

Abstract
Individuals who experience greater stimulation and less sedation from alcohol are at increased risk for alcohol-related problems. However, little is known regarding the neurobiological mechanisms underlying subjective response to alcohol. The current study examined the degree to which alcohol-induced brain activation correlates with ratings of stimulation and sedation, using a within-subjects, double-blind, placebo-controlled design. Participants (N = 34 healthy adults with no history of alcohol use disorder) completed three sessions: a calibration session to determine the duration of infusion needed to bring the breath alcohol to 80 mg/dl for each subject, and two counterbalanced fMRI sessions with placebo and alcohol administration. During the fMRI sessions, participants underwent 50 min scans, which included a 10 min baseline period, the IV infusion period needed to bring breath alcohol concentration (BrAC) to a peak 80 mg/dl (on the alcohol session), followed by a post-peak decline period. Participants rated their subjective stimulation and sedation at regular intervals throughout the scan. A priori VOI analyses showed that the time course of stimulation correlated with BOLD signal in the striatum. The time course of sedation did not correlate with BOLD signal in any VOIs. There were no correlations in primary visual cortex, which served as a control. These findings are the first to show that alcohol effects in the striatum are linked to the positive, stimulant-like effects of the drug and advance our understanding of the neurobiological mechanisms underlying individual differences in subjective responses to alcohol, and more broadly, risk for alcohol use disorders.
Introduction
Alcohol is one of the most widely used psychoactive substances in the world, yet only a subset of individuals who drink alcohol develop alcohol use problems. For example, 86.4% of Americans ages 18 or older reported that they drank alcohol at some point in their lifetime, while only 6.2% developed alcohol use disorders (AUDs) [1]. There is an urgent need to identify those factors that put some drinkers at greater risk for developing alcohol use problems, particularly neurobiological factors. One important approach is to study individual differences in behavioral or physiological responses to acute doses of alcohol [2]. Related to this, several investigators have studied individual differences in the subjective rewarding effects of the drug, which are typically related to its stimulant-like effects [3, 4]. Understanding the brain processes involved in individual differences in the subjective rewarding effects will set the stage for brain-based prevention and treatment efforts.
There is some evidence that self-report measures of subjective intoxication are related to alcohol-induced changes in brain function. Using positron emission tomography (PET), Yoder et al. [5, 6] showed that individuals who reported greater feelings of “intoxication” and “high” also displayed greater alcohol-induced increase in dopamine release in the striatum (although Boileau et al. [7] did not observe this relationship in a smaller sample). Using task-based functional magnetic resonance imaging (fMRI), participants who reported greater alcohol-induced subjective intoxication also exhibited greater alcohol-induced increase in striatal BOLD activation while viewing faces [8, 9]. Taken together, these studies suggest that greater activity in the striatum is related to subjective alcohol intoxication.
Despite these promising preliminary findings, little is known about brain responses in relation to either the quality (i.e., stimulant-like vs. sedative-like) of subjective effects of alcohol, or their time course. Alcohol produces both stimulant and sedative effects, each of which have been related, sometimes in conflicting ways, to risk for alcohol-related problems [2, 4, 10]. There is growing evidence that problematic alcohol use is associated with greater sensitivity to stimulant effects and lesser sensitivity to sedative effects [3, 11, 12]. It is also widely recognized that the effects of alcohol are biphasic and vary across the time course of brain exposure to alcohol. Stimulant responses tend to occur earlier in a drinking episode, as breath alcohol concentration (BrAC) is rising, whereas sedative responses tend to occur later in a drinking episode, while BrAC is falling [10]. To date, the studies of alcohol-induced brain activity have failed to characterize either the quality (stimulant vs. sedative) or time course of the alcohol response.
To address this gap, in the present study we administered alcohol intravenously (IV; [13]) during a 50 min fMRI scan. The IV route was used to maintain control over brain alcohol exposure by monitoring breath alcohol level, and to track the time course of effects during the onset, peak, and descending limb. Importantly, the alcohol administration protocol was individualized for each subject based on a pharmacokinetic (PK) determination of response to IV alcohol. Using an individualized protocol for reaching the peak level of alcohol at the targeted time in the scanner, we examined brain regions involved in subjective stimulation and sedation after alcohol compared to placebo. We focused our analyses within the striatum, as this region has been linked to subjective alcohol intoxication [5, 6, 8, 9]. Because striatal activation (using PET) is predominately associated with the positive, rewarding effects of stimulant drugs [14, 15], we hypothesized that the time course of striatal BOLD signal would be more closely linked to the stimulant effects of alcohol than to the sedative effects. Finally, we conducted exploratory, hypothesis-free, whole brain analyses to identify additional potential brain regions linked to the stimulant and sedative effects of alcohol.
Methods
Participants
Healthy men and women (N = 42, 74% male) were recruited without regard to race or ethnicity, using posters, newspaper and online advertisements. Inclusion criteria were: 21–35 years old, at least a high school education, fluency in English, body mass index between 19 and 26 kg/m2, no history of Major Axis I psychiatric disorders [16], including drug or alcohol dependence, no history of cardiac or liver disease, no serious medical conditions, normal range electrocardiogram and blood pressure, and consuming 7–30 drinks/week with at least one binge (5 or more drinks on a single occasion for men, 4 for women) in the past month; the latter to avoid any adverse effects of the experimental alcohol exposure. Exclusion criteria were: night shift work, left-handedness, smoking >5 cigarettes per day, contraindication for MRI (e.g., claustrophobia, pacemaker), and a pronounced “flushing” reaction to alcohol.
