

Abstract
Streptococcus agalactiae is one of the most important pathogens associated with streptococcosis outbreaks in Nile tilapia farms worldwide. High water temperature (above 27°C) has been described as a predisposing factor for the disease in fish. At low temperatures (below 25°C), fish mortalities are not usually observed in farms. Temperature variation can modulate the expression of genes and proteins involved in metabolism, adaptation, and bacterial pathogenicity, thus increasing or decreasing the ability to infect the host. This study aimed to evaluate the transcriptome and proteome of a fish-pathogenic S. agalactiae strain SA53 subjected to in vitro growth at different temperatures using a microarray and label-free shotgun LC-HDMSE approach. Biological triplicates of isolates were cultured in BHIT broth at 22 or 32°C for RNA and protein isolation and submitted for transcriptomic and proteomic analyses. In total, 1,730 transcripts were identified in SA53, with 107 genes being differentially expressed between the temperatures evaluated. A higher number of genes related to metabolism, mainly from the phosphotransferase system (PTS) and ATP-binding cassette (ABC) transport system, were upregulated at 32°C. In the proteome analysis, 1,046 proteins were identified in SA53, of which 81 were differentially regulated between 22 and 32°C. Proteins involved in defense mechanisms, lipid transport and metabolism, and nucleotide transport and metabolism were upregulated at 32°C. A higher number of interactions were observed in proteins involved in nucleotide transport and metabolism. We observed a low correlation between the transcriptome and proteome datasets. Our study indicates that the transcriptome and proteome of a fish-adapted S. agalactiae strain are modulated by temperature, particularly showing differential expression of genes/proteins involved in metabolism, virulence factors, and adaptation.
Keywords: GBS, temperature, fish, microarray, label-free shotgun proteome
Introduction
Streptococcus agalactiae (Lancefield's Group B Streptococcus, GBS) is one of the most important pathogens associated with disease outbreaks in farm-raised Nile tilapia in Brazil (Salvador et al., 2005; Mian et al., 2009; Chideroli et al., 2017), being responsible for significant economic losses annually (Mian et al., 2009). The pathogen causes septicemia and meningoencephalitis in different fish species from freshwater, estuarine, and marine environments worldwide (Evans et al., 2002), and commonly affects adult fish (Mian et al., 2009).
High water temperature, intensive husbandry, and high stock densities are considered as risk factors for streptococcal infection in tilapia (Zamri-Saad et al., 2014). A higher number of GBS outbreaks has been observed during the summer season when the water temperature is >27°C and a higher thermal amplitude is observed during the day (Mian et al., 2009; Kayansamruaj et al., 2014). Fish mortalities resulting from GBS infection are not usually observed when the water temperature is below 25°C (Rodkhum et al., 2011; Marcusso et al., 2015; Chideroli et al., 2017).
In an aquatic environment, fish are often exposed to spatial and temporal variations in temperature that affect the physiological traits and survival of the aquatic host (Boltaña et al., 2017). In addition, variation in water temperature can influence the fish immune response against bacterial infection as well as modify the morphology, metabolism, and pathogenicity of bacteria by increasing or decreasing the host susceptibility to infection (Mereghetti et al., 2008; Kayansamruaj et al., 2014; Zhao et al., 2015).
In this context, global changes in gene and protein expression resulting from adaptation to a particular niche in the host or environment can be evaluated through transcriptomic and proteomic approaches using different methods (Tian et al., 2013; Silva et al., 2014). The microarray approach allows analysis of global gene expression in a microorganism under a given experimental condition (Tian et al., 2013). In contrast, proteomic studies using liquid chromatography-mass spectrometry (LC-MS) allow evaluation of the global expression of the functional genome in a bacterial pathogen at the protein level, under given experimental conditions (Silva et al., 2014).
Regarding thermal stress, small temperature variations represent a challenge to pathogen survival. These effects can block the cell cycle, culminating in stagnation of bacterial growth and proliferation, or depending on the severity of heat stress, cause bacterial death (Richter et al., 2010). Cold shock proteins can alter the bacterial membrane fatty acid composition and global protein profile to prevent cold-induced decreases in membrane fluidity, reduced protein synthesis, inefficient protein folding, and changes in nucleic acid structures (Phadtare and Severinov, 2010). All microorganisms respond to temperature variation through increased expression of heat shock or cold shock proteins, which act as activators or repressors of several other proteins (Ehira et al., 2009). Evaluation of the temperature-induced transcriptome using microarray technology has been reported in Yersinia pestis (Han et al., 2004), Escherichia coli (Gadgil et al., 2005), Streptococcus pyogenes (Smoot et al., 2001), and GBS (Mereghetti et al., 2008), whereas shotgun proteomics analysis has been used to evaluate differential protein abundance induced by temperature in E. coli (Kocharunchitt et al., 2012), Ochrobactrum anthropi (Varano et al., 2016), and Bacillus weihenstephanensis (Stelder et al., 2015).
A previous study has demonstrated that when cultivated at 35°C, GBS shows raid growth, higher hemolytic activity, and a higher viability in tilapia whole blood compared to cultivation at 28°C. In addition, several virulence genes were upregulated at this temperature, inducing a higher mortality rate in infected fish (Kayansamruaj et al., 2014). However, there are no studies demonstrating the global expression of the functional genome of fish-adapted GBS strains at the transcript and protein levels under low or high temperature conditions.
Thus, this study aimed to evaluate the transcriptome and proteome of Streptococcus agalactiae, isolated from diseased fish and subjected to in vitro growth at different temperatures, using microarray and liquid chromatography-mass spectrometry label-free shotgun (LC-HDMSE) approaches.
Materials and methods
Bacterial strains and growth conditions
S. agalactiae SA53 (ST-260) isolated from diseased fish was used in this study. The strain was selected from a culture collection at the National Reference Laboratory for Aquatic Animal Diseases (AQUACEN), and belongs to the most frequent genotype occurring in Latin America (Evans et al., 2008; Godoy et al., 2013; Barato et al., 2015). The complete genome sequence is available in GenBank (accession number CP019802.1; Barony et al., 2017). The strain was streaked on 5% sheep blood agar and incubated at 28°C for 48 h. Colonies were then inoculated in triplicate cultures of 100 mL BHI broth (Brain Heart Infusion broth, Himedia, Mumbai, India) with 0.05% (v/v) Tween 80 (BHIT), and incubated at 32 or 22°C under low agitation. The cultures harvested at the mid-exponential phase of bacterial growth (OD600 = 0.2). Two aliquots (50 mL) of each biological replicate were harvested for RNA and protein extraction.
Transcriptomic approach
RNA extraction
For RNA extraction, a culture volume of 50 mL for each biological triplicate was immediately centrifuged at 12,000 × g for 30 min at 4°C. Bacterial pellets were resuspended in 2 mL of RNAlater (Life Technologies, Carlsbad, USA), incubated at room temperature for 5 min, and then stored at −70°C overnight. The mixture was centrifuged at 12,000 × g for 10 min at 4°C and the pellets were lysed mechanically using a pestle. Total RNA was extracted using TRIzol RNA Isolation Reagent (Invitrogen, Carlsbad, USA) according to the manufacturer's instructions. The extracted RNA of each replicate was treated using the Turbo DNA-free kit (Ambion, Carlsbad, USA) and 1 μL was subjected to GBS-specific PCR (Mata et al., 2004) to confirm the absence of genomic DNA. The extracted RNA was quantified using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, USA) and its quality and integrity were evaluated using TapeStation 2200 (Agilent Technologies, Santa Clara, USA). RNA samples with RNA integrity number (RIN) values ranging from 7.7 to 9.1 were used.
RNA labeling and cRNA synthesis
In total, 50 ng of RNA from each biological replicate was amplified and Cy3-labeled using the Agilent Quick Amp Labeling kit (Agilent Technologies) along with RNA spike-in controls according to the Agilent One-Color Microarray-Based Expression Analysis protocol (Agilent Technologies). The resulting cRNA of each replicate was purified using an RNAeasy Mini Kit (Qiagen, Valencia, USA) according to the manufacturer's instructions. The cRNA concentration was measured using a NanoDrop spectrophotometer.
Hybridization and microarray analysis
The cRNA fragmentation and hybridization were performed using an Agilent Gene Expression Hybridization kit (Agilent Technologies) according to the manufacturer's instructions. Briefly, 600 ng of Cy3-labeled cRNA, with specific activity ≥16.2 pmol Cy3/ng was fragmented at 60°C for 30 min in a mix composed of 5 μL of 10X Blocking agent, 1 μL of 25X Fragmentation Buffer, and nuclease-free water to reach a final volume of 25 μL. After fragmentation, 25 μL of 2X GEx Hybridization Buffer was added to each replicate, and centrifuged at 21,900x g for 2 min. Next, 45 μL of hybridization solution was dispensed onto a custom-made Agilent slide (8 × 60K) formulated based on a library of 4,673 non-redundant genes (Agilent.SingleColor.72627) from nine fish GBS strains (SA07, SA20, SA53, SA288, SA289, SA320, 138P, GD201008_001, ZQ0910) for microarray-based gene expression analysis. Each microarray was created with two probes for each gene. The oligonucleotide microarray slide was designed using the eArray server (https://earray.chem.agilent.com/earray/) with the objective to perform comparative transcriptomic studies with different fish GBS strains. The slide was incubated at 65°C for 18 h at 10 rpm in a hybridization oven (Agilent Technologies). The slides were then washed in two buffers (Gene Expression Wash Buffer 1 and 2, both Agilent Technologies) and scanned using the Agilent DNA Microarray Scanner (Agilent Technologies). The data obtained from array images were extracted using Agilent Features Extraction software version 11.5 (Agilent Technologies).
Data analysis was performed with GeneSpring GX version 11.0.2 software (Agilent Technologies) using the workflow type “find differentially expressed genes.” The processed raw signal intensity for all probes was adjusted with a percentile shift normalization (percentile target = 75). Sample quality was assessed using a box plot to compare the intensity distributions of all replicates. A correlation matrix was used to compare reproducibility across the biological replicates. Principal component analysis (PCA) was used to assess the variability in replicates among temperatures from all genes detected. These quality assessments of transcriptomic data were performed in the workflow analysis, which is incorporated in GeneSpring. Normalized data were then filtered to retain probe sets with present (acceptable flag) signal intensity values in at least two of the three biological replicates in any one out of two conditions. Statistical analyses were performed on filtered data using an unpaired t-test. Genes with p < 0.05 were considered statistically significant. A fold-change cut-off of 2.0 was set to identify a differentially expressed gene with statistical significance between the two temperatures evaluated. Hierarchical clustering analysis was performed with genes showing p < 0.05 to arrange replicates into groups based on gene expression levels. All microarray data were deposited to the Gene Expression Omnibus Database under accession number GSE112416.
qPCR validation analysis
Four differentially expressed genes (DEGs) were selected for further validation by qPCR (Supplementary Table 1). The primers were designed using Primer Express 3.0 software (Life Technologies) and synthesized by Integrated DNA Technologies (IDT, Coralville, USA). The same three biological replicates of treated RNA that were used for microarray analysis for both temperatures were reverse transcribed into cDNA using SuperScript III reverse transcriptase kit (Invitrogen), according to the manufacturer's instructions. qPCR was performed using a GoTaq qPCR Master Mix (Promega) intercalating dye kit in a final volume of 20 μL containing 10 μL of 1 × Master Mix, 0.5 μM of primers (Supplementary Table 1), 0.2 μL of CXR reference dye, and 50 ng of cDNA. The qPCR assay was performed using a ViiA 7 Real-Time PCR System (Life Technologies) with the following cycle protocol: an initial step at 50°C for 2 min followed by 1 cycle of 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. The relative mRNA expression of evaluated genes was normalized to the S. agalactiae reference genes gyrA and recA (Florindo et al., 2012; Faralla et al., 2014) using the ΔΔCt method (Livak and Schmittgen, 2001). Data acquisition and analysis were performed using ViiA 7 Software v.1.2.3 (Life Technologies). The Cohen/Kappa test was used to measure concordance between the microarray and qPCR results (Viera and Garrett, 2005).
Proteomic approach
Protein extraction and tryptic digestion
For protein extraction, a culture volume of 50 mL for each biological triplicate was immediately centrifuged at 16,100x g for 20 min at 4°C and washed thrice with 50 mM Tris-HCl (pH 7.5). The pellets were then resuspended in 1 mL of lysis buffer [42% (w/v) urea, 15% thiourea, 4% sodium deoxycholate (SDC), 12.5 mM Tris-HCl pH 7.5, 1.5% dithiothreitol (DTT)] containing 1% protease inhibitor mix (GE Healthcare, Pittsburgh, USA). The samples were then incubated on ice for 15 min and sonicated on ice using a cell ultrasonic disruptor (Unique, Indaiatuba, Brazil), which was run for 1 min at maximum power (495 W) and stopped for 1 min in cycles until 20 min. The lysates were centrifuged at 21,900x g for 40 min at 4°C. The supernatant was collected, loaded into a Vivaspin 500 column with a threshold of 10 kDa (GE HealthCare), concentrated, and washed five times with 50 mM NH4HCO3. After washing, the concentrated samples were quantified on a Qubit 2.0 fluorometer (Invitrogen, Oregon, USA) using a Qubit protein assay kit (Molecular Probes, Oregon, USA).
For tryptic digestion, 50 μL (2 μg/μL) of each protein extract was mixed with 10 μL of 50 mM NH4HCO3, denatured with 25 μL of 0.2% RapiGest SF surfactant (Waters, Milford, USA), and incubated at 80°C for 15 min. After this, 2.5 μL of 100 mM DTT was added and heated at 60°C for 30 min. Afterwards, 2.5 μL of 300 mM iodoacetamine was added and the samples were kept at room temperature in a dark chamber for 30 min. The proteins were then enzymatically digested with 10 μL (0.5 μg/μL) trypsin (Promega, Madison, USA) and incubated at 37°C for 18 h. Following incubation, 10 μL of 5% trifluoroacetic acid (TFA; Sigma Aldrich, Saint Louis, USA) was added to each sample and incubated at 37°C for 90 min. The resulting peptide extracts were centrifuged at 21,900x g for 30 min at 6°C. Removal of SDC was performed by two-phase solvent extraction with ethyl acetate (Sigma Aldrich; 2:1) followed by addition of 0.5% TFA and centrifugation at 15,000x g for 5 min at 20°C. After centrifugation, the aqueous phase was collected and desalted using C18 MacroSpin Columns (Harvard Apparatus, Holliston, USA), according to the manufacturer's instructions. The samples were dried under vacuum in a Vacufuge Concentrator (Eppendorf, Hamburg, Germany), resuspended in 100 μL of 20 mM ammonium formate (Sigma Aldrich), transferred to Waters Total Recovery vials (Waters), and stored at −70°C until use.
