chose HKY85 with gamma-distributed variation in the substitution rate and a fixed proportion of invariable sites as the most suitable model, which finally was used to construct maximum-likelihood tree using phyML [53]. We generally observed a small amount of undetermined sites, but the two African hunting dogs analyzed displayed poorer alignments and smaller genomes. This is most likely due to the reconstruction biases associated with using a distant reference and a lack of paired-end data to exploit the maximum potential of MtArchitect. Alignment visualization and tree inspection of the reconstructions confirmed that the phylogenetic clustering complied with previously reported data [4]. We observed, however, that the D-loop was particularly enriched in undetermined sites, and aligned notably worse than the remaining sequence. Given its potentially confounding nature and its small contribution to the phylogeny reconstruction when the rest of the sequence is well resolved [10], the D-loop, as well as minor positions containing the majority of the gaps, were manually discarded, resulting in a final 15.435 bp alignment.
D statistics
We used allele frequency-based D statistics as implemented in ADMIXTOOLS [23] to evaluate possible gene flow between the different lineages. D statistics are based on the observation that, if the given topology (((H1,H2), H3), Outgroup) is correct, then under the null hypothesis of no gene flow between any of the two lineages in the ingroup (H1, H2) and the lineage H3, the number of sites across the genome where the segregation patterns ABBA and BABA occur should be equal in number, as they can arise solely due to incomplete lineage sorting. But the presence of gene flow between H1 and H3 would lead to an increase in the number of BABA sites (H1 and H3 share the same allele B), while gene flow between H2 and H3 would lead to an increase in the number of ABBA sites (H2 and H3 share the same allele B). The D statistic measures the disparity between the number of ABBA and BABA sites across the genome to infer gene flow.
To account for the varying depth of coverage of the samples, we used a randomly sampled allele per site instead of called genotypes. Reads with mapping quality lower than 30, bases with quality lower than 20 and sites with coverage lower than 3 were discarded from the analysis. The significance of each test was estimated using a weighted block jackknife procedure over 1 Mb blocks. Deviations from D = 0 were presumed significant when the observed Z-score was above or below 3.3 (|Z|>3.3). To avoid inflating significance of the tests, only scaffolds 1 Mb or longer (∼70% of the genome) were used in the analysis. Tests were performed with combinations of samples as individuals and samples were grouped into categories representing the main genetic clusters (for details see Data S1).
TreeMix
TreeMix [22] was used to infer potential admixture edges in the phylogeny. TreeMix models the correlation of allele frequencies at variable positions across the genome. The correlations that do not fit well under the modeled tree are then corrected for using migration events. We used a randomly sampled allele for each sample and a similar filtering approach as the one described for the D statistics tests. Tests were with combinations of samples as individuals and samples grouped into categories representing the main genetic clusters (for details see Data S1). Sites with at least one individual with coverage per group were kept. The final dataset consisted of a total of 834,537 segregating sites. We ran TreeMix on the final dataset assuming 0 to 4 migration edges (m = 0-4). For each value of m, we ran 100 replicates starting in different seed values and evaluated the replicate with the highest likelihood. Figure S3B shows the best replicate obtained for the graph modeled with four migration edges.
qpGraph
We used qpGraph from the ADMIXTOOLS package [23] to evaluate the relationships between the different species in our samples. In particular, we addressed the question of whether the African golden wolf can be modeled as a hybrid species. qpGraph uses the correlation on all possible f statistic tests in a given admixture graph to evaluate its overall fit. The same dataset and filtering parameters used for the D statistics tests were used in this analysis. Samples were grouped into clusters representing the main lineages in the admixture graph as indicated in Data S1. First, we started with a tree including the coyote, Ethiopian wolf, gray wolf and Andean fox and evaluated the most likely branching point for the African golden wolf. Then, we modeled the African golden wolf as a sister clade to all possible internal and external nodes and as an admixed group from all possible node pairs. Finally, we extended our model with an admixture event to account for African golden wolf admixture in the Ethiopian wolf (Figure 4).
Data and Software Availability
The BioProject accession number for the short read sequences used in this paper is available at the NCBI short read archive under the accession PRJNA494815.
