

Abstract
Brain-derived neurotrophic factor (BDNF) is critical for establishing activity-related neural plasticity. There is increasing interest in the mechanisms of active avoidance and its extinction, but little is known about the role of BDNF in these processes. Using the platform-mediated avoidance task combined with local infusions of an antibody against BDNF, we show that blocking BDNF in either prelimbic (PL) or infralimbic (IL) medial prefrontal cortex during extinction training impairs subsequent recall of extinction of avoidance, differing from extinction of conditioned freezing. By combining retrograde tracers with BDNF immunohistochemistry, we show that extinction of avoidance increases BDNF expression in ventral hippocampal (vHPC) neurons, but not amygdala neurons, projecting to PL and IL. Using the CRISPR/Cas9 system, we further show that reducing BDNF production in vHPC neurons impairs recall of avoidance extinction. Thus, the vHPC may mediate behavioral flexibility in avoidance by driving extinction-related plasticity via BDNFergic projections to both PL and IL. These findings add to the growing body of knowledge implicating the hippocampal-prefrontal pathway in anxiety-related disorders and extinction-based therapies.
Introduction
There has been much interest in the neural mechanisms of extinction of conditioned fear. Fear extinction is thought to underlie exposure therapies for the treatment of anxiety-related disorders, and is deficient in post-traumatic stress disorder [1–3]. Extinction of freezing requires IL, basolateral amygdala (BLA), and vHPC [4–8]. Brain-derived neurotrophic factor (BDNF) is a key mediator of extinction-related neural plasticity. Pharmacological enhancement of BDNF signaling facilitates extinction of freezing [9] and impaired BDNF signaling is associated with deficient fear extinction and deficient response to extinction-based therapies [10, 11]. BDNF has been shown to be necessary for extinction of freezing in the amygdala [12], hippocampus [13], and infralimbic (IL), but not in the prelimbic (PL), prefrontal cortex [14]. Furthermore, extinction training increases the expression of BDNF in ventral hippocampal and basal amygdalar neurons [12, 14].
People suffering from PTSD repeatedly avoid stimuli associated with their traumatic experience. Persistent avoidance is considered a debilitating symptom of PTSD and other anxiety-related disorders because it interferes with attainment of goals, and reduces the opportunities for extinction [15–17]. Thus, circuits mediating extinction of active avoidance may also be compromised in PTSD. Similar to extinction of conditioned freezing, pharmacological inactivation of IL impairs extinction of platform-mediated avoidance [18]; however, conditioned fear and avoidance responses do not fully overlap. Avoidance learning reduces conditioned freezing [19], and avoidance responses can occur in the absence of freezing [20]. Furthermore, avoidance is mediated by inhibitory activity in PL whereas conditioned freezing is mediated by excitatory activity in PL [21, 22]. Moreover, circuits mediating expression of freezing vs. avoidance show some differences [18, 23, 24]. It is therefore an open question as to whether extinction of active avoidance depends on BDNFergic circuits. For this study, we characterized the role of BDNF in extinction of active avoidance using anti-body infusions, immunohistochemistry, and the CRISPR/Cas9 system to target BDNF expression.
Materials and methods
Subjects
A total of 127 male Sprague-Dawley rats (Harlan Laboratories, Indianapolis, IN) were housed and handled as previously described [25, 26]. Briefly, rats were restricted to 18 g/day of standard laboratory rat chow, and trained to press a bar for food on a variable interval schedule of reinforcement (VI-30 s). All procedures were approved by the University of Puerto Rico School of Medicine Animal Care and Use Committee, in compliance with the National Institutes of Health guidelines for the care of laboratory animals.
Surgery
Following bar-press training, rats were anesthetized under isoflurane anesthesia and 26-gauge guide bilateral cannulas (Plastics One, Roanoke, VA) were implanted at the infralimbic cortex (IL, coordinates: +2.8 AP; ±3.1 ML; −3.8 DV; angled at 30˚ [27]) or the prelimbic cortex (PL, coordinates: +2.8 AP; ±0.6 ML; −2.6 DV); For retrograde labeling experiments, the retrograde tracer cholera toxin subunit B conjugated to Alexa Fluor-555 (CTB; Life Technologies Corporation, Carslbad, CA) was injected into either IL or PL using the following coordinates relative to Bregma: +2.8 AP; ±0.6 ML; −4.0 DV or +2.8 AP; ±0.6 ML; −2.6 DV respectively. A 0.5 µl Hamilton syringe was used to inject a volume of 0.1 µl of CTB at a rate of 0.01 µl per minute across 10 min for IL or 0.2 µl of CTB at a rate of 0.02 µl per minute across 10 min for PL.