NanoUPLC-HDMSE Analysis
Biological replicates were analyzed by LC-MS using a nanoACQUITY ultra-performance liquid chromatography (UPLC) system coupled to a Synapt G2-Si HDMS mass spectrometer (Waters). Peptides were separated on an ACQUITY UPLC M-Class HSS T3 (1.8, 75 μm × 150 mm–pH 3) column used with a reversed-phase M-Class BEH C18 (5, 300 μm × 50 mm–pH 10) column (Waters). Analytical column temperature was maintained at 55°C. Bidimensional nanoUPLC tandem nano electrospray high definition mass spectrometry (nanoESI-HDMSE) using multiplexed data-independent acquisition (DIA) experiments were conducted using a reverse-phase gradient from 7 to 40% (v/v) acetonitrile (0.1% v/v formic acid) with a simulated 1D analysis and a delivery of 500 nL.min−1 in a nanoACQUITY UPLC 2D Technology system over 60 min (Gilar et al., 2005). Stoichiometric measurements based on scouting runs of the integrated total ion account (TIC) prior to analysis were performed to ensure standardized molar values across all samples. Typical on-column sample loads were 500 ng of total protein digests for each of the five fractions (500 ng per fraction/load).
For each measurement, the mass spectrometer was operated in resolution mode with a typical m/z resolving power of at least 25,000 full width at half maximum (FWHM), an ion mobility cell filled with helium gas, and a cross-section resolving power of at least 40 Ω/Δ Ω (Lalli et al., 2013). Analyses were performed using nano-electrospray ionization in the positive ion mode nanoESI (+) and a NanoLock-Spray ionization source (both from Waters). The lock mass channel was sampled every 30 s. The eluent was sprayed via PicoTip Emitters (Waters) at a spray voltage of 2.8 kv, sampling cone voltage of 30 v, and source offset of 30 v. The source temperature was set at 70°C. The time-of-flight analyzer of the mass spectrometry was calibrated with an MS/MS spectrum of GFP. Final instrument calibration was obtained by the double charged precursor ion [M + 2H]2+ = 785.8426 signal. The exact mass signals from multiplexed ion-mobility DIA scanning (HDMSE) were collected in an alternating low energy and elevated energy acquisition mode. Mass spectrometric analysis of tryptic peptides was performed using a mass spectrometer equipped with a T-Wave-IMS device in MSE and HDMSE modes as described previously (Distler et al., 2014). The radio frequency offset (MS profile) was adjusted such that the nanoESI-HDMSE data were effectively acquired from m/z 400 to 2,000 by the MassLynx v.4.1 software (Waters), ensuring that any masses that were observed in the high energy spectra of < m/z 400 arose from dissociations in the collision cell.
Protein identification and quantitation
HDMSE raw data of samples were processed using Progenesis QI for Proteomics (QIP) v.2.0 (Nonlinear Dynamics, Newcastle, UK), following previously described methods (Kuharev et al., 2015). Imported runs were used for automatic data processing for protein identification and quantitative information using dedicated algorithms in Progenesis QIP. The following parameters were used: peak picking limits = 5; maximum charge retention time limits = 8.
For peptide identification, data from three biological replicates of the strain in two conditions were searched against the S. agalactiae strain SA53 in-house database compiled from the annotated protein.fasta file. Using the database management tool of the ProteinLynx Global Server (PLGS) software v.3.0.2 (Waters), the sequence of each protein was reversed during the database queries and appended to the original database to assess the false positive rate during identification. The following parameters were used for peptide identification in Progenesis: digest reagent = trypsin; maximum missed cleavage = one; maximum protein mass = 600 kDa; modifications: carbamidomethyl of cysteine (fixed), acetyl N-terminal (variable), phosphoryl (variable), oxidation of methionine (variable); search tolerance parameters: peptide tolerance = 10 ppm, fragment tolerance = 20 ppm, maximum false discovery rate (FDR) = 4%; ion matching requirements used default parameters: fragments per peptide = 1, fragments per protein = 3, peptide per protein = 1.
Protein-level quantitation was performed by relative quantitation using an Hi-N algorithm incorporated in Progenesis. Peptides identified with a score ≤ 3, mass error ≥20 ppm, and sequence length ≤ 6 amino acids were removed. Proteins identified with at least two peptides (with ≥1 proteotypic peptide per protein) and present in at least two of three biological replicates for the GBS strain were considered. A protein was considered differentially expressed at 32°C in relation to 22°C if there was also a significant (p ≤ 0.05, ANOVA) change in expression with ≥ two-fold (log2 ratio ≥1.0). To obtain a general overview of protein expression among strains in different conditions, PCA and hierarchical clustering analyses were performed using ggbiplot version 0.55 (Vu, 2011) and gplots version 3.0.1 (Warnes et al., 2016) packages, respectively, in R software version 3.4.1 (R Core Team, 2013). PCA plot was performed with all the proteins detected in our data, whereas the heatmap plot only used proteins if there was significance (p ≤ 0.05, ANOVA). The MS proteomics data are available at the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al., 2016) partner repository under the identifier PXD009330.
Bioinformatic analyses
The transcripts and proteins identified for SA53 under both temperature conditions were analyzed using the prediction tools SurfG+ version 1.0.2 (Barinov et al., 2009) and Cluster of Orthologous Genes (COG) version 2014 db (Galperin et al., 2015) to predict subcellular localization and orthologous groups by functional category, respectively. COG database search was performed using an in-house script (available at: https://github.com/aquacen/blast_cog). Interactions among genes/proteins identified as differentially expressed at 32°C were analyzed using STRING web tool version 10.5 (Szklarczyk et al., 2017) with Streptococcus agalactiae NEM316 as reference and allowed experimental confidences and interaction score ≥0.700 (high confidence). The interaction networks obtained were visualized using Cytoscape version 3.5.1 (Shannon et al., 2003).
Correlation analysis between transcriptomic and proteomic data
To determine the correlation between expressed genes and proteins from our results, the expression level of a transcript with statistical significance (p < 0.05) was correlated with the abundance of the corresponding protein (also p < 0.05) present in the proteomic dataset. Pearson's correlation was calculated using R software version 3.4.1 (R Core Team, 2013).
Results
Transcriptome analysis
The effect of temperature on the GBS transcriptome was evaluated using a whole-genome DNA microarray. We identified a total of 1,730 transcripts in SA53, with at least 98% identity with the probes of the array, characterizing 94.9% of the predicted genome of the strain (Supplementary Table 2). The same transcripts were identified at both temperatures, but differences in transcriptome intensity values were observed during bacterial growth at 22 and 32°C. The quality of transcriptomic data showed small variations among replicates at intensity values (Supplementary Figure 1A) with a correlation coefficient >95% (Supplementary Figure 1B), demonstrating a high reproducibility of transcriptome profiles. Additionally, in the PCA analysis, data from 22 and 32°C could be discriminated (Supplementary Figure 1C).
The 1,730 transcripts originally identified in two out of three replicates were reduced to 579 transcripts with p ≤ 0.05. Hierarchical clustering of these transcripts showed that the strain has a distinct transcriptomic pattern influenced by temperature, demonstrating a relationship in gene expression profiling across replicates with temperature (Figure 1A). Among these transcripts, 75 were upregulated at 32°C, whereas 32 were downregulated at 32°C (Table 1). These differentially expressed genes represented ~6% of the genome of the SA53 strain.
Figure 1.
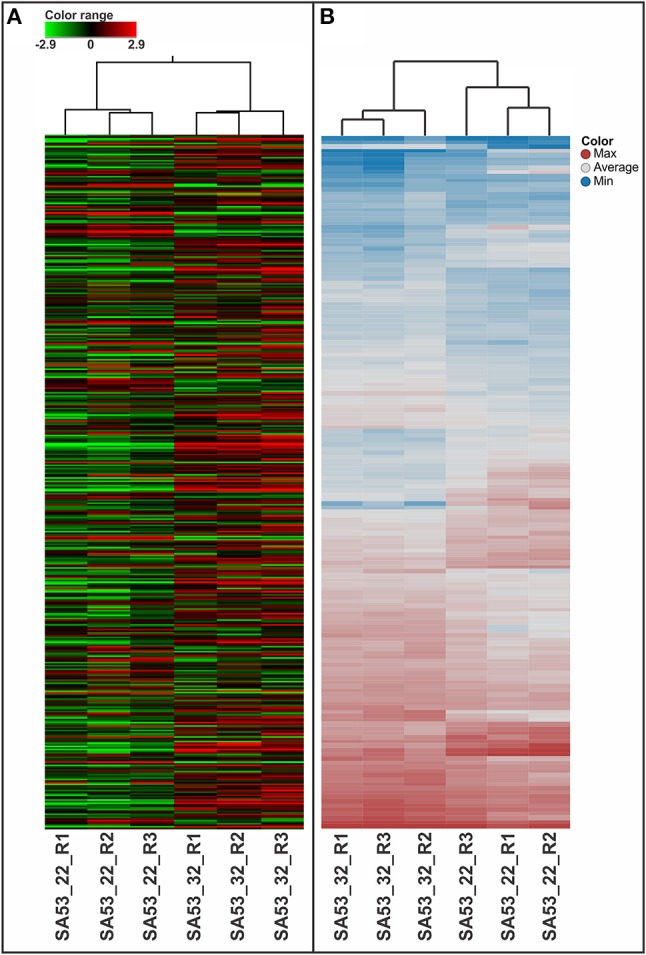
Heatmap analysis for biological triplicates of GBS strains tested at 22 and 32°C, as evaluated by microarray (A) and LC-HDMSE (B).
Table 1.
Transcripts identified as differentially regulated at 32°C compared to 22°C.
| Accession | Gene | Fold change | Functional categorya | Subcellular localizationb |
|---|---|---|---|---|
| SaSA53_0098 | Aspartokinase | −2.53 | E | CYT |
| SaSA53_0175 | Phosphoribosylformylglycinamidine synthase | −2.28 | F | CYT |
| SaSA53_0177 | purM Phosphoribosylformylglycinamidine cyclo-ligase | −2.08 | F | CYT |
| SaSA53_0180 | purH Bifunctional purine biosynthesis protein | −2.02 | F | CYT |
| SaSA53_0194 | purD Phosphoribosylamine–glycine ligase | −2.15 | F | CYT |
| SaSA53_0196 | purK N5-carboxyaminoimidazole ribonucleotide synthase | −2.11 | F | CYT |
| SaSA53_0583 | Phosphate ABC transporter ATP-binding protein | −4.17 | P | CYT |
| SaSA53_0584 | Membrane protein | −4.20 | P | MEM |
| SaSA53_0827 | noxE NADH oxidase | −2.10 | I | CYT |
| SaSA53_0850 | Hypothetical protein | −2.02 | K | CYT |
| SaSA53_0860 | Ribosomal RNA small subunit methyltransferase B | −2.07 | J | MEM |
| SaSA53_0930 | apbE Thiamine biosynthesis protein | −2.09 | H | CYT |
| SaSA53_0931 | NADPH-dependent FMN reductase | −2.11 | C | CYT |
| SaSA53_0932 | NADPH-dependent FMN reductase | −2.12 | S | CYT |
| SaSA53_0936 | guaC GMP reductase | −2.00 | F | CYT |
| SaSA53_1044 | Branched-chain amino acid ABC transporter permease | −2.04 | R | MEM |
| SaSA53_1244 | Glutamine ABC transporter permease | −2.18 | ET | PSE |
| SaSA53_1245 | Peptide ABC transporter ATP-binding protein | −2.30 | E | CYT |
| SaSA53_1300 | Peptidylprolyl isomerase | −2.27 | O | PSE |
| SaSA53_1432 | Isochorismatase | −2.06 | HR | CYT |
| SaSA53_1640 | Adhesion protein | −2.25 | P | PSE |
| SaSA53_1651 | PTS mannose transporter subunit IID | −4.15 | G | MEM |
| SaSA53_1652 | PTS mannose transporter subunit IIC | −4.18 | G | MEM |
| SaSA53_1653 | PTS mannose transporter subunit IIB | −4.77 | G | CYT |
| SaSA53_1654 | PTS mannose transporter subunit IIA | −5.44 | G | CYT |
| SaSA53_1706 | cAMP factor | −2.04 | R | SEC |
| SaSA53_1710 | metF Methylenetetrahydrofolate reductase | −4.84 | E | CYT |
| SaSA53_1711 | metE 5-methyltetrahydropteroyltriglutamate– homocysteine methyltransferase | −4.04 | E | CYT |
| SaSA53_1735 | ABC transporter ATP-binding protein | −3.81 | R | CYT |
| SaSA53_1736 | ABC transporter permease | −3.74 | R | MEM |
| SaSA53_1737 | ABC transporter substrate-binding protein | −2.96 | R | PSE |
| SaSA53_1745 | nrdD Anaerobic ribonucleoside-triphosphate reductase | −2.16 | F | CYT |
| SaSA53_0015 | Hypothetical protein | 2.05 | Q | CYT |
| SaSA53_0090 | PTS cellobiose transporter subunit IIA | 5.62 | G | CYT |
| SaSA53_0091 | Cytochrome C biogenesis protein CcmE | 5.88 | G | SEC |
| SaSA53_0092 | PTS system. cellobiose-specific IIC component | 7.00 | G | MEM |
| SaSA53_0095 | Competence protein | 2.02 | L | CYT |
| SaSA53_0100 | Enoyl-CoA hydratase | 3.20 | I | CYT |
| SaSA53_0104 | 2-nitropropane dioxygenase | 2.07 | R | CYT |
| SaSA53_0105 | fabD Malonyl CoA-acyl carrier protein transacylase | 2.08 | I | CYT |
| SaSA53_0265 | Single-stranded DNA-binding protein | 2.10 | L | CYT |
| SaSA53_0315 | comX Competence-specific sigma factor | 2.11 | K | CYT |
| SaSA53_0319 | hrcA Heat-inducible transcription repressor | 2.13 | K | CYT |
| SaSA53_0320 | grpE Protein | 2.03 | O | CYT |
| SaSA53_0336 | Pyridine nucleotide-disulfide oxidoreductase family protein | 2.11 | C | CYT |
| SaSA53_0344 | Permease | 2.12 | V | MEM |
| SaSA53_0373 | oppC Oligopeptide transport system permease protein | 2.17 | EP | MEM |
| SaSA53_0374 | oppD Oligopeptide transport ATP-binding protein | 2.17 | EP | CYT |
| SaSA53_0375 | oppF Oligopeptide transport ATP-binding protein | 2.24 | E | CYT |
| SaSA53_0387 | Hypothetical protein | 2.14 | R | CYT |
| SaSA53_0472 | MutT/nudix family protein | 2.75 | V | CYT |