Design
The study utilized a within-subjects, placebo-controlled design. There was a calibration session with single-blind administration of IV alcohol outside the scanner, followed by two scanning sessions with double-blind administration of placebo and alcohol in counterbalanced order. During the calibration session, we determined the infusion length needed to bring the breath alcohol to 80 mg/dl for each subject (infusion rate and infusate concentration were held constant for all subjects; see Infusion Procedure below). The sessions began at 12 pm and were at least 72 h apart (mean = 8.7 ± 1.2 days, max = 36 days). Connectivity analyses of fMRI data from the infusion end-point to10 min post-infusion have been presented elsewhere [17].
Experimental procedure
The protocol was approved by the University of Chicago Institutional Review Board for human participants. Experimental sessions were conducted at the Human Behavioral Pharmacology Laboratory (HBPL), the Clinical Research Center (CRC), and the Brain Research Imaging Center (BRIC) at the University of Chicago. Participants attended an orientation session to familiarize them with the study procedures. They gave written, informed consent. For blinding purposes subjects were told that the infusions could contain a stimulant (appetite suppressant), sedative (tranquilizer), opiate (analgesic), cannabinoid (marijuana-like drug), alcohol or placebo (inactive solution) [18]. Participants were instructed to abstain from alcohol and drugs for 24 h, and from caffeine and nicotine for 2 h, before the sessions, and to not eat after 10:00 am on the morning of sessions.
Participants provided breath and urine or saliva samples to detect recent alcohol and drug use or pregnancy in women (no one tested positive). They were then escorted to the CRC where a nurse inserted an intravenous catheter into an ante-cubital vein of the non-dominant arm for delivery of alcohol.
Infusion procedure
Infusion solutions were prepared by the Investigational Drug Service Pharmacy at the University of Chicago Medical Center and delivered using a pump (Hospira, Inc.) at the maximum rate (1998 ml/h). Alcohol infusate was 6% alcohol in 0.45% saline solution. Placebo infusate was 0.45% saline solution.
Calibration session
After pre-infusion vital signs were obtained, the pump was turned on and breath alcohol was sampled every 2 min until 80 mg/dl BrAC was attained, at which time the pump was turned off. To maintain the blind, subjects were told that breath samples taken with the “Drugalyzer” were tested for the presence of any drug metabolites. The infusion length needed to attain 80 mg/dl was recorded and used for the alcohol scanning session. The average infusion length for the sample was 14.1 ± 0.7 min (range = 8–26). Subjects completed questionnaires and vital signs were recorded every 10 min after infusions began, for 50 min. Finally, they were asked to indicate what drug they thought they received. BrAC was monitored and participants were allowed to leave the laboratory after it fell below 40 mg/dl (http://www.niaaa.nih.gov/Resources/ResearchResources/job22.htm#post).
Scanning sessions
Tubing from the infusion pump outside the scanning room passed through an opening in the wall and connected to the subject’s iv catheter. Subjects were told to lie still and relax with their eyes open, and to alert the research assistant if they experienced any nausea (no one reported feeling nauseated). During the first 10 min, baseline BOLD data were collected. At 5 min, participants rated their stimulation and sedation. Infusions began 10 min after the start of the scan. Participants completed ratings at the infusion mid-point (BrAC ascending), at the end of the infusion (BrAC peak), and then at 5 min intervals until the end of the scan (BrAC descending). After all experimental sessions were completed, participants attended a separate debriefing session, where they were informed of the sessions they had received alcohol. They were compensated $300 for participation.
Dependent measures
Drinking characteristics
Subjects reported past month drinking using the Timeline Follow Back (TLFB; [19]) and alcohol problems using the Alcohol Use Disorders Identification Test (AUDIT; [20]).
Subjective drug effects
Subjects rated their stimulation and sedation using the following scales: “I feel stimulated right now (i.e., energized, excited, up)”, and “I feel sedated right now (i.e., sedated, slow thoughts, sluggish).” The descriptors were taken from the Brief Biphasic Alcohol Effects Scale (B-BAES; [21]) that retains the strong psychometric properties of the Martin et al. [22] original 14-item scale [23]. Participants viewed the scales on a projection screen and used a button box in their right hand to indicate how they felt. The scale ranged from 1 (“not at all”) to 7 (“extremely”), with response options in increments of 1.
Imaging acquisition and processing
Images were acquired on a 3T Philips Achieva Quasar Dual 16-channel MRI scanner. A SENSE 8-channel head coil was used for reception of the MRI signal. A single-shot gradient echo EPI sequence was used with the following imaging parameters: TR/TE/flip angle = 4 s/30 ms/90°, slice thickness = 4 mm, slice gap = 0.5 mm, FOV = 228 mm, matrix size = 76 × 76, 35 slices covering the whole brain with five z-shimmed slices covering the orbital frontal cortex region [24]. High-resolution T1-weighted anatomical images with 1 mm3 isotropic resolution were acquired at the start of each scan.
Images were processed and analyzed using AFNI [25], FSL5.0.9 (FMRIB Software Library), and SPM12 (Wellcome Trust Centre for Neuroimaging). Time series BOLD images were volume registered to the first image in the time series to reduce motion artifact using 3dvolreg within AFNI. Additionally, motion outliers were identified based on framewise displacement (FD; >0.5 mm) using FSL’s motion outlier tool [26], and then censored from analyses. Subjects were excluded if at least one of their scans showed motion >10 mm total displacement in any one direction (N = 7), or if >50% of volumes within any time point window (see below) were censored (N = 1). Neither maximum motion nor mean FD differed between alcohol and placebo scans prior to exclusion of subjects (mean maximum motion difference = 0.89 ± 1.0 mm, t(41) = 0.9, p = 0.4 and mean FD difference = 0.03 ± 0.17 mm, t(41) = 1.03, p = 0.31).