Acknowledgments
The authors would like to acknowledge the assistance of the Danish National High-Throughput Sequencing Centre for assistance in Illumina data generation. We also gratefully acknowledge the Danish National Supercomputer for Life Sciences, Computerome (https://www.computerome.dk), for the computational resources to perform the sequence analyses. For making sample material available, we would like to thank Jörns Fickel from Leibniz-Institut für Zoo- und Wildtierforschung and Kristian Gregersen from the Natural History Museum of Denmark. We also acknowledge the following for funding our research: the Qimmeq project funded by The Velux Foundations and Aage og Johanne Louis-Hansens Fond; Carlsbergfondet grant CF14–0995 and Marie Skłodowska-Curie Actions grant 655732-WhereWolf to S.G.; grant 676154-ArchSci2020 to J.N.; NSFC grant 91531303 to G.-D.W.; Danish National Research Foundation grant DNRF94, Lundbeckfonden grant R52–5062, and ERC Consolidator grant 681396-Extinction Genomics to M.T.P.G.; and the Universities of Oslo and Copenhagen for a PhD stipend awarded to M.-H.S.S. T.M.-B. is supported by MINECO/FEDER, UE, grant BFU2017-86471-P, NIMH grant U01 MH106874, a Howard Hughes Medical Institute International Early Career grant, Obra Social “La Caixa,” and Secretaria d’Universitats i Recerca and CERCA Programme del Departament d’Economia i Coneixement de la Generalitat de Catalunya.
Author Contributions
S.G., M.-H.S.S., A.J.H., and M.T.P.G. conceived the study. M.-H.S.S. and C.C. did the DNA lab work for high-throughput sequencing. S.G., J.R.-M., J.N., J.A.S.C., F.G.V., M.d.M.M., L.K., A.S., V.M.G.-B., Y.-H.L., and S.M. performed analyses. C.F., P.G., K.-P.K., J.B., E.K.R., and M.P.H.-J. contributed with sample collection. B.P. and T.S.-P. provided computation expertise and support. L.B., Ø.W., T.M.-B., A.J.H., and M.T.P.G. supervised the work. S.G., M.-H.S.S., J.R.-M., and M.T.P.G. wrote the manuscript. All authors contributed to the preparation and editing of the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: October 18, 2018
Footnotes
Supplemental Information includes four figures, two tables, and one data file and can be found with this article online at https://doi.org/10.1016/j.cub.2018.08.041.
Supplemental Information
Table showing the catalog number, species information, average genomic sequencing coverage, identified genetic clusters, location and date of sample collection, and the publication associated with the publicly available data.
References
- 1.Viranta S., Atickem A., Werdelin L., Stenseth N.C. Rediscovering a forgotten canid species. BMC Zoology. 2017;2:6. [Google Scholar]
- 2.Rueness E.K., Asmyhr M.G., Sillero-Zubiri C., Macdonald D.W., Bekele A., Atickem A., Stenseth N.C. The cryptic African wolf: Canis aureus lupaster is not a golden jackal and is not endemic to Egypt. PLoS ONE. 2011;6:e16385. doi: 10.1371/journal.pone.0016385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaubert P., Bloch C., Benyacoub S., Abdelhamid A., Pagani P., Djagoun C.A.M.S., Couloux A., Dufour S. Reviving the African wolf Canis lupus lupaster in North and West Africa: a mitochondrial lineage ranging more than 6,000 km wide. PLoS ONE. 2012;7:e42740. doi: 10.1371/journal.pone.0042740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koepfli K.-P., Pollinger J., Godinho R., Robinson J., Lea A., Hendricks S., Schweizer R.M., Thalmann O., Silva P., Fan Z. Genome-wide evidence reveals that African and Eurasian golden jackals are distinct species. Curr. Biol. 2015;25:2158–2165. doi: 10.1016/j.cub.2015.06.060. [DOI] [PubMed] [Google Scholar]