| SaSA53_0477 | Phosphoglucomutase | 2.19 | G | CYT |
| SaSA53_0507 | Acetoin reductase | 2.49 | IQR | CYT |
| SaSA53_0551 | Hypothetical protein | 2.04 | M | CYT |
| SaSA53_0581 | DNA-entry nuclease | 2.57 | L | SEC |
| SaSA53_0589 | Glucuronide permease | 4.04 | G | MEM |
| SaSA53_0590 | 2-dehydro-3-deoxygluconokinase | 2.23 | G | CYT |
| SaSA53_0593 | uxaC Uronate isomerase | 2.98 | G | CYT |
| SaSA53_0594 | uxuA Mannonate dehydratase | 2.01 | G | CYT |
| SaSA53_0660 | MFS transporter | 2.26 | P | MEM |
| SaSA53_0691 | nagB Glucosamine-6-phosphate deaminase | 2.21 | G | CYT |
| SaSA53_0801 | msrB Peptide methionine sulfoxide reductase | 2.07 | O | CYT |
| SaSA53_0919 | Hypothetical protein | 2.09 | P | MEM |
| SaSA53_0954 | Hypothetical protein | 5.49 | I | CYT |
| SaSA53_0955 | Hypothetical protein | 5.92 | G | MEM |
| SaSA53_0956 | Hypothetical protein | 7.56 | G | MEM |
| SaSA53_0957 | Hypothetical protein | 7.95 | R | CYT |
| SaSA53_0982 | BCCT family transporter | 2.07 | M | MEM |
| SaSA53_1114 | ABC transporter | 2.09 | V | CYT |
| SaSA53_1215 | Ammonium transporter | 2.24 | P | MEM |
| SaSA53_1251 | Cell wall surface anchor protein | 2.04 | – | PSE |
| SaSA53_1268 | Transglutaminase | 2.18 | D | CYT |
| SaSA53_1269 | Hypothetical protein | 3.01 | P | CYT |
| SaSA53_1281 | Hypothetical protein | 2.22 | S | PSE |
| SaSA53_1287 | Hypothetical protein | 3.84 | EP | CYT |
| SaSA53_1288 | Peptide ABC transporter ATP-binding protein | 3.49 | EP | CYT |
| SaSA53_1289 | Peptide ABC transporter permease | 4.11 | EP | MEM |
| SaSA53_1290 | Peptide ABC transporter permease | 4.59 | EP | MEM |
| SaSA53_1291 | Nickel ABC transporter substrate-binding protein | 3.09 | E | PSE |
| SaSA53_1373 | Amidase | 2.26 | J | CYT |
| SaSA53_1406 | Dihydroxyacetone kinase | 2.21 | K | CYT |
| SaSA53_1407 | Dihydroxyacetone kinase subunit K | 2.99 | G | CYT |
| SaSA53_1408 | Hypothetical protein | 2.84 | R | CYT |
| SaSA53_1409 | PTS mannose transporter subunit IID | 2.89 | T | CYT |
| SaSA53_1410 | Glycerol transporter | 2.09 | G | MEM |
| SaSA53_1412 | Hypothetical protein | 2.45 | G | CYT |
| SaSA53_1431 | 3-hydroxybutyryl-CoA dehydrogenase | 2.90 | I | CYT |
| SaSA53_1435 | Universal stress protein | 2.16 | T | CYT |
| SaSA53_1460 | Multidrug MFS transporter | 3.00 | GEPR | MEM |
| SaSA53_1469 | trx Thioredoxin | 2.01 | O | CYT |
| SaSA53_1518 | Acid phosphatase | 3.63 | R | PSE |
| SaSA53_1549 | Glycine/betaine ABC transporter permease | 2.74 | E | PSE |
| SaSA53_1550 | Glycine/betaine ABC transporter ATP-binding protein | 3.33 | E | CYT |
| SaSA53_1561 | tal Transaldolase | 2.30 | G | CYT |
| SaSA53_1564 | ulaD 3-keto-L-gulonate-6-phosphate decarboxylase | 2.93 | G | CYT |
| SaSA53_1565 | PTS ascorbate transporter subunit IIA | 4.00 | GT | CYT |
| SaSA53_1566 | PTS ascorbate transporter subunit IIB | 4.49 | G | CYT |
| SaSA53_1567 | PTS ascorbate transporter subunit IIC | 3.76 | G | MEM |
| SaSA53_1598 | PTS system N-acetylgalactosamine-specific transporter subunit IIC | 2.45 | G | MEM |
| SaSA53_1599 | PTS system N-acetylgalactosamine-specific transporter subunit IIB | 2.12 | G | CYT |
| SaSA53_1600 | Glucuronyl hydrolase | 2.47 | G | CYT |
| SaSA53_1601 | PTS system N-acetylgalactosamine-specific transporter subunit IIA | 2.36 | G | CYT |
| SaSA53_1602 | Gluconate 5-dehydrogenase | 2.93 | IQR | CYT |
| SaSA53_1603 | Hypothetical protein | 3.79 | G | CYT |
| SaSA53_1604 | 2-keto-3-deoxygluconate kinase | 3.87 | G | CYT |
| SaSA53_1605 | 2-dehydro-3-deoxyphosphogluconate aldolase | 2.45 | G | CYT |
| SaSA53_1791 | sdhA L-serine dehydratase. iron-sulfur-dependent. alpha subunit | 2.02 | E | CYT |
Orthologous groups by functional category: C, Energy production and conversion; D, Cell cycle control, cell division, chromosome partitioning; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; G, Carbohydrate transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; J, Translation, ribosomal structure and biogenesis; K, Transcription; L, Replication, recombination and repair; M, Cell wall/membrane/envelope biogenesis; O, Post-translational modification, protein turnover, and chaperones; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport, and catabolism; R, General function prediction only; S, Function unknown; T, Signal transduction mechanisms; V, Defense mechanisms; -, not determined.
Subcellular localization: CYT, cytoplasmic; PSE, potentially surface-exposed; MEM, membrane; SEC, secreted.
In accordance with the subcellular localization analysis, these DEGs were classified as cytoplasmic (n = 73), membrane (n = 22), potentially surface-exposed (PSE) (n = 9), and secreted (n = 3), with ~34 and ~30% being bacterial surface-related when downregulated and upregulated at 32°C, respectively (Table 1). The DEGs were classified using COG on 18 functional categories (Table 1 and Figure 2A). A higher number of genes related to metabolism were upregulated at 32°C, except those involved in nucleotide metabolism (F) and coenzyme transport and metabolism (H), which were exclusively downregulated at 32°C. On the other hand, genes involved in cell cycle control (D), cell wall biogenesis (M), defense mechanisms (V), replication (L), and secondary metabolites biosynthesis (Q) were exclusively upregulated at 32°C.
Figure 2.

Prediction of COG functional categories of the DEGs (A) and DPAs (B) identified in strain SA53 at the two temperatures tested. Blue bar represents downregulation and red bar represents upregulation at 32°C.
From 38 genes considered to be associated with virulence (Glaser et al., 2002; Mereghetti et al., 2008) in the SA53 genome, 37 were detected by transcriptomic analysis (Table 2). Among them, 13 showed statistical significance, including cAMP factor, lmb, and noxE, which were downregulated at 32°C. The other putative virulence genes (cell wall surface anchor protein, bibA, fbsA, hylB, cpsB, cpsC, cpsD, rmlA, pbp2A, and pbpX) did not vary in expression between the evaluated temperatures (Table 2). In our transcriptomic dataset, we identified stress protein genes (hrcA, grpE, universal stress protein, and trx) that were upregulated at 32°C.
Table 2.
Known GBS virulence factors and their identification and regulation at 32°C compared to 22°C in transcriptomic and proteomic analysis.
| Virulence factor | Accession | Transcript detecteda | Regulationc | Protein detectedb | Regulationc |
|---|---|---|---|---|---|
| ADHESION | |||||
| Elongation factor Tu | SaSA53_0653 | Yes | NS | Yes | NS |
| Cell wall surface anchor protein | SaSA53_0662 | Yes | Unchanged | Yes | NS |
| GapC | SaSA53_1519 | Yes | NS | Yes | NS |
| GapN | SaSA53_0716 | Yes | NS | Yes | Unchanged |
| PavA | SaSA53_1046 | Yes | NS | Yes | NS |
| BibA | SaSA53_1722 | Yes | Unchanged | No | |
| FbsA | SaSA53_0903 | Yes | Unchanged | No | |
| Lmb | SaSA53_1640 | Yes | Down | Yes | NS |
| Pi-2b | SaSA53_1187 | Yes | NS | No | |
| INVASION | |||||
| CylE | SaSA53_0568 | No | No | ||
| cAMP factor | SaSA53_1706 | Yes | Down | Yes | Up |
| Hemolysin A | SaSA53_0483 | Yes | NS | Yes | NS |
| Eno | SaSA53_0533 | Yes | NS | Yes | NS |
| IagA | SaSA53_0601 | Yes | NS | Yes | Up |
| Internalin | SaSA53_0799 | Yes | NS | No | |
| HylB | SaSA53_1053 | Yes | Unchanged | Yes | NS |
| NoxE | SaSA53_0827 | Yes | Down | Yes | NS |
| IMMUNE EVASION | |||||
| Capsular polysaccharide–CpsG | SaSA53_1029 | Yes | NS | Yes | NS |
| Capsular polysaccharide–CpsF | SaSA53_1030 | Yes | NS | Yes | NS |
| Capsular polysaccharide–CpsE | SaSA53_1031 | Yes | NS | Yes | NS |
| Capsular polysaccharide–CpsD | SaSA53_1032 | Yes | NS | Yes | NS |
| Capsular polysaccharide–CpsC | SaSA53_1033 | Yes | Unchanged | Yes | NS |
| Capsular polysaccharide–CpsB | SaSA53_1034 | Yes | Unchanged | Yes | Up |
| SodA | SaSA53_0678 | Yes | Unchanged | Yes | NS |
| ScpB | SaSA53_0381 | Yes | NS | Yes | Down |
| Group B antigen–RmlB | SaSA53_1054 | Yes | NS | Yes | NS |
| Group B antigen–RmlC | SaSA53_1055 | Yes | NS | Yes | NS |
| Group B antigen–RmlA | SaSA53_1056 | Yes | Unchanged | Yes | NS |
| Sip | SaSA53_0182 | Yes | NS | Yes | NS |
| Serine protease | SaSA53_1720 | Yes | NS | Yes | NS |
| MULTIDRUG RESISTANCE | |||||
| DltD | SaSA53_1540 | Yes | NS | Yes | NS |
| DltB | SaSA53_1542 | Yes | NS | Yes | NS |
| DltA | SaSA53_1543 | Yes | NS | Yes | NS |
| Pbp2A | SaSA53_1724 | Yes | Unchanged | Yes | NS |
| PbpX | SaSA53_0050 | Yes | Unchanged | Yes | NS |
| Pbp1A | SaSA53_0061 | Yes | NS | Yes | NS |
| Pbp2B | SaSA53_0656 | Yes | NS | Yes | NS |
| Beta-lactamase | SaSA53_0562 | Yes | NS | Yes | NS |
Transcripts identified in microarray analysis.
Proteins identified in LC-HDMSE analysis.
Transcript or protein with p > 0.05 were considered as detected but not significant–NS; Transcript or protein with p < 0.05 were classified according to the fold-change as downregulated (Down), upregulated (Up), or unchanged.
To better understand the biological functions of transcripts identified as differentially expressed at 32°C, an interactome analysis was conducted, revealing 97 interactions (Supplementary Figure 2A). The greatest number of interactions were verified in proteins related to the phosphoenolpyruvate-dependent phosphotransferase system (PTS; cluster 1), ABC transport system (cluster 2), ascorbate and aldarate metabolism (cluster 3), purine metabolism (cluster 4), and metabolic pathways (cluster 5) that were both downregulated and upregulated at 32°C (Supplementary Figure 2A).
In our validation analysis, we observed a perfect agreement (kappa coefficient = 1) between microarray and qPCR results in the direction of regulation (downregulation and upregulation) for all genes evaluated (Supplementary Table 1). Therefore, this result demonstrated that the microarray data are valid.
Proteome analysis
The effect of temperature on the GBS proteome was evaluated using an LC-HDMSE approach. In total, 29,790 peptides with a normal distribution of 10 ppm error were identified (Supplementary Figure 3A). Peptides as source fragments, with a charge state of at least [M +2H]2+, and absence of decoys were considered to increase data quality. Therefore, our proteomics analysis allowed the identification and quantitation of 1,046 proteins for SA53 (Supplementary Table 3), with an average of 28 peptides per protein and a calculated FDR = 0% when decoy detection was set at an agreement with two of the three replicates. These data characterized ~62% of the predicted proteome of SA53. Predicted proteins smaller than 10 kDa were also identified in the proteome, despite the threshold of the column used in protein extraction (see Material and Methods). In total, 1,043 proteins were present in both temperatures tested. In relation to protein content, more than 99% of the identified proteins were found in all three replicates of each temperature, showing a high reproducibility among the biological replicates (Supplementary Figure 3B). In the PCA analysis there was no clear variability in protein abundance among the evaluated temperatures (Supplementary Figure 3C). The dynamic range of the identified proteins reached ~4.5 log orders of magnitude between the most and least abundant proteins in each temperature tested. Supplementary Table 4 summarizes the ten most and least abundant proteins.
Label-free quantification was applied to evaluate the relative abundance of the proteome of the strain at low and high temperature conditions. In summary, 163 proteins had p ≤ 0.05, and 81 (4.7% of the predicted proteome) of them showed a difference in level of abundance in SA53, with 37 and 44 proteins downregulated and upregulated at 32°C compared to 22°C, respectively (Table 3). Beyond that, two proteins were exclusively identified only at 32°C, and one protein was exclusively identified only at 22°C (Table 3). The hierarchical clustering of significant proteins (n = 163) demonstrated the arrangement of biological replicates by temperature condition (Figure 1B), indicating that the strain had a distinct proteomic pattern influenced by temperature.
Table 3.