Data analyses
Subjective response analyses
Linear mixed effects models for repeated measures [27] were conducted in SPSS24 to test the degree to which alcohol increased ratings of stimulation and sedation relative to placebo during the scanning sessions. The models included random intercept, drug, and time effects to allow for individual differences in drug response and time trends, and to account for the correlation between repeated measurements. In addition, we created summary measures of alcohol subjective responses (Net peak change=peak change Alcohol–peak change Placebo) to assess relationships between alcohol effects and average drinks per week, infusion length, and drug order using correlation analyses and independent samples t-tests. We also compared alcohol effects on subjective response between participants included in the final sample and those excluded due to excessive motion using Welch’s t-tests.
fMRI analyses
Imaging data were temporally downsampled to the behavioral data by taking a windowed average including the 35 time points before and after the time of the subjective response measurement, resulting in a downsampled dataset at the same temporal resolution as the behavioral data. Data were not further temporally filtered. We chose this relatively long time period because subjective responses were slow-changing and averaging over a larger window allowed for better signal to noise ratio. The windows did not overlap for any time point. The downsampled fMRI data were voxel wise correlated with the subjective response data (across all time points) using AFNI’s 3dfim+, with the resultant coefficients converted to z-scores using Fischer’s transformation. The correlation maps were then coregistered to the participant’s anatomical data and warped to MNI space, resampled to 2 mm isotropic voxels and smoothed with an 8 mm FWHM isotropic Gaussian kernel.
We compared correlation maps between the time course of subjective response ratings and the time course of BOLD signal following alcohol administration to those following placebo using paired t-tests. In order to control for typical alcohol consumption, we entered average drinks per week from the TLFB as a covariate. Based on previous associations between subjective alcohol response and striatal activity, we focused our analyses within three hypothesis-driven anatomically-focused bilateral (i.e., combination of left and right) striatal volumes of interest (VOIs): the nucleus accumbens, caudate, and putamen. We also included bilateral primary visual cortex to serve as a negative control VOI. The striatal VOIs were anatomically defined (AAL atlas) and created using MARINA (www.bion.de/eng/MARINA.php; [28]). The primary visual cortex VOI (Brodmann area 17) was created using the WFU PickAtlas tool [29, 30]. We extracted parameter estimates/ß weights representing the correlation between BOLD signal and subjective ratings in z-scores from each VOI following alcohol and placebo and conducted paired t-tests in SPSS to compare the two drug conditions. We applied a Bonferroni correction across the four VOIs tested, and so the p-value was set at p < 0.0125. We also conducted exploratory, whole-brain analyses of correlations between BOLD signal and stimulation and sedation ratings using SPM12 (cluster-forming voxel-wise height of p < 0.001, k = 50). Statistical inferences were based on cluster level significance, corrected for family wise error (FWE, p < 0.05).
Results
Participants
Most volunteers included in the analyses were male (76%) moderate drinkers (6.75–29 drinks/week) and in their mid-twenties (Table 1). They drank alcohol on average 16 days a month and engaged in binge drinking approximately once per week. Demographic characteristics of participants excluded due to motion did not differ significantly from those included in analyses.
Table 1.
Demographic and drug use characteristics of study participants. Data indicate mean±SEM unless otherwise indicated
| Characteristic | |
|---|---|
| N (male/female) | 34 (26/8) |
| Age | 26.0 ± 0.7 |
| BMI | 22.8 ± 0.3 |
| Racea (N) | |
| Asian | 1 |
| Black or African American | 3 |
| White | 27 |
| More than One Race | 2 |
| Unknown | 1 |
| Education (Years) | 15.2 ± 0.3 |
| Alcohol Use (past month) | |
| Drinks per week | 15.1 ± 1.0 |
| Binges | 4.7 ± 0.5 |
| AUDIT | 9.8 ± 0.7 |
| Other current drug useb | |
| Caffeine (N, cups per week) | 13.2 ± 1.8 (N = 31) |
| Cigarettes (N, per week) | 2.8 ± 0.6 (N = 16) |
| Marijuana (N, times in last month) | 4.5 ± 1.5 (N = 20) |
| Drug Use History (% ever used) | |
| Stimulants | 65 |
| Tranquilizers | 27 |
| Hallucinogens | 71 |
| Opiates | 29 |
| Inhalants | 21 |
| Cannabis | 91 |
aParticipants self-identified their Race by selecting from one or more of the following categories; “American Indian or Alaska Native”, “Asian”, “Black or African American”, “Native Hawaiian or Other Pacific Islander”, “White”, “More than one Race”. Individuals who declined to provide information were categorized as “Unknown”
bN represents number of individuals who reported current use
Infusion characteristics
Infusions lasted 14.1 ± 0.7 min inducing a mean peak BrAC of 81.9 ± 4.6 mg/dl. There was no significant difference between BrAC measured at the end of the laboratory (36.8 ± 1.4 mg/dL) and imaging sessions (37.2 ± 1.5 mg/dL) indicating similar peak BrACs and BrAC curves between sessions.
Correct guesses of drug received after each session were significantly greater than chance, i.e., 16.7% (calibration session = 41%, t(33) = 2.7, p < 0.05, scan 1 = 50%, t(33) = 3.8, p < 0.05; scan 2 = 62%, t(33) = 5.3, p < 0.05). Correct guesses after the alcohol scan (53%) were not significantly greater than after the alcohol familiarization session (41%, t(33) = 1.4, p = 0.18), and also did not significantly differ from correct guesses after the placebo session (59%, t(33) = 0.7, p = 0.5). Drug order did not significantly influence correct guesses after alcohol (t(32) = 0.7, p = 0.5) although correct guesses after placebo were significantly greater among individuals who received alcohol first (t(32) = 2.3, p < 0.05).