- 5.Lindblad-Toh K., Wade C.M., Mikkelsen T.S., Karlsson E.K., Jaffe D.B., Kamal M., Clamp M., Chang J.L., Kulbokas E.J., 3rd, Zody M.C. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 6.Fan Z., Silva P., Gronau I., Wang S., Armero A.S., Schweizer R.M., Ramirez O., Pollinger J., Galaverni M., Ortega Del-Vecchyo D. Worldwide patterns of genomic variation and admixture in gray wolves. Genome Res. 2016;26:163–173. doi: 10.1101/gr.197517.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gopalakrishnan S., Samaniego Castruita J.A., Sinding M.S., Kuderna L.F.K., Räikkönen J., Petersen B., Sicheritz-Ponten T., Larson G., Orlando L., Marques-Bonet T. The wolf reference genome sequence (Canis lupus lupus) and its implications for Canis spp. population genomics. BMC Genomics. 2017;18:495. doi: 10.1186/s12864-017-3883-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allman E.S., Degnan J.H., Rhodes J.A. Identifying the rooted species tree from the distribution of unrooted gene trees under the coalescent. J. Math. Biol. 2011;62:833–862. doi: 10.1007/s00285-010-0355-7. [DOI] [PubMed] [Google Scholar]
- 9.Freedman A.H., Gronau I., Schweizer R.M., Ortega-Del Vecchyo D., Han E., Silva P.M., Galaverni M., Fan Z., Marx P., Lorente-Galdos B. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 2014;10:e1004016. doi: 10.1371/journal.pgen.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lobon I., Tucci S., de Manuel M., Ghirotto S., Benazzo A., Prado-Martinez J., Lorente-Galdos B., Nam K., Dabad M., Hernandez-Rodriguez J. Demographic history of the genus Pan inferred from whole mitochondrial genome reconstructions. Genome Biol. Evol. 2016;8:2020–2030. doi: 10.1093/gbe/evw124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ostrander E.A., Wayne R.K. The canine genome. Genome Res. 2005;15:1706–1716. doi: 10.1101/gr.3736605. [DOI] [PubMed] [Google Scholar]
- 12.Wayne R.K., Ostrander E.A. Origin, genetic diversity, and genome structure of the domestic dog. BioEssays. 1999;21:247–257. doi: 10.1002/(SICI)1521-1878(199903)21:3<247::AID-BIES9>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 13.Gottelli D., Sillero-Zubiri C., Applebaum G.D., Roy M.S., Girman D.J., Garcia-Moreno J., Ostrander E.A., Wayne R.K. Molecular genetics of the most endangered canid: the Ethiopian wolf Canis simensis. Mol. Ecol. 1994;3:301–312. doi: 10.1111/j.1365-294x.1994.tb00070.x. [DOI] [PubMed] [Google Scholar]
- 14.Yumnam B., Negi T., Maldonado J.E., Fleischer R.C., Jhala Y.V. Phylogeography of the golden jackal (Canis aureus) in India. PLoS ONE. 2015;10:e0138497. doi: 10.1371/journal.pone.0138497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marino, J., and Sillero-Zubiri, C. (2011). Canis simensis. The IUCN Red List of Threatened Species 2011: e.T3748A10051312. 10.2305/IUCN.UK.2011-1.RLTS.T3748A10051312.en. [DOI]
- 16.vonHoldt B.M., Pollinger J.P., Earl D.A., Knowles J.C., Boyko A.R., Parker H., Geffen E., Pilot M., Jedrzejewski W., Jedrzejewska B. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 2011;21:1294–1305. doi: 10.1101/gr.116301.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galov A., Fabbri E., Caniglia R., Arbanasić H., Lapalombella S., Florijančić T., Bošković I., Galaverni M., Randi E. First evidence of hybridization between golden jackal (Canis aureus) and domestic dog (Canis familiaris) as revealed by genetic markers. R. Soc. Open Sci. 2015;2:150450. doi: 10.1098/rsos.150450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.vonHoldt B.M., Cahill J.A., Fan Z., Gronau I., Robinson J., Pollinger J.P., Shapiro B., Wall J., Wayne R.K. Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2016;2:e1501714. doi: 10.1126/sciadv.1501714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.vonHoldt B.M., Kays R., Pollinger J.P., Wayne R.K. Admixture mapping identifies introgressed genomic regions in North American canids. Mol. Ecol. 2016;25:2443–2453. doi: 10.1111/mec.13667. [DOI] [PubMed] [Google Scholar]
- 20.Skotte L., Korneliussen T.S., Albrechtsen A. Estimating individual admixture proportions from next generation sequencing data. Genetics. 2013;195:693–702. doi: 10.1534/genetics.113.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lorenzen E.D., Heller R., Siegismund H.R. Comparative phylogeography of African savannah ungulates. Mol. Ecol. 2012;21:3656–3670. doi: 10.1111/j.1365-294X.2012.05650.x. [DOI] [PubMed] [Google Scholar]