Proteins identified as differentially regulated at 32°C compared to 22°C.
| Accession | Peptides | Score | Product | Fold change | Functional categorya | Subcellular localizationb |
|---|---|---|---|---|---|---|
| SaSA53_0162 | 7 | 39.04 | rnpA Ribonuclease P protein component | −4.34 | J | CYT |
| SaSA53_0262 | 4 | 29.12 | Thioredoxin | −2.78 | O | CYT |
| SaSA53_0373 | 5 | 29.93 | oppC Oligopeptide transport system permease protein | −4.08 | EP | MEM |
| SaSA53_0381 | 20 | 111.21 | Reticulocyte binding protein | −20.82 | D | PSE |
| SaSA53_0468 | 4 | 20.23 | RNA-binding protein | −9.57 | R | CYT |
| SaSA53_0494 | 11 | 62.19 | UDP-N-acetylmuramoylpentapeptide-lysine N(6)-alanyltransferase MurM | −2.73 | M | CYT |
| SaSA53_0504 | 27 | 167.67 | ftsX Cell division protein | −2.23 | D | PSE |
| SaSA53_0506 | 7 | 44.70 | Hypothetical protein | −2.94 | R | CYT |
| SaSA53_0535 | 20 | 107.34 | aroA 3-phosphoshikimate 1-carboxyvinyltransferase | −2.56 | E | CYT |
| SaSA53_0591 | 22 | 161.80 | GntR family transcriptional regulator | −3.38 | K | CYT |
| SaSA53_0621 | 31 | 227.07 | N5.N10-methylenetetrahydromethanopterin reductase | −2.08 | HR | CYT |
| SaSA53_0627 | 14 | 95.62 | lgt Prolipoprotein diacylglyceryl transferase | −3.53 | M | MEM |
| SaSA53_0667 | 67 | 570.69 | RNA helicase | −2.08 | L | CYT |
| SaSA53_0689 | 25 | 183.91 | queA S-adenosylmethionine:tRNA ribosyltransferase-isomerase | −2.49 | J | CYT |
| SaSA53_0758 | 60 | 443.06 | atpG ATP synthase gamma chain | −2.11 | C | CYT |
| SaSA53_0767 | 94 | 702.74 | pheT Phenylalanyl–tRNA ligase beta subunit | −3.45 | J | CYT |
| SaSA53_0837 | 11 | 57.66 | GNAT family acetyltransferase | −3.05 | J | CYT |
| SaSA53_0855 | 45 | 342.98 | Phosphate import ATP-binding protein PstB | −3.03 | P | CYT |
| SaSA53_0857 | 6 | 30.89 | Phosphate ABC transporter. permease protein PstA | −2.08 | P | PSE |
| SaSA53_0910 | 5 | 28.06 | Appr-1-p processing protein | −24.25 | J | CYT |
| SaSA53_1178 | 23 | 151.58 | Hypothetical protein | −2.77 | S | CYT |
| SaSA53_1182 | 5 | 26.28 | SAM-dependent methyltransferase | −74.54 | J | CYT |
| SaSA53_1276 | 14 | 92.26 | ybeY Endoribonuclease | −4.19 | J | CYT |
| SaSA53_1291 | 8 | 39.72 | Nickel ABC transporter substrate-binding protein | −3.22 | E | PSE |
| SaSA53_1348 | 10 | 66.78 | tRNA [cytidine(34)-2′-O]-methyltransferase | −2.31 | J | CYT |
| SaSA53_1381 | 30 | 208.69 | Primosomal protein DnaI | −2.53 | L | CYT |
| SaSA53_1392 | 9 | 53.24 | Cobalt ABC transporter permease | −13.08 | H | MEM |
| SaSA53_1476 | 25 | 131.84 | ATP-dependent DNA helicase RecD-like protein | −4.22 | L | CYT |
| SaSA53_1484 | 6 | 46.76 | Glycerol uptake permease | −14.72 | G | MEM |
| SaSA53_1487 | 6 | 33.13 | Crp/Fnr family transcriptional regulator | −4.53 | T | CYT |
| SaSA53_1504 | 22 | 140.11 | Hypothetical protein | −2.42 | R | CYT |
| SaSA53_1522 | 30 | 242.33 | rpsG 30S ribosomal protein S7 | −2.80 | J | CYT |
| SaSA53_1580 | 113 | 845.26 | ATP-dependent Clp protease ATP-binding protein | −2.80 | O | CYT |
| SaSA53_1624 | 10 | 54.84 | dexB Glucan 1.6-alpha-glucosidase | −7.97 | G | CYT |
| SaSA53_1675 | 30 | 187.15 | Ribosomal RNA small subunit methyltransferase E | −2.41 | J | CYT |
| SaSA53_1679 | 7 | 47.08 | GNAT family acetyltransferase | −3.63 | J | CYT |
| SaSA53_1734 | 18 | 190.32 | groS 10 kDa chaperonin | −2.25 | O | CYT |
| SaSA53_0041 | 77 | 778.26 | tkt Transketolase | 2.67 | G | CYT |
| SaSA53_0177 | 14 | 90.81 | purM Phosphoribosylformylglycinamidine cyclo-ligase | 3.68 | F | CYT |
| SaSA53_0180 | 44 | 342.70 | purH Bifunctional purine biosynthesis protein | 2.51 | F | CYT |
| SaSA53_0196 | 16 | 108.09 | purK N5-carboxyaminoimidazole ribonucleotide synthase | 2.34 | F | CYT |
| SaSA53_0197 | 22 | 136.77 | Hypothetical protein | 3.07 | O | CYT |
| SaSA53_0268 | 5 | 24.57 | lytR Sensory transduction protein | 13.26 | KT | CYT |
| SaSA53_0358 | 39 | 322.36 | Amino acid ABC transporter ATP-binding protein | 2.34 | E | CYT |
| SaSA53_0466 | 17 | 142.16 | sepF Cell division protein | 2.90 | D | CYT |
| SaSA53_0478 | 28 | 186.28 | folD Bifunctional protein | 2.20 | H | CYT |
| SaSA53_0481 | 12 | 118.73 | xseB Exodeoxyribonuclease 7 small subunit | 3.22 | L | CYT |
| SaSA53_0522 | 5 | 74.86 | Flavodoxin | 3.45 | C | CYT |
| SaSA53_0601 | 15 | 92.29 | Family 1 glycosyl transferase | 4.11 | M | CYT |
| SaSA53_0730 | 5 | 44.15 | Hypothetical protein | 3.36 | M | SEC |
| SaSA53_0736 | 36 | 274.28 | Phosphomethylpyrimidine kinase | 3.31 | H | CYT |
| SaSA53_0740 | 5 | 32.94 | GNAT family acetyltransferase | 2.07 | JO | CYT |
| SaSA53_0811 | 106 | 1000.67 | Pyruvate kinase | 2.47 | G | CYT |
| SaSA53_0815 | 5 | 26.89 | Amino acid ABC transporter permease | 3.05 | E | MEM |
| SaSA53_0867 | 14 | 71.90 | Hypothetical protein | 2.98 | S | CYT |
| SaSA53_0890 | 7 | 90.07 | ESAT-6-like protein | 2.00 | S | CYT |
| SaSA53_0896 | 6 | 43.51 | pyrC Dihydroorotase | 2.88 | F | CYT |
| SaSA53_0932 | 10 | 63.85 | NADPH-dependent FMN reductase | 3.55 | S | CYT |
| SaSA53_0975 | 19 | 117.47 | folP Dihydropteroate synthase | 2.14 | H | CYT |
| SaSA53_1034 | 32 | 245.18 | cpsB Tyrosine-protein phosphatase | 2.09 | T | CYT |
| SaSA53_1091 | 3 | 23.67 | Amino acid transporter | 3.70 | E | MEM |
| SaSA53_1101 | 39 | 260.34 | 3-hydroxy-3-methylglutaryl coenzyme A reductase | 2.17 | I | CYT |
| SaSA53_1156 | 22 | 219.87 | Hypothetical protein | 2.05 | T | CYT |
| SaSA53_1239 | 7 | 33.05 | Beta-1,6-galactofuranosyltransferase | 2.54 | M | CYT |
| SaSA53_1245 | 47 | 463.68 | Peptide ABC transporter ATP-binding protein | 2.02 | E | CYT |
| SaSA53_1277 | 6 | 28.64 | Tetracenomycin polyketide synthesis O-methyltransferase TcmP | 2.04 | Q | CYT |
| SaSA53_1325 | 2 | 11.05 | PTS lactose transporter subunit IIC | 2.28 | G | MEM |
| SaSA53_1346 | 30 | 281.46 | upp Uracil phosphoribosyltransferase | 2.31 | F | CYT |
| SaSA53_1383 | 9 | 84.65 | nrdR Transcriptional repressor | 2.07 | K | CYT |
| SaSA53_1416 | 6 | 37.90 | SAM-dependent methyltransferase | 2.56 | H | CYT |
| SaSA53_1440 | 27 | 181.27 | recG ATP-dependent DNA helicase | 2.96 | L | CYT |
| SaSA53_1469 | 23 | 221.64 | trx Thioredoxin | 6.23 | O | CYT |
| SaSA53_1502 | 10 | 77.30 | Hypothetical protein | 2.86 | L | CYT |
| SaSA53_1585 | 19 | 157.05 | Alkyl hydroperoxide reductase subunit C | 2.07 | V | CYT |
| SaSA53_1641 | 11 | 65.99 | dtd D-aminoacyl-tRNA deacylase | 4.00 | J | CYT |
| SaSA53_1682 | 7 | 52.91 | Hypothetical protein | 2.42 | E | CYT |
| SaSA53_1706 | 7 | 40.62 | cAMP factor | 2.75 | R | SEC |
| SaSA53_1735 | 48 | 374.57 | ABC transporter ATP-binding protein | 3.53 | R | CYT |
| SaSA53_1737 | 54 | 484.23 | ABC transporter substrate-binding protein | 4.53 | R | PSE |
| SaSA53_1796 | 9 | 46.73 | Energy-coupling factor transporter ATP-binding protein EcfA1 | 2.39 | PR | CYT |
| SaSA53_1812 | 34 | 354.77 | arcC Carbamate kinase | 2.39 | E | CYT |
| SaSA53_0353 | 2 | 17.02 | Hypothetical protein | S | CYT | |
| SaSA53_1139 | 5 | 32.18 | Macrolide ABC transporter ATP-binding protein | M | CYT | |
| SaSA53_1378 | 6 | 35.03 | Hypothetical protein | R | CYT |
Orthologous groups by functional category: C, Energy production and conversion; D, Cell cycle control, cell division, chromosome partitioning; E, Amino acid transport and metabolism; F, Nucleotide transport and metabolism; G, Carbohydrate transport and metabolism; H, Coenzyme transport and metabolism; I, Lipid transport and metabolism; J, Translation, ribosomal structure and biogenesis; K, Transcription; L, Replication, recombination and repair; M, Cell wall/membrane/envelope biogenesis; O, Post-translational modification, protein turnover, and chaperones; P, Inorganic ion transport and metabolism; Q, Secondary metabolites biosynthesis, transport, and catabolism; R, General function prediction only; S, Function unknown; T, Signal transduction mechanisms; V, Defense mechanisms.
Subcellular localization: CYT, cytoplasmic; PSE, potentially surface-exposed; MEM, membrane; SEC, secreted.
In total, 67, 7, 5, and 2 proteins with differential protein abundances (DPAs) were predicted as cytoplasmic, membrane, PSE, and secreted, respectively; of these, ~21 and ~13% were bacterial surface proteins downregulated and upregulated at 32°C, respectively (Table 3). According to COG analysis, the DPAs were classified into 18 categories (Table 3 and Figure 2B). Proteins involved in translation, ribosomal structure, and biogenesis (J), and inorganic ion metabolism (P) were the most abundant proteins that were downregulated at 32°C in relation to upregulated proteins. On the other hand, proteins involved in defense mechanisms (V), lipid metabolism (I), nucleotide metabolism, and secondary metabolites biosynthesis (Q) were exclusively identified as upregulated at 32°C.
We identified 33 known SA53 virulence factors by proteomic analysis (Table 2). Reticulocyte binding protein was downregulated at 32°C; IagA (Family 1 glycosyl transferase), CpsB, and cAMP factor were upregulated; and GapN did not vary in abundance between temperatures (Table 2). The other 28 putative virulence proteins, although detected, did not show differential abundance in our study. In addition, thioredoxin, which is a protein involved in oxidative stress, was also identified as downregulated at 32°C.
In order to study the main interactions among proteins identified as more abundant at 32°C compared to 22°C, we performed an interactome analysis, which revealed 26 protein-protein interactions. The greatest number of interactions was observed as upregulated at 32°C: PurH (SaSA53_0180) and PurM (SaSA53_0177), involved in purine metabolism; alkyl hydroperoxide reductase subunit C (SaSA53_1585) and thioredoxin (SaSA53_1469), involved in oxidative stress; pyruvate kinase (SaSA53_0811), related to glycolysis; and amino acid ABC transporter permease (SaSA53_0815), involved in amino acid metabolism (Supplementary Figure 2B).
Correlation analysis
The Pearson correlation coefficient between gene expression levels and protein abundance for SA53 was 0.3390. This result showed that there was a low correlation between the transcriptome and proteome datasets in regard to the genes and proteins that were downregulated or upregulated in the same direction, for a given condition (low or high temperature).
Discussion
Unlike human-adapted GBS, fish-adapted GBS strains are exposed to constant and abrupt changes in temperature during adaptation to aquatic environments and during the infection process as fish are poikilothermic animals and water in the environment can undergo rapid temperature changes due to short-term weather events. To better understand the characteristics of metabolism, adaptation, and pathogenicity of this agent in response to temperature variation, we analyzed the transcriptome and the proteome of a fish-adapted GBS strain subjected to two growth temperatures using microarray and LC-HDMSE approaches.
The isolate selected for these analyses was the GBS SA53 strain. This strain was previously isolated from diseased Nile tilapia in Brazil (Mian et al., 2009) and belongs to ST-260, one of the most frequently identified genotypes in Latin America (Evans et al., 2008; Delannoy et al., 2013; Godoy et al., 2013; Barato et al., 2015; Barony et al., 2017). SA53 belongs to a group of fish-adapted genotypes that descend from a single branch unlike human GBS strains, and are going through reductive genome evolution (Barony et al., 2017). Its LD50 was not previously determined but isolates obtained from diseased fish have been demonstrated as highly virulent in experimental assays with Nile tilapia, with an LD50 ranging from 101 to 105 cfu mL−1 (Mian et al., 2009; Evans et al., 2015). Therefore, due the characteristics described above, SA53 was chosen to elucidate the gene expression and protein abundance in fish-adapted GBS strains at low (22°C) and high (32°C) temperature conditions, corresponding to temperatures showing low and high mortality rates in fish, respectively (Salvador et al., 2005; Mian et al., 2009; Amal et al., 2015; Al-Harbi, 2016; Mainardi et al., 2016; Chideroli et al., 2017).