Subjective response
Linear mixed effects models tested the degree to which alcohol increased ratings of stimulation and sedation relative to placebo during the scanning sessions (Table S1). Alcohol increased both stimulation and sedation, depending upon the time after administration (indicated by significant drug×time interactions; ts(33) > 2.7; ps < 0.05). Figure 1 shows the time course of stimulation and sedation following alcohol and placebo across the scanning sessions, along with BrACs from the calibration session. The time course of stimulation and sedation behavioral ratings closely followed changes in BrAC, with peak increases at the end of infusions followed by decreasing effects across descending BrAC.
Fig. 1.

Mean BrACs during the calibration session (left panel), and mean ratings of stimulation (middle panel) and sedation (right panel) following alcohol and placebo across the scanning sessions. BL = baseline, Asc = ascending breath alcohol concentration (mid-point of infusion), Peak = peak breath alcohol concentration (end of infusion); Post5, Post10, Post15, Post20 = 5, 10, 15, and 20 min post infusion. Capped vertical lines indicate standard error of the mean
It seems paradoxical that the same drug would increase ratings of both stimulation and sedation. Therefore, we examined the patterns of these effects within subjects to determine whether there were separate subgroups of individuals who reported mainly one or the other effect. The result was mixed (Figure S1). Some individuals reported mainly stimulation and some reported mainly sedation, whereas the majority reported both effects. Thus, it appears that stimulation and sedation are not simple opposites, but may refer to different components of alcohol effects that can occur concurrently.
There were no significant relationships between summary measures of alcohol subjective response and drinking frequency (stimulation: r = −0.19, p = 0.3, sedation: r = −0.07, p = 0.7) or infusion length (stimulation: r = 0.23, p = 0.2, sedation: r = 0.03, p = 0.9). In addition, drug order did not significantly influence subjective responses (stimulation mean difference = 1.4 ± 1.5; t(32) = 0.9, p = 0.4; sedation mean difference = −2.1 ± 1.6; t(32) = 1.3, p = 0.2). There were no significant differences in subjective response between participants who were included (N = 34) and excluded (N = 8) from analyses (sedation mean difference = −0.3 ± 2.0, t(10.1) = 0.2, p = 0.9; stimulation mean difference = −0.6 ± 2.3, t(8.7) = 0.2, p = 0.8).
fMRI analyses
A priori VOI analyses: stimulation
Time course of stimulation was more highly correlated with BOLD signal following alcohol compared to placebo administration in two of the three a priori striatal VOIs (nucleus accumbens and putamen: ts(33) > 3.7, ps < 0.0125, but not caudate; Fig. 2). Figure 2 shows that correlations between striatal activity and stimulation ratings were observed only in the alcohol condition, and not following placebo. Importantly, stimulation ratings were not correlated with BOLD signal in primary visual cortex (p > 0.0125; Fig. 2), suggesting that stimulation ratings were specifically correlated with striatal regions known to be implicated in drug reward.
Fig. 2.

Mean BOLD activation (z-scores) correlated with stimulation ratings following alcohol and placebo in a priori striatal volumes of interest (VOIs), as well as the primary visual cortex, included as a control VOI. * indicates p < 0.0125. Capped vertical lines indicate standard error of the mean
A priori VOI analyses: sedation
Correlations between sedation ratings and BOLD signal did not differ following alcohol compared to placebo in any of the a priori striatal or control VOIs (ps > 0.0125).
Whole brain analyses: stimulation
Exploratory whole brain analyses of correlations between BOLD signal and stimulation ratings revealed several significant brain regions (pFWE < 0.05) (Table 2; Fig. 3) in which correlations were stronger following alcohol compared to placebo. These included regions in the insula, inferior frontal gyrus, superior temporal gyrus, thalamus, cingulate gyrus, and cerebellum. Figure 3 illustrates the right insula and cingulate activation, demonstrating a stronger correlation between the time course of stimulation ratings and the time course of BOLD signal in these regions following alcohol compared to placebo. There were no regions where stimulation ratings were more strongly correlated with BOLD signal following placebo compared to alcohol. When the alcohol and placebo conditions were analyzed separately, correlations were observed in the alcohol condition only, and not in the placebo condition.
Table 2.
Locations of significant correlations between time course of stimulation response and brain activation for alcohol>placebo, whole-brain cluster-corrected at pFWE < 0.05
| Region | MNI Coordinates | Cluster Level | Peak Level | ||||
|---|---|---|---|---|---|---|---|
| x | y | z | pFWE | k | Z | ||
| R | Superior temporal gyrus/Insula/Inferior frontal gyrus | 58 | −8 | 4 | <0.001 | 2932 | 5.65* |
| 40 | 10 | −6 | 4.89 | ||||
| 26 | −30 | 0 | 4.88 | ||||
| L | Insula | −38 | −10 | 4 | <0.001 | 726 | 4.81 |
| −18 | −24 | 2 | 4.45 | ||||
| −34 | 12 | −4 | 4.31 | ||||
| R | Cerebellum/pons | 8 | −32 | −32 | <0.001 | 1446 | 4.70 |
| 40 | −38 | −28 | 4.46 | ||||
| 8 | −34 | −40 | 4.45 | ||||
| R/L | Posterior cingulate | 4 | −52 | 12 | <0.001 | 196 | 4.56 |
| 10 | −40 | 14 | 4.51 | ||||
| −4 | −48 | 14 | 4.22 | ||||
| R/L | Cingulate gyrus | 0 | −22 | 26 | 0.001 | 163 | 4.56 |
| 10 | −24 | 24 | 3.81 | ||||
| −10 | −26 | 16 | 3.59 | ||||
| R | Anterior cingulate | 2 | 18 | 34 | 0.001 | 174 | 4.54 |
| 4 | 26 | 28 | 4.29 | ||||
| 2 | 8 | 32 | 3.51 | ||||
| L | Inferior frontal gyrus | −34 | 26 | −12 | 0.003 | 135 | 4.45 |
| −32 | 18 | −18 | 4.16 | ||||
| −34 | 28 | −2 | 4.00 | ||||
| R | Thalamus | 10 | −18 | 0 | <0.001 | 342 | 4.27 |
| 16 | −38 | −10 | 4.11 | ||||
| 10 | −6 | 6 | 3.86 | ||||
| R | Middle frontal gyrus | 34 | 40 | 24 | 0.029 | 93 | 4.21 |
| 22 | 36 | 24 | 4.31 | ||||
| R | Parahippocampa gyrus/cerebellum | 14 | −50 | 2 | <0.001 | 187 | 3.86 |
| 8 | −56 | −2 | 3.83 | ||||
| 16 | −58 | 14 | 3.79 | ||||
Display threshold = p < 0.001; k = 50; * indicates peak-level pFWE < 0.05
Fig. 3.