For the CRISPR/Cas9 experiments, lentiviri at a concentration of 109 particles/mL co-expressing GFP and the CRISPR/Cas9 system targeting the bdnf gene or expressing a non-targeting sequence (obtained from Sigma-Aldrich) were injected intracranially. Our surgery method was adapted from a previous study [28]. A 2.5 μL Hamilton syringe which was coated with 1% BSA before virus loading. The animals received a set of 2 pairs of bilateral injections targeting the vHPC at the following coordinates: −4.7 AP; ±5.0 ML; −7.5 DV and −5.2 AP; ±5.3 ML; −7.5 DV. The syringe was left to rest in the brain for 2 min before injecting 2 μl of virus per injection site (0.2 μl/min across 10 min) using a motorized stereotaxic injector (Stoelting, Wood Dale, IL). Following injections, the needle was left in place for 10 min before suturing the skin. Animals were allowed to recover for at least 7 days following surgery.
Behavior
Conditioning and extinction of platform-mediated avoidance was performed as previously described [18]. Throughout all the behavioral sessions, food pellets were available in the conditioning chambers and delivered at a VI-30 reward schedule during all instances. Bars in the floor delivered a scrambled electric foot-shock (Coulbourn Instruments, Allentown, PA). In the quadrant opposite of the lever, there was a plexiglass escape platform. For conditioning, rats were presented with 9 presentations of a pure tone (30 s, 4 kHz, 75 dB; 3 min intertrial interval) co-terminating with a shock delivered through the floor grids (2 s, 0.4 mA), across 10 days. On Day 11, rats were infused with anti-BDNF into either IL or PL and placed into the conditioning chambers for extinction training consisting of 15 tone presentations in the absence of foot shocks. In the platform task, rats are continually drawn to the food-bar opposite to the platform, to receive food pellets. Even when on the platform, rats periodically test the grid with their paws for the presence of shock. Sheep anti-BDNF antibody dissolved in physiologic saline (EMD Millipore, Billerica, MA) was administered at a dose of 0.5 µg/0.5 µl per side [29]. On Days 12 and 13, rats were returned to the chambers for additional extinction trials. For the immunocytochemistry experiments, rats were sacrificed 2 h following the last behavioral session and processed for immunohistochemistry. For the CRISPR/Cas9 experiments, rats were either sacrificed and processed for western blots to measure BDNF levels 2 h after the extinction session or returned to the chambers for the next 2 days for additional extinction sessions.
Histology
After behavioral experiments, rats were deeply anesthetized with sodium pentobarbital (450 mg/kg i.p.) and transcardially perfused with 0.9% saline followed by 10% buffered formaldehyde. For cryoprotection, brains were removed and stored in a 30% sucrose/10% formaldehyde solution for at least 48 h before sectioning. Coronal sections were cut 40 µm thick, mounted on slides, and stained for Nissl bodies.
Immunohistochemistry
Our immunohistochemical procedure for BDNF and NeuN followed our previous study [14], blocking with 10% NGS for 1.5 h and incubating with primary antibody at 1:100 dilution. To visualize expression of our CRISPR/Cas9 system we conducted immunohistochemistry against GFP with mouse anti-GFP antibody (1:800; ab1218, Abcam, Cambridge, MA) as primary antibody, and anti-mouse fluorescent secondary antibody (1:500, Alexa Fluor 546, Life Technologies Corporation, Carslbad, CA). Immunohistochemistry images were obtained using a microscope (Model BX51, Olympus, Tokyo, Japan) at 20x magnification coupled to a fluorescent lamp (X-Cite Series 120Q, Ontario, Canada) and a digital camera (Model DP72, Olympus, Tokyo, Japan). Images were generated for PL, IL, vHPC, basal nucleus of the amygdala (BA), and mediodorsal thalamus (MD) using appropriate filter sets for Alexa 488, Alexa 555, and Alexa 594. Images were processed in semi-automated fashion using commercial software (Metamorph version 6.1; Molecular Devices, Sunnyvale, CA) as previously described [30], to measure colocalization of BDNF with NeuN or retrograde tracer CTB. The percentage of overlapping area was averaged from regions ipsilateral to the CTB injection side at 2–3 distinct rostro-caudal levels. A similar number of CTB- labeled neurons were counted in both Extinction and No Extinction groups, for each structure. In the IL experiment, Ext and No-Ext cell counts were as follows: 71.8 and 73.7 in vHPC, 67.5 and 77.2 in BA, and 36.6 and 35.9 in MD. In the PL experiment, Ext and No-Ext cell counts were as follows: 43.1 and 44.2 in vHPC, 169.4 and 128.1 in BA, and 83.5 and 65.0 in MD.
Lentiviral vectors and CRISPR/Cas9
Lentiviral vectors at a concentration of 109 particles/mL expressing the CRISPR/Cas 9 system were obtained from Sigma-Aldrich. Vectors co-expressed the Puro antibiotic resistance element, Cas9 enzyme, and GFP under control of tEF1a promoter, and the custom gRNA sequence (guide RNA sequence) under control of the U6 promoter. The custom gRNA sequence (RatBDNF 0_18 _CGG) targeting bdnf expressed by the vector is CGGCTATGGTTATTTCATACTTCGG. Non-target control vectors expressed exactly the same elements previously mentioned with the exception that the gRNA sequence expressed a targeting sequence against a non-existing element (non-target) within the rat genome, and was therefore unable to initiate the knockout process. The custom non-target sequence expressed by the vector is GAGTCCGAGCAGAAGAAGAA.