For the temperatures analyzed in this study, the effect on transcriptome and proteome modification was not extensive, corroborating a previous study that compared the transcriptional response to temperature in one fish-adapted Lactococcus garvieae strain (Aguado-Urda et al., 2013). This suggests that fish-adapted GBS strains may be physiologically stable in the global gene expression and protein synthesis, even with rapid variation in environmental temperature, thus supporting the idea of possible adaptation of bacteria to the aquatic environment and the fish host. However, the expression of some important proteins was strongly modulated by the temperature shift, providing insights about the interaction of GBS with the fish host.
Transcriptional response is modulated by temperature
In our transcriptomic analysis, we observed that temperature could influence the expression of genes involved mainly in cellular metabolism (Figure 3A). Among genes that were downregulated at 32°C, we detected genes involved in purine metabolism: purD, purH, purK, purM, and guaC. These genes grouped together in our interactome analysis and are related to the purine biosynthetic pathway, described by Mereghetti et al. (2008) as responsible for the synthesis of inosine monophosphate, a compound important to satisfy the purine auxotrophic requirements of GBS (Rajagopal et al., 2005). A previous study demonstrated that purine metabolism in GBS is influenced by incubation temperature, being upregulated at 40°C compared to 30°C (Mereghetti et al., 2008). In our study, these transcripts were identified at 22°C and may be associated with an adaptive condition where the bacterium presumably increases its requirements of adenine and guanine during growth at low temperatures in an aquatic environment. The increase in uptake is achieved by either bacterial hydrolytic degradation of nucleic acids and nucleotides, or uptake of these nucleotides from the environment through utilization of the purine salvage pathway (Rajagopal et al., 2005). Utilization of this pathway is corroborated by detection of salvage enzymes apt (adenine phosphoribosyltransferase), xpt (xanthine phosphoribosyltransferase), hypoxanthine phosphoribosyltransferase, inosise-uride nucleoside N-ribohydrolase, and add (adenosine deaminase) in the transcriptome dataset.
Figure 3.

Schematic summarizing the transcriptional (A) and proteomic (B) response of fish-adapted GBS to incubation at different temperatures. Red circles indicate upregulation at 32°C, whereas blue circles represent downregulation at 32°C.
Four genes involved in PTS mannose transport (subunit IIA, IIB, IIC, and IID) related to carbohydrate metabolism were downregulated at 32°C. This PTS system is considered important to the transport of mannose, fructose, and glucose for the species of genus Streptococcus (Pelletier et al., 1998; Abranches et al., 2003; Bidossi et al., 2012), through a mechanism that couples translocation (subunits IIC and IID) with phosphorylation (subunits IIA and IIB) of the carbon sources (Gutknecht et al., 1999). Mutation in subunits IIAB of Streptococcus mutans led to lower uptake of glucose and mannose in medium containing glucose as the sole carbohydrate source, whereas fructose uptake was not affected (Abranches et al., 2003). Fish-adapted GBS strains need to adjust their metabolism in response to nutrient availability in aquatic and host environments, especially in relation to glucose availability. At high temperature, a greater affinity for glucose is required for the growth of psychrotolerant and mesophilic bacteria (Nedwell, 1999). Exposure of GBS strains to glucose permits modulation of genes involved in cell envelope biogenesis and metabolism and transport of amino acids, ions, and other carbohydrates. However, high glucose availability can lead to decreased expression of genes involved in the uptake of carbohydrates and putative virulence factors (Di Palo et al., 2013). Thus, one can argue that fish-adapted GBS has a lower affinity for glucose at 22°C, increasing the expression of the PTS system, involved in mannose transport, which in turn promotes an increased capacity to import glucose for the bacterial cell. Nevertheless, future research needs to be conducted to evaluate the PTS mannose activity in fish-adapted GBS strains cultured in media containing different carbohydrate sources, and at different temperatures.
Genes involved in methionine metabolism, such as metE and metF, were also downregulated at 32°C. These genes are involved in the methionine synthesis pathway through methylation of homocysteine by metE in conjunction with metF, which then receives a methyl group from folD (methylenetetrahydrofolate dehydrogenase), to form methionine (Afzal et al., 2016). Genes involved in methionine synthesis have been considered critical for virulence in GBS (Shelver et al., 2003). However, in Brucella melintesis, mutation in one gene related to methionine synthesis culminated in reduced bacterial colonization in the spleen of a murine model (Lestrate et al., 2000). Together, these findings demonstrate that genes involved in methionine metabolism allow bacterial survival during infection. Methionine availability in fish blood, as well as fish body temperature, might influence the growth rate of GBS, predisposing the expression of virulence factors, and allowing the bacterium to resist clearance by the immune system, as previously verified by in vitro and in vivo assays with a human GBS strain (Shelver et al., 2003). However, further studies are needed to determine if metE and metF contribute to GBS virulence in fish and if they are essential for the growth of this bacterium in fish blood.
Metabolic pathways involved in the uptake of cellobiose, ascorbate, N-acetylgalactosamine, and amino acids were upregulated at 32°C. GBS strains also use cellobiose, ascorbate, and N-acetylgalactosamine as a carbon source (Glaser et al., 2002). Fish-adapted GBS strains do not contain cellulase genes in their genome and cellobiose uptake may occur during the infectious process in the gastrointestinal epithelium of fish, as the gastrointestinal tract is one of the major routes of GBS entry in fish (Iregui et al., 2016), or during adaptation to aquatic environments where cellobiose excreted via feces can be used as an alternative energy source. The genes involved in this type of metabolism (PTS cellobiose transporter subunit IIA and IIC) show a change in expression level of 5- to 7-fold when cultured at high temperature, thus interacting with each other in our interactome analysis.
Ascorbate can also be used as an alternative carbon source for many bacterial species including E. coli, Klebsiella pneumoniae, and Streptococcus pneumoniae, and its entry into bacterial cells occurs through the ascorbate-specific PTS system (Afzal et al., 2015). In our transcriptomic analysis, we identified four genes involved in ascorbic acid uptake (ulaD, PTS ascorbate transporter subunit IIA, IIB, and IIC) that interact with each other. Therefore, during the infectious process, the ascorbate contained in fish tissues may be used by GBS strains as an energy source for their growth and dissemination in this host, especially at high temperature.
N-acetylglucosamine is an important polysaccharide component in the cell wall of GBS strains (Pereira et al., 2013), and its catabolism provides organisms with a carbon and nitrogen source (Moye et al., 2014). N-acetylglucosamine was described as important for GBS adhesion to fish cells (Barato et al., 2016). Thus, expression of these genes at high temperature may contribute to adhesion and immune system evasion by fish-adapted GBS strains in aquatic hosts.
Additionally, among the genes upregulated at 32°C, six genes were involved in the transport of peptides including the Opp transport system (oppCDF), an important superfamily of conserved ATP-binding cassette transporters involved in bacterial nutrition, signaling, and virulence though internalization of peptides from the extracellular environment (Silva et al., 2017). In GBS, the Opp transport system is responsible for the uptake of oligopeptides in a nutritionally rich environment and contributes to infection by stimulating the adherence of pathogens to human cells and modulating fibrinogen-binding adhesin expression (Samen et al., 2004). However, there are no studies that demonstrate the participation of Opp genes in the pathogenesis of fish-adapted GBS strains.
Variations in temperature during bacterial growth can induce adaptive response mechanisms to environment change, such as upregulation of heat and cold shock proteins (Fayet et al., 1989; Schulz and Schumann, 1996; Zhang and Griffiths, 2003; Varcamonti et al., 2006). In our transcriptome analysis, we solely identified hrcA and grpE as genes related to heat shock, both of which were upregulated at 32°C. Detection of these important genes in fish-adapted GBS strains suggests their participation in bacterial resistance to high temperature in both aquatic and host environments. The universal stress protein (usp) involved in natural defense mechanisms against stress conditions was also upregulated at 32°C. It is noteworthy that cold shock is one of the few conditions which cause repression of usp expression in bacteria (Kvint et al., 2003). And as usp was induced at high temperature, we can speculate that it may induce thermal tolerance in fish-adapted GBS strains. Moreover, in GBS, usp has been associated with long term survival during nutrient stress (Yang et al., 2012). Therefore, the expression of usp in fish-adapted GBS might be essential to its survival during adaptation in aquatic environments, where nutrient deprivation and competition with other species for the same substrates occurs. And at high temperature the bacterial species have a higher affinity for nutrients, allowing the perseverance of pathogens in this stress conditions (Nedwell, 1999).
Genes involved in oxidative stress control were divergently downregulated and upregulated at 32°C. GBS strains are able to induce reactive oxygen species (ROS) production in host cells during the infectious process (Costa et al., 2016). ROS are generated by host phagocytes as part of defense mechanisms against the invasion of different microorganisms to damage essential components of bacterial cells (Zheng et al., 2017). Previous studies have suggested that GBS strains can adapt to an oxidative environment (Yamamoto et al., 2006) and persist and survive within macrophages (Shabayek and Spellerberg, 2017). In our study, the gene trx was upregulated at 32°C. Trx and other oxidoreductase proteins are important for maintaining the thiol state in bacterial cells through reduction of oxidized cysteine residues in cytoplasm (Ezraty et al., 2017). Higher expression of genes involved in oxidative stress at elevated temperatures coincides with the greater potential of fish-adapted GBS strain to infect and cause disease in the aquatic host. Also, the temperature increase for some fish species induces a higher production of ROS as a response to the heat stress (Banh et al., 2016). Thus, the expression of trx in GBS should facilitate bacterial survival in the host. On the other hand, during winter, at low temperature, the demand for anti-oxidants may be lower as the bacterium occurs more in the environment than in the host. Corroborating this statement, noxE (NADH oxygenase) was downregulated at 32°C. This gene assists in oxygen tolerance in GBS strains, either by decreasing the intracellular NADH/NAD+ ratio or by direct elimination of oxygen, and contributes to the bacterial infection process in the blood, liver, and brains of mice (Yamamoto et al., 2006). Moreover, noxE also contributes to growth of Streptococcus sp. in carbohydrate-limited environments (Gibson et al., 2000), indicating that the higher expression of this gene at low temperature is more related with adaptation to nutrient availability in aquatic environment than with protection against oxidative stress.
Two putative virulence genes were downregulated at 32°C: cAMP factor and adhesion protein, which is homologous to lmb (laminin-binding surface protein). cAMP factor is a protein secreted by GBS strains with pore-forming and hemolytic proprieties (Rajagopal, 2009). The high expression of this gene has been described at high temperature conditions (35°C) in fish GBS strains (Kayansamruaj et al., 2014). However, our results contradict this statement as higher expression of cAMP factor was detected at low temperature. Lmb promotes binding between GBS and host laminin, a glycoprotein of the basement membrane, and induces GBS invasion into brain tissues of human hosts (Al Safadi et al., 2010). Few fish-adapted GBS strains showed the presence of lmb in their genome when screened by PCR (Godoy et al., 2013). However, lmb has not yet been studied in terms of its biological functions in these strains.
Changes in the proteome are modulated by temperature
In our proteomic analysis, we observed that temperature could influence metabolic pathway protein abundance (Figure 3B). Of proteins downregulated at 32°C, we identified proteins involved in carbohydrate metabolism including DexB and glycerol uptake permease. DexB (Glucan 1,6-alpha-glucoside) participates in the starch and sucrose metabolic pathway and releases glucose from the non-reducing terminus of alpha-1,6-linked dextran or isomaltosaccharides (Whiting et al., 1993). The released free glucose units may be used by fish-adapted GBS strains in the gluconeogenesis pathway, even during adaptation to low temperature when glucose affinity is decreased (Nedwell, 1999). Glycerol uptake permease is an aquaporin responsible for conducting water and small hydrophilic solutes, particularly glycerol, into the bacterial cell (Lu et al., 2003). Glycerol uptake at low temperatures can also contribute to obtain an alternative energy source for bacterial growth in this condition.
Proteins involved in inorganic ion transport and metabolism, such as phosphate ABC transporters (PtsAB), nickel ABC transporters, and cobalt ABC transporters were downregulated at 32°C. Inorganic ions, like iron, cobalt, nickel, copper are required by bacteria to carry out biological processes and are important enzymatic cofactors acquired mainly from the environment, that contribute to the biological activities of many bacterial proteins (Hohle et al., 2011; Schreur et al., 2011). Expression of some of these proteins has been previously shown to be upregulated at high temperatures (Mereghetti et al., 2008), contradicting our findings. Some genes involved in inorganic ion metabolism are absent or inactive in fish-adapted GBS strains, affecting ion exchange and reducing the bacterial ability to maintain homeostasis in unfavorable environmental conditions (Rosinski-Chupin et al., 2013). However, for these strains, proteins involved in inorganic and metallic ion metabolism were identified in a label-free pan-proteomics analysis and were considered important for the growth and survival of pathogens in aquatic environment (unpublished data). PstAB are responsible for bacterial growth in environment with limited inorganic phosphate concentration and by adhesion of S. mutans in abiotic surfaces (Ferreira et al., 2016). Nickel uptake by ABC transporters is essential for maintaining a neutral bacterial cytosolic pH and to allow colonization of S. aureus and Helicobacter pylori in human cells (Tanaka et al., 2018). Cobalt is important for bacterial energy metabolism and anabolism, being found in the corrin ring of coenzyme B12. This metallic ion competes with iron in several metabolic pathways and adaptive changes in response to environmental cobalt concentration (Majtan et al., 2011). Therefore, the differential protein abundance of this functional category at low temperature may contribute to the bacterial viability in an environment limited in essential metal ions.
A change in regulatory direction was observed in proteins related to purine metabolism and oligopeptide uptake, when compared to transcriptome data. In the proteome, OppC protein was downregulated at 32°C, whereas PurH, PurK, and PurM were upregulated at 32°C. This change may have occurred because the samples were collected at the same time point. However, transcripts and proteins have different half-lives, and changes in transcript expression only affect protein levels after a certain temporal delay, as protein synthesis takes time (Liu et al., 2016).
In our study, RNA helicase was identified as downregulated at 32°C with its abundance altered by 2.08-fold. RNA helicase is a cold shock protein involved in mRNA processing, transport, or degradation, ribosome biogenesis, and translation initiation during cold acclimation (Phadtare and Severinov, 2010). Similarly, the RNA helicase deaD gene was upregulated at 30°C relative to 40°C in a previous study on a GBS strain during the stationary phase of bacterial growth (Mereghetti et al., 2008). In our proteomic dataset, GroS and ClpL (both heat shock proteins) were downregulated at 32°C. Clp ATPases, like ClpL, are involved in the folding, assembly, and proteolysis of proteins (Nair et al., 2003), whereas GroS prevents inactivation of cellular proteins and assists in the degradation of non-repairable denatured proteins, which may accumulate during bacterial growth in normal or stress conditions (Yura et al., 1993). A previous study showed that ClpL protein synthesis is enhanced in Streptococcus thermophilus cells stored at high and low temperature conditions (Varcamonti et al., 2006). In GBS cells, however, there was no differential expression of GroS and ClpL during growth at 30 and 40°C, suggesting that proteins are always required during bacterial growth, regardless of growth temperature (Mereghetti et al., 2008). Therefore, our results indicate that fish-adapted GBS presented the same phenomenon of expressing heat shock proteins at both temperatures. However, higher expression of heat and cold shock proteins at low temperature can represent an important mechanism of protection and homeostasis in this condition.