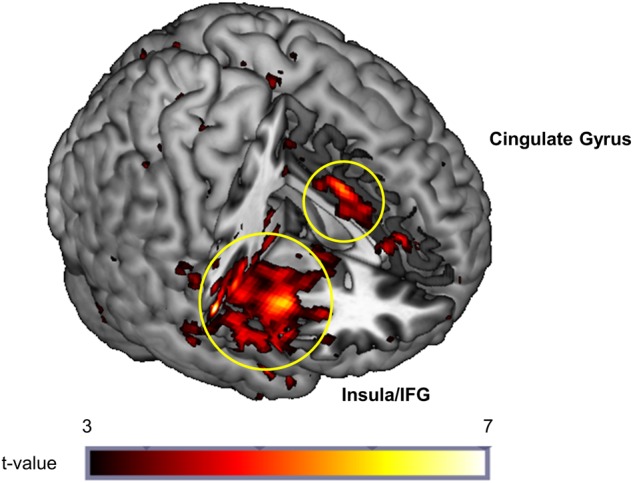
Whole-brain activation map showing greater correlation among time course of BOLD activation and stimulation ratings following alcohol compared to placebo in the right insula, inferior frontal gyrus, and cingulate gyrus (pFWE < 0.05, cluster level corrected)
Whole brain analyses: sedation
Exploratory whole brain analyses of correlations between BOLD signal and sedation ratings revealed one significant cluster (pFWE < 0.05, k = 196) for the alcohol>placebo contrast. The cluster was located in the parietal lobe and included the inferior and superior parietal lobules. There were no significant findings for the placebo>alcohol contrast.
Discussion
In this study, we sought to determine the neurobiological bases of individual differences in subjective responses to alcohol. We focused our analyses on the striatum, given the putative involvement of this region in drug reward [31, 32]. As hypothesized, BOLD signal strength in the striatum was correlated with subjective ratings of stimulation, but unrelated to sedation. Additional hypothesis-free whole brain analyses showed associations between stimulation and BOLD signal in the insula, inferior frontal gyrus, superior temporal gyrus, thalamus, cingulate gyrus, and cerebellum. The regions identified in the whole brain analyses will provide the basis for future hypothesis-driven tests of associations with alcohol-induced stimulation. Together, these findings have important implications for understanding neurobiological mechanisms underlying individual differences in subjective responses to alcohol, and more broadly, risk for AUD.
The finding that alcohol-induced striatal BOLD signal was related to stimulation but not sedation supports the notion that alcohol effects in the striatum give rise to the subjective rewarding effects of the drug. Indeed, the reinforcing effects of all drugs of abuse are thought to be due in part to drug-induced dopamine release in the striatum [31, 32], suggesting a potential relationship between striatal activity and subjective drug reward. In line with this theory, both animal and human studies show that alcohol increases dopamine activity in the striatum [33], and previous studies have linked striatal activation to alcohol intoxication [5, 6, 8, 9]. However, this is the first study to show that alcohol effects on striatal BOLD signal are specifically linked to the positive, stimulant-like effects of the drug, and not to the sedative effects, which are often rated as unpleasant [34]. Taken together, these findings strongly implicate activity in the striatum in alcohol reward in humans.
These findings provide important clues regarding how alcohol action in the brain leads to alcohol-related problems. There is considerable evidence that risk for AUD is greater in individuals who show heightened sensitivity to the stimulant effects of alcohol. Specifically, heavy drinkers show greater alcohol-induced stimulation [35, 36], and greater stimulation predicts subsequent binge drinking and number of AUD symptoms [3, 37]. Even among young adults who have not yet developed AUD, greater stimulant-like subjective effects after alcohol are associated with greater preference for alcohol in a choice test [38]. However, until now, the neurobiological mechanisms underlying individual differences in stimulant responses were unknown. Here we show that striatal BOLD signal directly correlates with stimulant responses, thus identifying a neural correlate of alcohol-induced stimulation. Although future studies are needed to determine the degree to which alcohol effects in the striatum predict severity of alcohol use, it is possible that greater striatal activity following alcohol may be a biological mechanism that increases risk for AUD.
Given the observed link between alcohol effects on striatal activity and subjective ratings of stimulation, it will be important for future studies to examine the factors that influence individual differences in striatal sensitivity to alcohol. One likely candidate is variability due to genetic factors. Indeed, Ramchandani et al. [39] found that variation in the mu opioid receptor gene predicted magnitude of dopamine release in the striatum following an acute dose of alcohol. Another potential candidate is the dopamine D2 receptor system. Individual variation in striatal D2 receptors has been linked to the subjective effects of both alcohol and stimulant drugs [6, 40], and this association is likely mediated by drug-induced activity in the striatum. In addition, there could be variability in the effects of alcohol in the striatum within individuals. For instance, striatal activity following alcohol may differ across the menstrual cycle, as estradiol levels are directly linked to striatal dopamine activity [41]. Finally, it is possible that the link between striatal activity and stimulation may be due to upstream modulation and/or downstream consequences of activity in other brain regions. Future studies using functional connectivity and network analyses might help to identify networks of regions involved in subjective experiences of alcohol stimulation. Studies testing these possibilities will provide important information regarding who might be at increased biological risk for alcohol-related problems.