- 22.Pickrell J.K., Pritchard J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:e1002967. doi: 10.1371/journal.pgen.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patterson N., Moorjani P., Luo Y., Mallick S., Rohland N., Zhan Y., Genschoreck T., Webster T., Reich D. Ancient admixture in human history. Genetics. 2012;192:1065–1093. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ripoll M.P., Morales Pérez J.V., Sanchis Serra A., Aura Tortosa J.E., Montañana I.S. Presence of the genus Cuon in upper Pleistocene and initial Holocene sites of the Iberian Peninsula: new remains identified in archaeological contexts of the Mediterranean region. J. Archaeol. Sci. 2010;37:437–450. [Google Scholar]
- 25.Werhahn G., Senn H., Kaden J., Joshi J., Bhattarai S., Kusi N., Sillero-Zubiri C., Macdonald D.W. Phylogenetic evidence for the ancient Himalayan wolf: towards a clarification of its taxonomic status based on genetic sampling from western Nepal. R. Soc. Open Sci. 2017;4:170186. doi: 10.1098/rsos.170186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gronau I., Hubisz M.J., Gulko B., Danko C.G., Siepel A. Bayesian inference of ancient human demography from individual genome sequences. Nat. Genet. 2011;43:1031–1034. doi: 10.1038/ng.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurtén B. A history of coyote-like dogs (Canidae, Mammalia) Acta Zool. Fenn. 1974;140:1–38. [Google Scholar]
- 28.Nowak R.M. Museum of Natural History, University of Kansas; 1979. North American Quaternary Canis. [Google Scholar]
- 29.Tedford R.H., Wang X., Taylor B.E. Phylogenetic systematics of the North American fossil Caninae (Carnivora: Canidae) Bull. Am. Mus. Nat. Hist. 2009;325:1–218. [Google Scholar]
- 30.Jónsson H., Schubert M., Seguin-Orlando A., Ginolhac A., Petersen L., Fumagalli M., Albrechtsen A., Petersen B., Korneliussen T.S., Vilstrup J.T. Speciation with gene flow in equids despite extensive chromosomal plasticity. Proc. Natl. Acad. Sci. USA. 2014;111:18655–18660. doi: 10.1073/pnas.1412627111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Figueiró H.V., Li G., Trindade F.J., Assis J., Pais F., Fernandes G., Santos S.H.D., Hughes G.M., Komissarov A., Antunes A. Genome-wide signatures of complex introgression and adaptive evolution in the big cats. Sci. Adv. 2017;3:e1700299. doi: 10.1126/sciadv.1700299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar V., Lammers F., Bidon T., Pfenninger M., Kolter L., Nilsson M.A., Janke A. The evolutionary history of bears is characterized by gene flow across species. Sci. Rep. 2017;7:46487. doi: 10.1038/srep46487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Auton A., Rui Li Y., Kidd J., Oliveira K., Nadel J., Holloway J.K., Hayward J.J., Cohen P.E., Greally J.M., Wang J. Genetic recombination is targeted towards gene promoter regions in dogs. PLoS Genet. 2013;9:e1003984. doi: 10.1371/journal.pgen.1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campana M.G., Parker L.D., Hawkins M.T.R., Young H.S., Helgen K.M., Szykman Gunther M., Woodroffe R., Maldonado J.E., Fleischer R.C. Genome sequence, population history, and pelage genetics of the endangered African wild dog (Lycaon pictus) BMC Genomics. 2016;17:1013. doi: 10.1186/s12864-016-3368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y.-H., Wang L., Xu T., Guo X., Li Y., Yin T.-T., Yang H.-C., Yang H., Adeola A.C., Sanke J.O. Whole-genome sequencing of African dogs provides insights into adaptations against tropical parasites. Mol. Biol. Evol. 2017;35:287–298. doi: 10.1093/molbev/msx258. [DOI] [PubMed] [Google Scholar]
- 36.Wang G.-D., Zhai W., Yang H.-C., Fan R.-X., Cao X., Zhong L., Wang L., Liu F., Wu H., Cheng L.-G. The genomics of selection in dogs and the parallel evolution between dogs and humans. Nat. Commun. 2013;4:1860. doi: 10.1038/ncomms2814. [DOI] [PubMed] [Google Scholar]