Proteins involved in oxidative stress control were upregulated at 32°C. Alkyl hydroperoxide reductase subunit C (AhpC), Trx and flavodoxin showed a change in abundance level of 2.07-, 6.2- and 3.4-fold, respectively. AhpC is an important protein involved in defense against different substrates as H2O2 and organic peroxides, participating of detoxification of ROS formed in bacterial cells or derived from the host. This protein is responsible for the induction and maintenance of the viable but non-culturable state in Vibrio parahaemolyticus (Wang et al., 2013). Similar to Trx (described above), flavodoxin defends against ROS and contributes to Pseudomonas aeruginosa survival in macrophagic cells and in Drosophila melanogaster (Moyano et al., 2014). Thus, AhpC and flavodoxin might also contribute to the survival of fish-adapted GBS strains in fish cells during the infection process. Notably, in Nile tilapia, the respiratory burst activity and phagocytic activity of macrophages decreases with both reduction in temperature and exposure time to the aquatic environment (Qiang et al., 2018). In this manner, a higher macrophage activity at high temperature could decrease the sensitivity of fish to pathogens. However, in this condition, fish-adapted GBS showed higher abundance of proteins involved in oxidative stress control, which leads us to speculate that at 32°C, the GBS isolate required greater protection against ROS to survive under environment or during infection in the aquatic host.
Four proteins related to bacterial pathogenicity showed differential protein abundance between 22 and 32°C (Table 2). Reticulocyte binding protein, homologous to C5a peptidase (ScpB), was downregulated at 32°C, and showed a change in abundance level of 20.8-fold. This protein is a serine protease involved in adhesion and host immune evasion (Rajagopal, 2009). On the other hand, IagA (4.1-fold), CpsB (2-fold), and cAMP factor (2.7-fold) were upregulated at 32°C. In human infections by GBS, IagA contributes to blood-brain barrier invasion (Doran et al., 2005), whereas CpsB, a protein related to capsule synthesis, protects the bacterium though prevention of complement deposition (factor C3b) and opsonophagocytosis (Glaser et al., 2002). cAMP factor also switched its direction of regulation compared to the transcriptome analysis, being upregulated at 32°C in proteome analysis. As previously reported for the lmb gene, neither of these virulence factors have been their rules evaluated in fish infection models. However, identification of reticulocyte binding protein, IagA, CpsB, and cAMP factor indicates that these proteins may contribute to the adhesion, dissemination, and survival of GBS in fish tissues at both temperature conditions, with a pathogenesis landscape similar to human GBS infections.
Temperature and pathogenicity
It is known that GBS can be isolated from diseased fish at low temperatures (Chideroli et al., 2017). It has been clearly demonstrated that fish can become infected under these conditions, and that GBS strains should therefore use virulence factors such as reticulocyte binding protein and lmb, under these conditions to cause disease in the aquatic host. In field conditions, when tilapia are cultured in water at high temperatures, the GBS load in the tissue of infected fish can increase, causing extensive tissue damage with a massive inflammatory response, thus increasing the expression of several virulence genes, allowing GBS to transcend the host-defense mechanism, and increasing the incidence of death in fish (Kayansamruaj et al., 2014). An experimental infection assay with GBS in Nile tilapia cultured at 32 and 22°C (the same temperatures used in our in vitro assay) showed that all fish died after three days and in the second week post-challenge, respectively (Wang et al., 2016). Another experimental infection showed cumulative mortality rates of 70, 50, and 0% in tilapia adapted to water temperatures of 30, 25, or 20°C, respectively (Zhao et al., 2015). In Brazil, an experimental challenge with GBS performed in tilapia acclimated to water at 22 and 31°C showed mortality rates of 10 and 80%, respectively (Chideroli et al., 2017). In our study, we expected to find several proteins related to pathogen-host interaction being highly abundant at 32°C compared to 22°C, because there are higher mortality rates at this temperature.
For several bacterial pathogens of mammals, the main trigger for the expression of virulence factors in pathogenic bacteria is temperature (Guijarro et al., 2015). At high temperature (37°C), Shigella spp. (Maurelli et al., 1984), Bordetella pertussis (Rappuoli et al., 1992), Yersinia pestis (Karlyshev et al., 1992), and E. coli (Falconi et al., 1998) showed virulence gene expression to be stimulated by environmental factors, becoming virulent for the mammalian host. On the other hand, in fish-pathogenic bacteria, upregulation of virulence factors is commonly verified at low temperature, as observed in Yersinia ruckeri (18 vs. 28°C; Méndez et al., 2009), Flavobacterium psychrophilum (12 vs. 18°C; Gómez et al., 2012), L. garvieae (18 vs. 37°C; Aguado-Urda et al., 2013), Aeromonas hydrophila (25 vs. 37°C; Yu et al., 2007), and Edwardsiella tarda (25 vs. 37°C; Srinivasa Rao et al., 2004). In our study, genes/proteins involved in the virulence of GBS were detected at both temperature conditions, indicating that their expression is not totally temperature-dependent and the bacterial virulence machinery always active. Non-differentiation of some virulence factors expression may be related to the fact that fish GBS is frequently exposed to the environment (water and fish tissues), conferring an adaptive phenotypic plasticity necessary to maintain infectivity in a broad range of body temperatures. Nevertheless, genes/proteins that play a key role in pathogenicity were detected with differential abundance among the temperatures. Although the fish-adapted GBS strains had evolved in divergent branch from mammalian GBS isolates (Barony et al., 2017), several genes are shared among piscine and mammalian strains (Rosinski-Chupin et al., 2013; Kawasaki et al., 2018), including those identified as differentially expressed in our study. Therefore, upregulation at 32°C of proteins IagA, CpsB and cAMP factor may be important for the onset of streptococosis in tilapia during the warmer months. However, in Brazil, GBS have been considered endemic in tilapia farms, being detected in diseased and carrier fish in field condition throughout the year, even during the winter (Delphino et al., 2019). In this way, virulence factors detected in the proteome of GBS in low temperature may contribute to the maintenance of the bacteria in carrier (apparently healthy) fish during the cold months.
Low correlation between transcriptomic and proteomic datasets
A weak relationship between transcriptomic and proteomic profiles was observed in our analysis. Correspondence among these datasets has been reported as low for different bacterial pathogens (Dressaire et al., 2010; Margalef-Català et al., 2016), including bacteria of the Streptococcus genus (Ahn et al., 2017), due to different regulatory mechanisms involved in gene expression and protein synthesis (Haider and Pal, 2013) and technical limitations among the approaches used (Lundberg et al., 2010). Among the physiological mechanisms involved in this low correlation between transcriptome and proteome, half-lives of mRNAs and proteins, and post-transcriptional machinery such as translation and protein degradation and modification have been described (Haider and Pal, 2013; Ahn et al., 2017). The half-life of mRNA in bacteria is short, varying from seconds to over an hour (Laalami et al., 2014) and is much shorter than their corresponding proteins, which have stable half-lives (>20 h; Deller et al., 2016). In addition, regulatory factors may be responsible for the low correlation between transcriptomic and proteomic datasets, especially of protein nature, where some proteins were not detected in our analysis due of limitations of detection capacity of the method employed.
Conclusions
The present study is the first to characterize the functional genome of fish-adapted GBS strains at transcriptomic and proteomic levels under different temperature conditions. The data indicate that the transcriptome and proteome of fish-adapted GBS are modulated by temperature. A greater variation in the expression of the functional genome was observed in transcriptome analysis, especially at 32°C. Our comparison analysis demonstrated that fish-adapted GBS regulates the differential expression of transcripts and proteins involved in metabolism and adaptation to aquatic environment and to the fish host at low and high temperatures. Temperature was also able to influence the differential expression of some virulence factors at both low and high temperatures, which indicates the ability of colonization of the aquatic host in both conditions. The results obtained in this study open prospects for future investigations on the expression trends of genes and proteins enabling bacterial adaptation to aquatic environments or their survival and growth inside the fish host.
Author contributions
GT performed microbiological analyses and sample preparation for transcriptomic and proteomic analyses. GT and AC conducted the transcriptomic analysis. GT and CR conducted the proteomic analysis. GT and FP performed bioinformatics analysis of data. GT wrote the manuscript. FP, CL, and VA contributed substantially to data interpretation and revisions. HF coordinated all analyses of the project and critically reviewed the manuscript. All authors read and approved the final manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Coordination for the Improvement of Higher Education Personnel (CAPES), the National Council of Technological and Scientific Development (CNPq), FAPEMIG and the Ministry of Agriculture, Livestock and Food Supply for financing this study.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02639/full#supplementary-material
Genes selected and primers used for validation of microarray results.
Complete list of transcripts identified by microarray analysis during GBS growth at 22°C and 32°C.
Complete list of proteins identified by LC-HDMSE analysis during GBS growth at 22°C and 32°C.
List of ten most and least expressed proteins identified by LC-HDMSE in GBS growth at 22°C and 32°C.
Quality assessment of the biological replicates used in transcriptome analysis. (A) Distribution of the intensities evaluated by box plot; (B) Correlation analysis matrix. (C) PCA plot, red circles represent samples grown at 32°C, whereas blue circles represent samples grown at 22°C.
Interaction networks of DEGs (A) and DPAs (B) identified in strain SA53 between the two temperatures. Red circles represent proteins upregulated at 32°C, whereas blue circles represent proteins downregulated at 32°C. (1) PTS systems; (2) ABC transport system; (3) Ascorbate and aldarate metabolism; (4) Purine metabolism; (5) metabolic pathways.
Quality assessment of biological replicates used in proteome analysis. (A) Normal distribution of 10 ppm error in the total identified peptides; (B) repeat rate indicating the number of times that an identified protein appears in the replicates: 3 of 3 (blue) and 2 of 3 (red). (C) PCA plot, red circles represent samples grown at 32°C, whereas blue circles represent samples grown at 22°C.
References
- Abranches J., Chen Y.-Y. M., Burne R. A. (2003). Characterization of Streptococcus mutans strains deficient in EIIAB (Man) of the sugar phosphotransferase system. Appl. Environ. Microbiol. 69, 4760–4769. 10.1128/AEM.69.8.4760-4769.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzal M., Shafeeq S., Kuipers O. P. (2015). Ascorbic acid-dependent gene expression in Streptococcus pneumoniae and the activator function of the transcriptional regulator UlaR2. Front. Microbiol. 6:72. 10.3389/fmicb.2015.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzal M., Shafeeq S., Kuipers O. P. (2016). Methionine-mediated gene expression and characterization of the CmhR regulon in Streptococcus pneumoniae. Microb. Genomics 2:e000091. 10.1099/mgen.0.000091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado-Urda M., Gibello A., Blanco M. D., Fernández-Garayzábal J. F., López-Alonso V., López-Campos G.H. (2013). Global transcriptome analysis of Lactococcus garvieae strains in response to temperature. PLoS ONE 8:e79692. 10.1371/journal.pone.0079692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S.-J., Gu T., Koh J., Rice K. C. (2017). Remodeling of the Streptococcus mutans proteome in response to LrgAB and external stresses. Sci. Rep. 7:14063. 10.1038/s41598-017-14324-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Safadi R., Amor S., Hery-Arnaud G., Spellerberg B., Lanotte P., Mereghetti L., et al. (2010). Enhanced expression of lmb gene encoding laminin-binding protein in Streptococcus agalactiae strains harboring IS1548 in scpB-lmb intergenic region. PLoS ONE 5:e10794. 10.1371/journal.pone.0010794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Harbi A. H. (2016). Phenotypic and genotypic characterization of Streptococcus agalactiae isolated from hybrid tilapia (Oreochromis niloticus × O. aureus). Aquaculture 46, 515–520. 10.1016/j.aquaculture.2016.07.036 [DOI] [Google Scholar]
- Amal M. N. A., Saad M. Z., Zahrah A. S., Zulkafli A. R. (2015). Water quality influences the presence of Streptococcus agalactiae in cage cultured red hybrid tilapia, Oreochromis niloticus × Oreochromis mossambicus. Aquacult. Res. 46, 313–323. 10.1111/are.12180 [DOI] [Google Scholar]
- Banh S., Wiens L., Sotiri E., Treberg J. R. (2016). Mitochondrial reactive oxygen species production by fish muscle mitochondria: potential role in acute heat-induced oxidative stress. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 191, 99–107. 10.1016/j.cbpb.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Barato P., Martins E. R., Melo-Cristino J., Iregui C. A., Ramirez M. (2015). Persistence of a single clone of Streptococcus agalactiae causing disease in tilapia (Oreochromis sp.) cultured in Colombia over 8 years. J Fish Dis. 38, 1083–1087. 10.1111/jfd.12337 [DOI] [PubMed] [Google Scholar]