In addition, whole brain exploratory analyses identified the insula, cingulate gyrus, and superior and middle temporal gyri as regions related to stimulation ratings following alcohol. The insula is strongly implicated in interoception and awareness, and it receives direct incentive signals from the striatum, among other regions, which contribute to awareness of hedonic conditions [42, 43]. Thus, it is not surprising that activity in this region is correlated with subjective responses to alcohol. The insula is also an integral part of the Salience Network, along with the anterior cingulate and striatum. Our finding that activity in these regions is correlated with alcohol-induced stimulation suggests that individuals who experience greater stimulating, rewarding effects from alcohol perceive these effects as more salient, potentially leading to greater likelihood of ongoing and future alcohol consumption. Regarding the superior and middle temporal gyri, prospective studies show that both gray matter volume and BOLD activation in these regions predict the development of alcohol-related problems [44–46]. Although speculative, it could be that altered structure or function of the temporal cortex puts individuals at risk in part because they experience greater alcohol-induced stimulation.
It is not clear why correlations were not observed between BOLD signal and sedation ratings. One possibility is that we did not select the correct VOIs to identify associations with sedation. However, we also did not observe any associations in the exploratory whole brain analyses, suggesting that the sedative effects of alcohol might not be localized to specific brain regions or networks of regions, but rather reflect a general decrease of activity throughout the brain [47]. It is also possible that the lack of findings are due to the relatively small magnitude of alcohol effect on sedation ratings. This could be due in part to the lack of a clear definition of sedation. Participants were given two additional descriptors (i.e., “slow thoughts, sluggish”), but these may have been insufficient. It will be important for future studies to examine neural correlates of sedative effects in individuals who report pronounced sedation following alcohol.
This study has several limitations. First, we did not assess changes in cerebral blood flow (CBF) following alcohol. Given that alcohol increases brain perfusion [48, 49] we cannot disambiguate whether the observed findings reflect changes in neuronal activity per se, or non-specific, vasoactive changes in CBF. Future studies should incorporate CBF measurements in order to disentangle these alternatives [49]. Second, we cannot fully rule out the influence of expectancy effects on our results, as over half of the sample correctly guessed when they had received alcohol and placebo. Third, it is unfortunate that we did not obtain a measure of impulsive behavior, as stimulating drug responses have been related to impulsive behavior [50, 51]. Fourth, we focused on intra-subject analyses to determine the degree to which BOLD signal tracked subjective response across the full time course of alcohol exposure within individuals. It will be important for future studies to examine inter-subject correlations to determine the degree to which BOLD signal correlates with subjective response between individuals as well. Finally, we did not control for smoking status, as smoking was low (N = 8) and infrequent (<5 cigarettes per day). It will be important for future studies to investigate the effects of nicotine satiation and withdrawal on alcohol effects in the brain in smokers.
In sum, this study provides the first direct evidence of a neurobiological mechanism underlying individual differences in sensitivity to the stimulant effects of alcohol in humans. These findings are an important first step towards identifying neural mechanisms underlying alcohol reward sensitivity, and therefore increased risk for alcohol use disorders. Ultimately, it is hoped that such an understanding will lead to targeted prevention and treatment efforts for AUD.
Electronic supplementary material
{kind=link}
Acknowledgements
This research was supported by the National Institute on Alcohol Abuse and Alcoholism Grant R21 AA017502 (E.C.). J.W. was supported by National Institute on Alcohol Abuse and Alcoholism Grant K01 AA024519. T.J.R. and E.A.S. were supported by the National Institute on Drug Abuse Intramural Research Program. S.O. was supported by National Institute on Alcohol Abuse and Alcoholism grant P60 AA007611. The funding agencies had no involvement in the research other than financial support.
Competing interests
H.d.W. received support for a research study from Insys Therapeutics and a donation of a research drug from Indivior. Neither of these were related to the research reported here. The remaining authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41386-018-0166-x).
References
- 1.Substance Abuse and Mental Health Services Administration (SAMHSA). National Survey on Drug Use and Health (NSDUH) (2015).