- 37.Wang G.-D., Zhai W., Yang H.-C., Wang L., Zhong L., Liu Y.-H., Fan R.-X., Yin T.-T., Zhu C.-L., Poyarkov A.D. Out of southern East Asia: the natural history of domestic dogs across the world. Cell Res. 2016;26:21–33. doi: 10.1038/cr.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meyer M., Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010;2010:t5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 39.Schubert M., Ermini L., Der Sarkissian C., Jónsson H., Ginolhac A., Schaefer R., Martin M.D., Fernández R., Kircher M., McCue M. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 2014;9:1056–1082. doi: 10.1038/nprot.2014.063. [DOI] [PubMed] [Google Scholar]
- 40.Schubert M., Lindgreen S., Orlando L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes. 2016;9:88. doi: 10.1186/s13104-016-1900-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korneliussen T.S., Albrechtsen A., Nielsen R. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mirarab S., Warnow T. ASTRAL-II: coalescent-based species tree estimation with many hundreds of taxa and thousands of genes. Bioinformatics. 2015;31:i44–i52. doi: 10.1093/bioinformatics/btv234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Capella-Gutiérrez S., Silla-Martínez J.M., Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–1973. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Price M.N., Dehal P.S., Arkin A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sayyari, E., Whitfield, J.B., and Mirarab, S. (2017). DiscoVista: interpretable visualizations of gene tree discordance. arXiv, arXiv: 1709.09305, https://arxiv.org/abs/1709.09305. [DOI] [PubMed]
- 50.Yamada K.D., Tomii K., Katoh K. Application of the MAFFT sequence alignment program to large data-reexamination of the usefulness of chained guide trees. Bioinformatics. 2016;32:3246–3251. doi: 10.1093/bioinformatics/btw412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Darriba D., Taboada G.L., Doallo R., Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods. 2012;9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 54.Gilbert M.T.P., Tomsho L.P., Rendulic S., Packard M., Drautz D.I., Sher A., Tikhonov A., Dalén L., Kuznetsova T., Kosintsev P. Whole-genome shotgun sequencing of mitochondria from ancient hair shafts. Science. 2007;317:1927–1930. doi: 10.1126/science.1146971. [DOI] [PubMed] [Google Scholar]
- 55.Carøe C., Gopalakrishnan S., Vinner L., Mak S.S.T., Sinding M.-H.S., Samaniego J.A., Wales N., Sicheritz-Pontén T., Gilbert M.T.P. Single-tube library preparation for degraded DNA. Methods Ecol. Evol. 2017;9:410–419. [Google Scholar]
- 56.Allentoft M.E., Sikora M., Sjögren K.-G., Rasmussen S., Rasmussen M., Stenderup J., Damgaard P.B., Schroeder H., Ahlström T., Vinner L. Population genomics of Bronze Age Eurasia. Nature. 2015;522:167–172. doi: 10.1038/nature14507. [DOI] [PubMed] [Google Scholar]
- 57.Dabney J., Knapp M., Glocke I., Gansauge M.-T., Weihmann A., Nickel B., Valdiosera C., García N., Pääbo S., Arsuaga J.-L., Meyer M. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc. Natl. Acad. Sci. USA. 2013;110:15758–15763. doi: 10.1073/pnas.1314445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nielsen R., Paul J.S., Albrechtsen A., Song Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sayyari E., Mirarab S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 2016;33:1654–1668. doi: 10.1093/molbev/msw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schlebusch C.M., Malmström H., Günther T., Sjödin P., Coutinho A., Edlund H., Munters A.R., Vicente M., Steyn M., Soodyall H. Southern African ancient genomes estimate modern human divergence to 350,000 to 260,000 years ago. Science. 2017;358:652–655. doi: 10.1126/science.aao6266. [DOI] [PubMed] [Google Scholar]
- 61.Busing F.M.T.A., Meijer E., Van Der Leeden R. Delete-M jackknife for unequal M. Stat. Comput. 1999;9:3–8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table showing the catalog number, species information, average genomic sequencing coverage, identified genetic clusters, location and date of sample collection, and the publication associated with the publicly available data.