- Barato P., Martins E. R., Vasquez G. M., Ramirez M., Melo-Cristino J., Martínez N., et al. (2016). Capsule impairs efficient adherence of Streptococcus agalactiae to intestinal epithelium in tilapias Oreochromis sp. Microb. Pathog. 100, 30–36. 10.1016/j.micpath.2016.08.040 [DOI] [PubMed] [Google Scholar]
- Barinov A., Loux V., Hammani A., Nicolas P., Langella P., Ehrlich D., et al. (2009). Prediction of surface exposed proteins in Streptococcus pyogenes, with a potential application to other Gram-positive bacteria. Proteomics 9, 61–73. 10.1002/pmic.200800195 [DOI] [PubMed] [Google Scholar]
- Barony G. M., Tavares G. C., Pereira F. L., Carvalho A. F., Dorella F. A., Leal C. A. G., et al. (2017). Large-scale genomic analyses reveal the population structure and evolutionary trends of Streptococcus agalactiae strains in Brazilian fish farms. Sci. Rep. 7:13538. 10.1038/s41598-017-13228-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidossi A., Mulas L., Decorosi F., Colomba L., Ricci S., Pozzi G., et al. (2012). A functional genomics approach to establish the complement of carbohydrate transporters in Streptococcus pneumoniae. PLoS ONE 7:e33320. 10.1371/journal.pone.0033320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boltaña S., Sanhueza N., Aguilar A., Gallardo-Escarate C., Arriagada G., Valdes J. A., et al. (2017). Influences of thermal environment on fish growth. Ecol. Evol. 7, 6814–6825. 10.1002/ece3.3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chideroli R. T., Amoroso N., Mainardi R. M., Suphoronski S. A., de Padua S. B., Alfieri A. F., et al. (2017). Emergence of a new multidrug-resistant and highly virulent serotype of Streptococcus agalactiae in fish farms from Brazil. Aquaculture 479, 45–51. 10.1016/j.aquaculture.2017.05.013 [DOI] [Google Scholar]
- Costa A. F. E., Moraes J. A., Oliveira J. S. S., Santos M. H. B., Santos G. S., Barja-Fidalgo C., et al. (2016). Reactive oxygen species involved in apoptosis induction of human respiratory epithelial (A549) cells by Streptococcus agalactiae. Microbiol. 162, 94–99. 10.1099/mic.0.000202 [DOI] [PubMed] [Google Scholar]
- Delannoy C. M., Crumlish M., Fontaine M. C., Pollock J., Foster G., Dagleish M. P., et al. (2013). Human Streptococcus agalactiae strains in aquatic mammals and fish. BMC Microbiol. 13, 1–9. 10.1186/1471-2180-13-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller M. C., Kong L., Rupp B. (2016). Protein stability: a crystallographer's perspective. Acta Crystallogr. Sect. F. Struct. Biol. Commun. 72, 72–95. 10.1107/S2053230X15024619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delphino M. K. V. C., Leal C. A. G., Garder I. A., Assis G. B. N., Roriz G. D., Ferreira F., et al. (2019). Seasonal dynamics of bacterial pathogens of Nile tilapia farmed in a Brazilian reservoir. Aquaculture 498, 100–108. 10.1016/j.aquaculture.2018.08.023 [DOI] [Google Scholar]
- Di Palo B., Rippa V., Santi I., Brettoni C., Muzzi A., Metruccio M. M. E., et al. (2013). Adaptive response of group B Streptococcus to high glucose conditions: new insights on the CovRS regulation network. PLoS ONE 8:e61294. 10.1371/journal.pone.0061294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distler U., Kuharev J., Navarro P., Levin Y., Schild H., Tenzer S. (2014). Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Meth. 11, 167–170. 10.1038/nmeth.2767 [DOI] [PubMed] [Google Scholar]
- Doran K. S., Engelson E. J., Khosravi A., Maisey H. C., Fedtke I., Equils O., et al. (2005). Blood-brain barrier invasion by group B Streptococcus depends upon proper cell-surface anchoring of lipoteichoic acid. J. Clin. Investig. 115, 2499–2507. 10.1172/JCI23829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressaire C., Laurent B., Loubiere P., Besse P., Cocaign-Bousquet M. (2010). Linear covariance models to examine the determinants of protein levels in Lactococcus lactis. Mol. Biosyst. 6, 1255–1264. 10.1039/c001702g [DOI] [PubMed] [Google Scholar]
- Ehira S., Teramoto H., Inui M., Yukawa H. (2009). Regulation of Corynebacterium glutamicum heat shock response by the extracytoplasmic-function sigma factor SigH and transcriptional regulators HspR and HrcA. J. Bacteriol. 191, 2964–2972. 10.1128/jb.00112-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. J., Bohnsack J. F., Klesius P. H., Whiting A. A., Garcia J. C., Shoemaker C. A., et al. (2008). Phylogenetic relationships among Streptococcus agalactiae isolated from piscine, dolphin, bovine and human sources: a dolphin and piscine lineage associated with a fish epidemic in Kuwait is also associated with human neonatal infections in Japan. J. Med. Microbiol. 57, 1369–1376. 10.1099/jmm.0.47815-0 [DOI] [PubMed] [Google Scholar]
- Evans J. J., Klesius P. H., Gilbert P. M., Shoemaker C. A., Al Sarawi M. A., Landsberg J., et al. (2002). Characterization of β-haemolytic group B Streptococcus agalactiae in cultured seabream, Sparus auratus L., and wild mullet, Liza klunzingeri (Day), in Kuwait. J. Fish Dis. 25, 505–513. 10.1046/j.1365-2761.2002.00392.x [DOI] [Google Scholar]
- Evans J. J., Pasnik D. J., Klesius P. H. (2015). Differential pathogenicity of five Streptococcus agalactiae isolates of diverse geographic origin in Nile tilapia (Oreochromis niloticus L.). Aquacult. Res. 46, 2374–2381. 10.1111/are.12393 [DOI] [Google Scholar]
- Ezraty B., Gennaris A., Barras F., Collet J.-F. (2017). Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 15, 385–396. 10.1038/nrmicro.2017.26 [DOI] [PubMed] [Google Scholar]
- Falconi M., Colonna B., Prosseda G., Micheli G., Gualerzi C. O. (1998). Thermoregulation of Shigella and Escherichia coli EIEC pathogenicity. a temperature-dependent structural transition of DNA modulates accessibility of virF promoter to transcriptional repressor H-NS. EMBO J. 17, 7033–7043. 10.1093/emboj/17.23.7033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faralla C., Metruccio M. M., De Chiara M., Mu R., Patras K. A., Muzzi A., et al. (2014). Analysis of two-component systems in group B Streptococcus shows that RgfAC and the novel FspSR modulate virulence and bacterial fitness. MBio 5:e00870–14. 10.1128/mBio.00870-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayet O., Ziegelhoffer T., Georgopoulos C. (1989). The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures. J. Bacteriol. 171, 1379–1385. 10.1128/jb.171.3.1379-1385.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira E. L., Batista M. T., Cavalcante R. C., Pegos V. R., Passos H. M., Silva D. A., et al. (2016). Sublingual immunization with the phosphate-binding-protein (PstS) reduces oral colonization by Streptococcus mutans. Mol. Oral Microbiol. 31, 410–422. 10.1111/omi.12142 [DOI] [PubMed] [Google Scholar]
- Florindo C., Ferreira R., Borges V., Spellerberg B., Gomes J. P., Borrego M. J. (2012). Selection of reference genes for real-time expression studies in Streptococcus agalactiae. J. Microbiol. Meth. 90, 220–227. 10.1016/j.mimet.2012.05.011 [DOI] [PubMed] [Google Scholar]
- Gadgil M., Kapur V., Hu W.-S. (2005). Transcriptional response of Escherichia coli to temperature shift. Biotechnol. Progr. 21, 689–699. 10.1021/bp049630l [DOI] [PubMed] [Google Scholar]
- Galperin M. Y., Makarova K. S., Wolf Y. I., Koonin E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. 10.1093/nar/gku1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson C. M., Mallett T. C., Claiborne A., Caparon M. G. (2000). Contribution of NADH oxidase to aerobic metabolism of Streptococcus pyogenes. J. Bacteriol. 182, 448–455. 10.1128/JB.182.2.448-455.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilar M., Olivova P., Daly A. E., Gebler J. C. (2005). Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Separat. Sci. 28, 1694–1703. 10.1002/jssc.200500116 [DOI] [PubMed] [Google Scholar]
- Glaser P., Rusniok C., Buchrieser C., Chevalier F., Frangeul L., Msadek T., et al. (2002). Genome sequence of Streptococcus agalactiae, a pathogen causing invasive neonatal disease. Mol. Microbiol. 45, 1499–1513. 10.1046/j.1365-2958.2002.03126.x [DOI] [PubMed] [Google Scholar]
- Godoy D. T., Carvalho-Castro G. A., Leal C. A. G., Pereira U. P., Leite R. C., Figueiredo H. C. P. (2013). Genetic diversity and new genotyping scheme for fish pathogenic Streptococcus agalactiae. Lett. Appl. Microbiol. 57, 476–483. 10.1111/lam.12138 [DOI] [PubMed] [Google Scholar]
- Gómez E., Perez-Pascual D., Fernandez L., Mendez J., Reimundo P., Navais R., et al. (2012). Construction and validation of a GFP-based vector for promoter expression analysis in the fish pathogen Flavobacterium psychrophilum. Gene 497, 263–268. 10.1016/j.gene.2012.01.069 [DOI] [PubMed] [Google Scholar]
- Guijarro J. A., Cascales D., García-Torrico A. I., García-Domínguez M., Méndez J. (2015). Temperature-dependent expression of virulence genes in fish-pathogenic bacteria. Front. Microbiol. 6:700. 10.3389/fmicb.2015.00700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutknecht R., Flükiger K., Lanz R., Erni B. (1999). Mechanism of phosphoryl transfer in the dimeric IIABMan subunit of the Escherichia coli mannose transporter. J. Biol. Chem. 274, 6091–6096. 10.1074/jbc.274.10.6091 [DOI] [PubMed] [Google Scholar]
- Haider S., Pal R. (2013). Integrated analysis of transcriptomic and proteomic data. Curr. Genomics 14, 91–110. 10.2174/1389202911314020003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Zhou D., Pang X., Song Y., Zhang L., Bao J., et al. (2004). Microarray analysis of temperature-induced transcriptome of Yersinia pestis. Microbiol. Immunol. 48, 791–805. 10.1111/j.1348-0421.2004.tb03605.x [DOI] [PubMed] [Google Scholar]
- Hohle T. H., Franck W. L., Stacey G., O'Brian M. R. (2011). Bacterial outer membrane channel for divalent metal ion acquisition. Proc. Natl. Acad. Sci. U. S. A. 108, 15390–15395. 10.1073/pnas.1110137108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iregui C. A., Comas J., Vásquez G. M., Verján N. (2016). Experimental early pathogenesis of Streptococcus agalactiae infection in red tilapia Oreochromis spp. J. Fish Dis. 39, 205–215. 10.1111/jfd.12347 [DOI] [PubMed] [Google Scholar]
- Karlyshev A. V., Galyov E. E., Smirnov O., Guzayev A. P., Abramov V. M., Zav'yalov V. P. (1992). A new gene of the f1 operon of Y. pestis involved in the capsule biogenesis. FEBS Lett. 297, 77–80. 10.1016/0014-5793(92)80331-A [DOI] [PubMed] [Google Scholar]
- Kawasaki M., Delamare-Deboutteville J., Bowater R. O., Walker M. J., Beatson S., Ben Zakour N. L., et al. (2018). Microevolution of Streptococcus agalactiae ST-261 from Australia indicates dissemination via imported tilapia and ongoing adaptation to marine hosts or environment. Appl. Environ. Microbiol. 84:e00859–18. 10.1128/AEM.00859-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayansamruaj P., Pirarat N., Hirono I., Rodkhum C. (2014). Increasing of temperature induces pathogenicity of Streptococcus agalactiae and the up-regulation of inflammatory related genes in infected Nile tilapia (Oreochromis niloticus). Vet. Microbiol. 172, 265–271. 10.1016/j.vetmic.2014.04.013 [DOI] [PubMed] [Google Scholar]
- Kocharunchitt C., King T., Gobius K., Bowman J. P., Ross T. (2012). Integrated transcriptomic and proteomic analysis of the physiological response of Escherichia coli O157:H7 sakai to steady-state conditions of cold and water activity stress. Mol. Cell. Proteomics 11:9019 10.1074/mcp.M111.009019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuharev J., Navarro P., Distler U., Jahn O., Tenzer S. (2015). In-depth evaluation of software tools for data-independent acquisition based label-free quantification. Proteomics 15, 3140–3151. 10.1002/pmic.201400396 [DOI] [PubMed] [Google Scholar]
- Kvint K., Nachin L., Diez A., Nyström T. (2003). The bacterial universal stress protein: function and regulation. Curr. Opin. Microbiol. 6, 140–145. 10.1016/S1369-5274(03)00025-0 [DOI] [PubMed] [Google Scholar]
- Laalami S., Zig L., Putzer H. (2014). Initiation of mRNA decay in bacteria. Cell. Mol Life Sci. 71, 1799–1828. 10.1007/s00018-013-1472-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalli P. M., Corilo Y. E., Fasciotti M., Riccio M. F., Sa G. F., Daroda R. J., et al. (2013). Baseline resolution of isomers by traveling wave ion mobility mass spectrometry: investigating the effects of polarizable drift gases and ionic charge distribution. J. Mass Spectrom. 48, 989–997. 10.1002/jms.3245 [DOI] [PubMed] [Google Scholar]
- Lestrate P., Delrue R. M., Danese I., Didembourg C., Taminiau B., Mertens P., et al. (2000). Identification and characterization of in vivo attenuated mutants of Brucella melitensis. Mol. Microbiol. 38, 543–551. 10.1046/j.1365-2958.2000.02150.x [DOI] [PubMed] [Google Scholar]
- Liu Y., Beyer A., Aebersold R. (2016). On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550. 10.1016/j.cell.2016.03.014 [DOI] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using Real-time quantitative PCR and the 2–ΔΔCT method. Methods 25, 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Lu D., Grayson P., Schulten K. (2003). Glycerol conductance and physical asymmetry of the Escherichia coli glycerol facilitator GlpF. Biophys. J. 85, 2977–2987. 10.1016/S0006-3495(03)74718-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg E., Fagerberg L., Klevebring D., Matic I., Geiger T., Cox J., et al. (2010). Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol. 6, 450–450. 10.1038/msb.2010.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainardi R. M., Lima Júnior E. A., Ribeiro Júnior J. C., Beloti V., Carmo A. O., Kalapothakis E., et al. (2016). Complete genome sequence of Streptococcus agalactiae strain S25 isolated from peritoneal liquid of Nile tilapia. Genome Announc. 4:e00784–16. 10.1128/genomeA.00784-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majtan T., Frerman F. E., Kraus J. P. (2011). Effect of cobalt on Escherichia coli metabolism and metalloporphyrin formation. Biometals 24, 335–347. 10.1007/s10534-010-9400-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcusso P. F., Aguinaga J. Y., Claudiano G. S., Eto S. F., Fernandes D. C., Mello H., et al. (2015). Influence of temperature on Streptococcus agalactiae infection in Nile tilapia. Braz. J. Vet. Res. Anim. Sci. 52, 57–62. 10.11606/issn.1678-4456.v52i1p57-62 [DOI] [Google Scholar]