- 2.Schuckit MA. Low level of response to alcohol as a predictor of future alcoholism. Am J Psychiatry. 1994;151:184–9. doi: 10.1176/ajp.151.2.184. [DOI] [PubMed] [Google Scholar]
- 3.King AC, McNamara PJ, Hasin DS, Cao DC. Alcohol challenge responses predict future alcohol use disorder symptoms: a 6-year prospective study. Biol Psychiatry. 2014;75:798–806. doi: 10.1016/j.biopsych.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinn PD, Fromme K. Subjective response to alcohol challenge: a quantitative review. Alcohol Clin Exp Res. 2011;35:1759–70. doi: 10.1111/j.1530-0277.2011.01521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoder KK, Constantinescu CC, Kareken DA, Normandin MD, Cheng TE, O’Connor SJ, et al. Heterogeneous effects of alcohol on dopamine release in the striatum: a PET study. Alcohol Clin Exp Res. 2007;31:965–73. doi: 10.1111/j.1530-0277.2007.00390.x. [DOI] [PubMed] [Google Scholar]
- 6.Yoder KK, Kareken DA, Seyoum RA, O’Connor SJ, Wang C, Zheng QH, et al. Dopamine D(2) receptor availability is associated with subjective responses to alcohol. Alcohol Clin Exp Res. 2005;29:965–70. doi: 10.1097/01.ALC.0000171041.32716.42. [DOI] [PubMed] [Google Scholar]
- 7.Boileau I, Assaad JM, Pihl RO, Benkelfat C, Leyton M, Diksic M, et al. Alcohol promotes dopamine release in the human nucleus accumbens. Synapse. 2003;49:226–31. doi: 10.1002/syn.10226. [DOI] [PubMed] [Google Scholar]
- 8.Gilman JM, Ramchandani VA, Crouss T, Hommer DW. Subjective and neural responses to intravenous alcohol in young adults with light and heavy drinking patterns. Neuropsychopharmacology. 2012;37:467–77. doi: 10.1038/npp.2011.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilman JM, Ramchandani VA, Davis MB, Bjork JM, Hommer DW. Why we like to drink: a functional magnetic resonance imaging study of the rewarding and anxiolytic effects of alcohol. J Neurosci. 2008;28:4583–91. doi: 10.1523/JNEUROSCI.0086-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newlin DB, Thomson JB. Alcohol challenge with sons of alcoholics: a critical review and analysis. Psychol Bull. 1990;108:383–402. doi: 10.1037/0033-2909.108.3.383. [DOI] [PubMed] [Google Scholar]
- 11.King AC, Hasin D, O’Connor SJ, McNamara PJ, Cao DC. A prospective 5-year re-examination of alcohol response in heavy drinkers progressing in alcohol use disorder. Biol Psychiatry. 2016;79:489–98. doi: 10.1016/j.biopsych.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corbin WR, Gearhardt A, Fromme K. Stimulant alcohol effects prime within session drinking behavior. Psychopharmacology. 2008;197:327–37. doi: 10.1007/s00213-007-1039-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O’Connor S, Ramchandani VA, Li TK. PBPK modeling as a basis for achieving a steady BrAC of 60 +/- 5 mg% within ten minutes. Alcohol Clin Exp Res. 2000;24:426–7. [PubMed] [Google Scholar]
- 14.Leyton M, Boileau I, Benkelfat C, Diksic M, Baker G, Dagher A. Amphetamine-induced increases in extracellular dopamine, drug wanting, and novelty seeking: A PET/[C-11]raclopride study in healthy men. Neuropsychopharmacology. 2002;27:1027–35. doi: 10.1016/S0893-133X(02)00366-4. [DOI] [PubMed] [Google Scholar]
- 15.Oswald LM, Wong DF, McCaul M, Zhou Y, Kuwabara H, Choi L, et al. Relationships among ventral striatal dopamine release, cortisol secretion, and subjective responses to amphetamine. Neuropsychopharmacology. 2005;30:821–32. doi: 10.1038/sj.npp.1300667. [DOI] [PubMed] [Google Scholar]
- 16.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th edn. Washington, D.C: American Psychiatric Association; 2000. [Google Scholar]
- 17.Gorka SM, Phan KL, Childs E. Acute calming effects of alcohol are associated with disruption of the salience network. Addict Biol. 2018;23:921–30. doi: 10.1111/adb.12537. [DOI] [PubMed] [Google Scholar]
- 18.Conrad M, McNamara P, King A. Alternative substance paradigm: effectiveness of beverage blinding and effects on acute alcohol responses. Exp Clin Psychopharmacol. 2012;20:382–9. doi: 10.1037/a0029261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sobell, LC, Sobell, MB. Alcohol consumption measures. In: Allen JP, Wilson V editors. Assessing Alcohol Problems, 2nd edn. Rockville, MD: National Institute on Alcohol Abuse and Alcoholism; 2003. p. 75–99.
- 20.Babor TF, Kranzler HR, Lauerman RJ. Early detection of harmful alcohol consumption: comparison of clinical, laboratory, and self-report screening procedures. Addict Behav. 1989;14:139–57. doi: 10.1016/0306-4603(89)90043-9. [DOI] [PubMed] [Google Scholar]
- 21.Rueger SY, McNamara PJ, King AC. Expanding the utility of the Biphasic Alcohol Effects Scale (BAES) and initial psychometric support for the Brief-BAES (B-BAES) Alcohol Clin Exp Res. 2009;33:916–24. doi: 10.1111/j.1530-0277.2009.00914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin CS, Earleywine M, Musty RE, Perrine MW, Swift RM. Development and validation of the Biphasic Alcohol Effects Scale. Alcohol Clin Exp Res. 1993;17:140–6. doi: 10.1111/j.1530-0277.1993.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 23.Rueger SY, King AC. Validation of the brief Biphasic Alcohol Effects Scale (B-BAES) Alcohol Clin Exp Res. 2013;37:470–6. doi: 10.1111/j.1530-0277.2012.01941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du YP, Dalwani M, Wylie K, Claus E, Tregellas JR. Reducing susceptibility artifacts in fMRI using volume-selective z-shim compensation. Magn Reson Med. 2007;57:396–404. doi: 10.1002/mrm.21150. [DOI] [PubMed] [Google Scholar]
- 25.Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 1996;29:162–73. doi: 10.1006/cbmr.1996.0014. [DOI] [PubMed] [Google Scholar]
- 26.Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. NeuroImage. 2012;59:2142-2154. [DOI] [PMC free article] [PubMed]
- 27.Hedeker D, Gibbons RD. Longitudinal Data Analysis. Hoboken, NJ, USA: Wiley; 2006. [Google Scholar]
- 28.Walter B, Blecker C, Kirsch P, Sammer G, Schienle A, Stark R, et al. MARINA: an easy to use tool for the creation of masks for region of interest analyses. Nature 2003;19:e1899.
- 29.Maldjian JA, Laurienti PJ, Burdette JH. Precentral gyrus discrepancy in electronic versions of the Talairach atlas. Neuroimage. 2004;21:450–5. doi: 10.1016/j.neuroimage.2003.09.032. [DOI] [PubMed] [Google Scholar]
- 30.Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH. An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage. 2003;19:1233–9. doi: 10.1016/S1053-8119(03)00169-1. [DOI] [PubMed] [Google Scholar]
- 31.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–38. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, Cadoni C, et al. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology. 2004;47:227–41. doi: 10.1016/j.neuropharm.2004.06.032. [DOI] [PubMed] [Google Scholar]
- 33.Volkow ND, Wiers CE, Shokri-Kojori E, Tomasi D, Wang GJ, Baler R. Neurochemical and metabolic effects of acute and chronic alcohol in the human brain: studies with positron emission tomography. Neuropharmacology. 2017;122:175–88. doi: 10.1016/j.neuropharm.2017.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Holdstock L, King AC, de Wit H. Subjective and objective responses to ethanol in moderate/heavy and light social drinkers. Alcohol Clin Exp Res. 2000;24:789–94. doi: 10.1111/j.1530-0277.2000.tb02057.x. [DOI] [PubMed] [Google Scholar]
- 35.Marczinski CA, Combs SW, Fillmore MT. Increased sensitivity to the disinhibiting effects of alcohol in binge drinkers. Psychol Addict Behav. 2007;21:346–54. doi: 10.1037/0893-164X.21.3.346. [DOI] [PubMed] [Google Scholar]
- 36.Roche DJ, Palmeri MD, King AC. Acute alcohol response phenotype in heavy social drinkers is robust and reproducible. Alcohol Clin Exp Res. 2014;38:844–52. doi: 10.1111/acer.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King AC, de Wit H, McNamara PJ, Cao D. Rewarding, stimulant, and sedative alcohol responses and relationship to future binge drinking. Arch Gen Psychiatry. 2011;68:389–99. doi: 10.1001/archgenpsychiatry.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holdstock L, de Wit H. Individual differences in the biphasic effects of ethanol. Alcohol Clin Exp Res. 1998;22:1903–11. doi: 10.1111/j.1530-0277.1998.tb05897.x. [DOI] [PubMed] [Google Scholar]
- 39.Ramchandani VA, Umhau J, Pavon FJ, Ruiz-Velasco V, Margas W, Sun H, et al. A genetic determinant of the striatal dopamine response to alcohol in men. Mol Psychiatry. 2011;16:809–17. doi: 10.1038/mp.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volkow ND, Wang GJ, Fowler JS, Thanos P, Logan J, Gatley SJ, et al. Brain DA D2 receptors predict reinforcing effects of stimulants in humans: replication study. Synapse. 2002;46:79–82. doi: 10.1002/syn.10137. [DOI] [PubMed] [Google Scholar]
- 41.Witt ED. Puberty, hormones, and sex differences in alcohol abuse and dependence. Neurotoxicol Teratol. 2007;29:81–95. doi: 10.1016/j.ntt.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 42.Craig AD. Significance of the insula for the evolution of human awareness of feelings from the body. Ann N Y Acad Sci. 2011;1225:72–82. doi: 10.1111/j.1749-6632.2011.05990.x. [DOI] [PubMed] [Google Scholar]
- 43.Craig AD. How do you feel–now? The anterior insula and human awareness. Nat Rev Neurosci. 2009;10:59–70. doi: 10.1038/nrn2555. [DOI] [PubMed] [Google Scholar]
- 44.O’Halloran L, Nymberg C, Jollans L, Garavan H, Whelan R. The potential of neuroimaging for identifying predictors of adolescent alcohol use initiation and misuse. Addiction. 2016;112:719–26. doi: 10.1111/add.13629. [DOI] [PubMed] [Google Scholar]
- 45.Squeglia LM, Ball TM, Jacobus J, Brumback T, McKenna BS, Nguyen-Louie TT, et al. Neural predictors of initiating alcohol use during adolescence. Am J Psychiatry. 2017;174:172–85. doi: 10.1176/appi.ajp.2016.15121587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whelan R, Watts R, Orr CA, Althoff RR, Artiges E, Banaschewski T, et al. Neuropsychosocial profiles of current and future adolescent alcohol misusers. Nature. 2014;512:185–9. doi: 10.1038/nature13402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hendler RA, Ramchandani VA, Gilman J, Hommer DW. Stimulant and sedative effects of alcohol. Curr Top Behav Neurosci. 2013;13:489–509. doi: 10.1007/978-3-642-28720-6_135. [DOI] [PubMed] [Google Scholar]
- 48.Marxen M, Gan G, Schwarz D, Mennigen E, Pilhatsch M, Zimmermann US, et al. Acute effects of alcohol on brain perfusion monitored with arterial spin labeling magnetic resonance imaging in young adults. J Cereb Blood Flow Metab. 2014;34:472–9. doi: 10.1038/jcbfm.2013.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strang NM, Claus ED, Ramchandani VA, Graff-Guerrero A, Boileau I, Hendershot CS. Dose-dependent effects of intravenous alcohol administration on cerebral blood flow in young adults. Psychopharmacology. 2015;232:733–44. doi: 10.1007/s00213-014-3706-z. [DOI] [PubMed] [Google Scholar]
- 50.Weafer J, Gorka SM, Hedeker D, Dzemidzic M, Kareken DA, Phan KL, et al. Associations between behavioral and neural correlates of inhibitory control and amphetamine reward sensitivity. Neuropsychopharmacology. 2017;42:1905–13. doi: 10.1038/npp.2017.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wolf DH, Pinkham AE, Satterthwaite TD, Ruparel K, Elliott MA, Valdez J, et al. Oral alprazolam acutely increases nucleus accumbens perfusion. Mol Psychiatry. 2013;18:960–1. doi: 10.1038/mp.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.