- Margalef-Català M., Araque I., Bordons A., Reguant C., Bautista-Gallego J. (2016). Transcriptomic and proteomic analysis of Oenococcus oeni adaptation to wine stress conditions. Front. Microbiol. 7:1554. 10.3389/fmicb.2016.01554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata A. I., Gibello A., Casamayor A., Blanco M. M., Dominguez L., Fernandez-Garayzabal J. F. (2004). Multiplex PCR assay for detection of bacterial pathogens associated with warm-water streptococcosis in fish. Appl. Environ. Microbiol. 70, 3183–3187. 10.1128/AEM.70.5.3183-3187.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurelli A. T., Blackmon B., Curtiss R. (1984). Temperature-dependent expression of virulence genes in Shigella species. Infect. Immun. 43, 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez J., Fernández L., Menéndez A., Reimundo P., Pérez-Pascual D., Navais R., et al. (2009). A chromosomally located traHIJKCLMN operon encoding a putative type IV secretion system is involved in the virulence of Yersinia ruckeri. Appl. Environ. Microbiol. 75, 937–945. 10.1128/aem.01377-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereghetti L., Sitkiewicz I., Green N. M., Musser J. M. (2008). Remodeling of the Streptococcus agalactiae transcriptome in response to growth temperature. PLoS ONE 3:e2785. 10.1371/journal.pone.0002785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian G. F., Godoy D. T., Leal C. A. G., Yuhara T. Y., Costa G. M., Figueiredo H. C. P. (2009). Aspects of the natural history and virulence of S. agalactiae infection in Nile tilapia. Vet. Microbiol. 136, 180–183. 10.1016/j.vetmic.2008.10.016 [DOI] [PubMed] [Google Scholar]
- Moyano A. J., Tobares R. A., Rizzi Y. S., Krapp A. R., Mondotte J. A., Bocco J. L., et al. (2014). A long-chain flavodoxin protects Pseudomonas aeruginosa from oxidative stress and host bacterial clearance. PLos Genet. 10:e1004163. 10.1371/journal.pgen.1004163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moye Z. D., Burne R. A., Zeng L. (2014). Uptake and metabolism of N-acetylglucosamine and glucosamine by Streptococcus mutans. Appl. Environm. Microbiol. 80, 5053–5067. 10.1128/AEM.00820-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair S., Poyart C., Beretti J.-L., Veiga-Fernandes H., Berche P., Trieu-Cuot P. (2003). Role of the Streptococcus agalactiae ClpP serine protease in heat-induced stress defence and growth arrest. Microbiol 149, 407–417. 10.1099/mic.0.25783-0 [DOI] [PubMed] [Google Scholar]
- Nedwell D. B. (1999). Effect of low temperature on microbial growth: lowered affinity for substrates limits growth at low temperature. FEMS Microbiol. Ecol. 30, 101–111. 10.1111/j.1574-6941.1999.tb00639.x [DOI] [PubMed] [Google Scholar]
- Pelletier M., Lortie L.-A., Frenette M., Vadeboncoeur C. (1998). The phosphoenolpyruvate:mannose phosphotransferase system of Streptococcus salivarius. functional and biochemical characterization of IIABLMan and IIABHMan. Biochem. 37, 1604–1612. 10.1021/bi9721647 [DOI] [PubMed] [Google Scholar]
- Pereira U. P., Santos A. R., Hassan S. S., Aburjaile F. F., Soares S. C., Ramos R. T., et al. (2013). Complete genome sequence of Streptococcus agalactiae strain SA20-06, a fish pathogen associated to meningoencephalitis outbreaks. Stand. Genomic Sci. 8, 188–197. 10.4056/sigs.3687314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadtare S., Severinov K. (2010). RNA remodeling and gene regulation by cold shock proteins. RNA Biol. 7, 788–795. 10.4161/rna.7.6.13482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang J., Cui Y. T., Tao F. Y., Bao W. J., He J., Li X. H., et al. (2018). Physiological response and microRNA expression profiles in head kidney of genetically improved farmed tilapia (GIFT, Oreochromis niloticus) exposed to acute cold stress. Sci. Rep. 8:172. 10.1038/s,41598-017-18512-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Rajagopal L. (2009). Understanding the regulation of Group B Streptococcal virulence factors. Fut. Microbiol. 4, 201–221. 10.2217/17460913.4.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal L., Vo A., Silvestroni A., Rubens C. E. (2005). Regulation of purine biosynthesis by a eukaryotic-type kinase in Streptococcus agalactiae. Mol. Microbiol. 56, 1329–1346. 10.1111/j.1365-2958.2005.04620.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappuoli R., Arico B., Scarlato V. (1992). Thermoregulation and reversible differentiation in Bordetella: a model for pathogenic bacteria. Mol. Microbiol. 6, 2209–2211. 10.1111/j.1365-2958.1992.tb01395.x [DOI] [PubMed] [Google Scholar]
- Richter K., Haslbeck M., Buchner J. (2010). The heat shock response: life on the verge of death. Mol. Cell 40, 253–266. 10.1016/j.molcel.2010.10.006 [DOI] [PubMed] [Google Scholar]
- Rodkhum C., Kayansamruaj P., Pirarat N. (2011). Effect of water temperature on susceptibility to Streptococcus agalactiae serotype Ia infection in Nile tilapia (Oreochromis niloticus). Thai J. Vet. Med. 41, 309–314. [Google Scholar]
- Rosinski-Chupin I., Sauvage E., Mairey B., Mangenot S., Ma L., Da Cunha V., et al. (2013). Reductive evolution in Streptococcus agalactiae and the emergence of a host adapted lineage. BMC Genomics 14, 1–15. 10.1186/1471-2164-14-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador R., Muller E. E., Freitas J. C. d, Leonhadt J. H., Pretto-Giordano L. G., Dias J. A. (2005). Isolation and characterization of Streptococcus spp. group B in Nile tilapias (Oreochromis niloticus) reared in hapas nets and earth nurseries in the northern region of Parana State, Brazil. Cienc. Rural 35, 1374–1378. 10.1590/S0103-84782005000600023 [DOI] [Google Scholar]
- Samen U., Gottschalk B., Eikmanns B. J., Reinscheid D. J. (2004). Relevance of peptide uptake systems to the physiology and virulence of Streptococcus agalactiae. J. Bacteriol. 186, 1398–1408. 10.1128/JB.186.5.1398-1408.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreur P. J. W., Rebel J. M. J., Smits M. A., van Putten J. P. M., Smith H. E. (2011). TroA of Streptococcus suis is required for manganese acquisition and full virulence. J. Bacteriol. 193, 5073–5080. 10.1128/JB.05305-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz A., Schumann W. (1996). hrcA, the first gene of the Bacillus subtilis dnaK operon encodes a negative regulator of class I heat shock genes. J. Bacteriol. 178, 1088–1093. 10.1128/jb.178.4.1088-1093.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabayek S., Spellerberg B. (2017). Acid stress response mechanisms of group B Streptococci. Front. Cell. Infect. Microbiol. 7:395. 10.3389/fcimb.2017.00395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelver D., Rajagopal L., Harris T. O., Rubens C. E. (2003). MtaR, a regulator of methionine transport, is critical for survival of group B Streptococcus in vivo. J. Bacteriol. 185, 6592–6599. 10.1128/jb.185.22.6592-6599.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva W. M., Carvalho R. D., Soares S. C., Bastos I. F. S., Folador E. L., Souza G. H. M. F., et al. (2014). Label-free proteomic analysis to confirm the predicted proteome of Corynebacterium pseudotuberculosis under nitrosative stress mediated by nitric oxide. BMC Genomics 15:1065 10.1186/1471-2164-15-1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva W. M., Carvalho R. D. D. O., Dorella F. A., Folador E. L., Souza G. H. M. F., Pimenta A. M. C., et al. (2017). Quantitative proteomic analysis reveals changes in the benchmark Corynebacterium pseudotuberculosis biovar Equi exoproteome after passage in a murine host. Front. Cell. Infect. Microbiol. 7:325. 10.3389/fcimb.2017.00325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot L. M., Smoot J. C., Graham M. R., Somerville G. A., Sturdevant D. E., Migliaccio C. A. L., et al. (2001). Global differential gene expression in response to growth temperature alteration in group A Streptococcus. Proc. Natl. Acad. Sci. U. S. A. 98, 10416–10421. 10.1073/pnas.191267598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasa Rao P. S., Yamada Y., Tan Y. P., Leung K. Y. (2004). Use of proteomics to identify novel virulence determinants that are required for Edwardsiella tarda pathogenesis. Mol. Microbiol. 53, 573–586. 10.1111/j.1365-2958.2004.04123.x [DOI] [PubMed] [Google Scholar]
- Stelder S. K., Mahmud S. A., Dekker H. L., de Koning L. J., Brul S., de Koster C. G. (2015). Temperature dependence of the proteome profile of the psychrotolerant pathogenic food spoiler Bacillus weihenstephanensis type strain WSBC 10204. J. Proteome Res. 14, 2169–2176. 10.1021/pr501307t [DOI] [PubMed] [Google Scholar]
- Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., et al. (2017). The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368. 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K. J., Song S., Mason K., Pinkett H. W. (2018). Selective substrate uptake: The role of ATP-binding cassete (ABC) importers in pathogenesis. Biochim. Biophys. Acta 1860, 868–877. 10.1016/j.bbamem.2017.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian M., Qu J., Han X., Zhang M., Ding C., Ding J., et al. (2013). Microarray-based identification of differentially expressed genes in intracellular Brucella abortus within RAW264.7 cells. PLoS ONE 8:e67014. 10.1371/journal.pone.0067014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varano M., Gaspari M., Quirino A., Cuda G., Liberto M. C., Focà A. (2016). Temperature-dependent regulation of the Ochrobactrum anthropi proteome. Proteomics 16, 3019–3024. 10.1002/pmic.201600048 [DOI] [PubMed] [Google Scholar]
- Varcamonti M., Arsenijevic S., Martirani L., Fusco D., Naclerio G., De Felice M. (2006). Expression of the heat shock gene clpL of Streptococcus thermophilus is induced by both heat and cold shock. Microb. Cell Fact. 5, 6–6. 10.1186/1475-2859-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viera A. J., Garrett J. M. (2005). Understanding interobserver agreement: the kappa statistic. Fam. Med. 37, 360–363. [PubMed] [Google Scholar]
- Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456. 10.1093/nar/gkv1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu V. Q. (2011). ggbiplot: A ggplot2 Based Biplot. R package version 0.55. Available online at: https://github.com/vqv/ggbiplot
- Wang H.-W., Chung C.-W., Ma T.-W., Wong H.-C. (2013). Roles of alkyl hydroperoxide reductase subunit C (AhpC) in viable but nonculturable Vibrio parahaemolyticus. Appl. Environm. Microbiol. 79, 3734–3743. 10.1128/AEM.00560-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Liu P., Wan Z. Y., Huang S. Q., Wen Y. F., Lin G., et al. (2016). RNA-Seq revealed the impairment of immune defence of tilapia against the infection of Streptococcus agalactiae with simulated climate warming. Fish Shellfish Immunol. 55, 679–689. 10.1016/j.fsi.2016.06.058 [DOI] [PubMed] [Google Scholar]
- Warnes G. R., Bolker B., Bonebakker L., Gentleman R., Liaw W. H. A., Lumley T., et al. (2016). Various R Programming Tools for Plotting Data. R package version 3.0.1. Available online at: https://CRAN.R-project.org/package=gplots
- Whiting G. C., Sutcliffe I. C., Russell R. R. B. (1993). Metabolism of polysaccharides by the Streptococcus mutans dexB gene product. Microbiol 139, 2019–2026. 10.1099/00221287-139-9-2019 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y., Pargade V., Lamberet G., Gaudu P., Thomas F., Texereau J., et al. (2006). The Group B Streptococcus NADH oxidase Nox-2 is involved in fatty acid biosynthesis during aerobic growth and contributes to virulence. Mol. Microbiol. 62, 772–785. 10.1111/j.1365-2958.2006.05406.x [DOI] [PubMed] [Google Scholar]
- Yang Q., Porter A. J., Zhang M., Harrington D. J., Black G. W., Sutcliffe I. C. (2012). The impact of pH and nutrient stress on the growth and survival of Streptococcus agalactiae. Antonie Van Leeuwenhoek 102, 277–287. 10.1007/s10482-012-9736-9 [DOI] [PubMed] [Google Scholar]
- Yu H. B., Kaur R., Lim S., Wang X. H., Leung K. Y. (2007). Characterization of extracellular proteins produced by Aeromonas hydrophila AH-1. Proteomics 7, 436–449. 10.1002/pmic.200600396 [DOI] [PubMed] [Google Scholar]
- Yura T., Nagai H., Mori H. (1993). Regulation of the heat-shock response in bacteria. Ann. Rev. Microbiol. 47, 321–350. 10.1146/annurev.mi.47.100193.001541 [DOI] [PubMed] [Google Scholar]
- Zamri-Saad M., Amal M. N. A., Siti-Zahrah A., Zulkafli A. R. (2014). Control and prevention of streptococcosis in cultured tilapia in Malaysia: a review. Pertanika J. Trop. Agricult. Sci. 37, 389–410. [Google Scholar]
- Zhang Y. I., Griffiths M. W. (2003). Induced expression of the heat shock protein genes uspA and grpE during starvation at low temperatures and their influence on thermal resistance of Escherichia coli O157:H7. J. Food Protect. 66, 2045–2050. 10.4315/0362-028X-66.11.2045 [DOI] [PubMed] [Google Scholar]
- Zhao X.-,l., Han Y., Ren S.-,t., Ma Y.-,m., Li H., Peng X.-,x. (2015). l-proline increases survival of tilapias infected by Streptococcus agalactiae in higher water temperature. Fish Shellfish Immunol. 44, 33–42. 10.1016/j.fsi.2015.01.025 [DOI] [PubMed] [Google Scholar]
- Zheng C., Ren S., Xu J., Zhao X., Shi G., Wu J., et al. (2017). Contribution of NADH oxidase to oxidative stress tolerance and virulence of Streptococcus suis serotype 2. Virulence 8, 53–65. 10.1080/21505594.2016.1201256 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genes selected and primers used for validation of microarray results.
Complete list of transcripts identified by microarray analysis during GBS growth at 22°C and 32°C.
Complete list of proteins identified by LC-HDMSE analysis during GBS growth at 22°C and 32°C.
List of ten most and least expressed proteins identified by LC-HDMSE in GBS growth at 22°C and 32°C.
Quality assessment of the biological replicates used in transcriptome analysis. (A) Distribution of the intensities evaluated by box plot; (B) Correlation analysis matrix. (C) PCA plot, red circles represent samples grown at 32°C, whereas blue circles represent samples grown at 22°C.
Interaction networks of DEGs (A) and DPAs (B) identified in strain SA53 between the two temperatures. Red circles represent proteins upregulated at 32°C, whereas blue circles represent proteins downregulated at 32°C. (1) PTS systems; (2) ABC transport system; (3) Ascorbate and aldarate metabolism; (4) Purine metabolism; (5) metabolic pathways.
Quality assessment of biological replicates used in proteome analysis. (A) Normal distribution of 10 ppm error in the total identified peptides; (B) repeat rate indicating the number of times that an identified protein appears in the replicates: 3 of 3 (blue) and 2 of 3 (red). (C) PCA plot, red circles represent samples grown at 32°C, whereas blue circles represent samples grown at 22°C.