Cell culture
Rat neuroblastoma B35 cells (ATCC® CRL-2754™; Manassas, VA) were cultured in 6-well plates at a density of 5 × 105cells/well and transduced with 3.33 × 106 TU/mL in polybrene-containing media (8 µg/mL). Transduced cells were selected in culture with puromycin (1.5 µg/mL). Puromycin concentration was selected from a kill curve (0.5–10 µg/mL puromycin), measuring cell viability by MTT (Sigma-Aldrich; St. Louis, MO) assay after 9 days of treatment. Cells cultured in polybrene-containing media (as vehicle) and untreated cells were used as negative controls and puromycin selection indicators. At day 6, cells were lysed in RIPA buffer (Abcam; Cambridge, UK) with protease inhibitor cocktail (Sigma-Aldrich; St. Louis, MO) for total protein measurement and western blotting.
We performed a bicinchoninic acid (BCA) assay (Pierce, Thermo Scientific; Waltham, MA and Bio-Rad; Hercules, CA) to determine the total protein concentration in B35 lysates. For the SDS Page, 30 µg of each sample was reduced and denatured in Laemli buffer/5% β-mercaptoethanol, heated at 70 °C for 10 min and then loaded onto a 4–20% tris/glycine (TGX; Bio-Rad) gel. The running was conducted at 150 V for 60 min. The proteins were transferred to a polyvinylidene fluoride (PVDF) membrane and blocked with 5% bovine serum albumin (BSA) in tween tris buffered saline (TTBS). Primary antibodies: polyclonal sheep anti-BDNF (1:500) and mouse monoclonal anti-GAPDH (1:500) were incubated at 4 °C overnight diluted in 5% BSA/TTBS. After three washes with TTBS, the membranes were incubated with HRP-conjugated secondary antibodies: donkey anti-sheep or goat anti-mouse (1:15,000) for 1 h at room temperature. The bands corresponding to each protein were detected in a Gel Doc™ XR+ (Bio-Rad; Hercules, CA) instrument using chemiluminescence SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Scientific) and Image Lab™ software (Bio-Rad) for acquisition of pictures and band volume densitometry analysis. Volumes of each band were calculated and normalized against the loading control GAPDH on each sample. Experiment was performed in duplicate.
Prefrontal and hippocampal tissue for western blots
Rats were deeply anesthetized with sodium pentobarbital (450 mg/Kg i.p.) 2 h after concluding the corresponding experiment. Ventromedial prefrontal cortex (vmPFC) and vHPC was dissected bilaterally, frozen, and homogenized using lysis buffer. Protein in brain lysates was measured by BCA assay. For each sample, 50 µg of total protein was reduced and denatured in Laemli Buffer with 5% 2-mercaptoethanol, heated for 5 min at 95 °C, and loaded into a TGX 4–20% gel (Bio-Rad). After SDS Page at 150 V for 1 h, proteins were transferred to a PVDF membrane in a Trans Turbo Blot equipment (Bio-Rad). The membrane was blocked for 1 h in 5% bovine serum albumin (BSA) at room temperature. Primary antibodies were diluted in 1% BSA and incubated overnight at 4 °C in the following concentrations: Rabbit monoclonal anti-BDNF (Abcam ab108319; 1:500), rabbit monoclonal anti-phospho-TrkB (Boster P01388), mouse monoclonal anti-GAPDH (Santa Cruz Biotechnology #365062; 1:500), and mouse monoclonal anti-GFP (Abcam ab1218; 1:500). Secondary antibodies used were HRP-conjugated goat anti-mouse and anti-rabbit, at 1:15,000 dilution in blocking buffer (Sigma-Aldrich). Membranes were developed with chemiluminescence in a Gel Doc™ XR+ equipment (Bio-Rad). Picture acquisition and volume intensity densitometry analyses were performed using Image Lab™ software (Bio-Rad).
Data analysis
Behavior was recorded with digital video cameras and quantified using commercial software (AnyMaze, Stoelting Co., Wood Dale, IL). Trials were averaged in blocks of two and subjected to repeated-measures analysis of variance (ANOVA), followed by Tukey’s post hoc comparisons as appropriate (STATISTICA; Statsoft, Tulsa, OK).
Results
Blocking BDNF in IL or PL delays recall of avoidance extinction
We previously observed that BDNF signaling in the IL (but not PL) was necessary for extinction of conditioned freezing [14]. Therefore, we first assessed if BDNF signaling in IL is necessary for the extinction of platform-mediated avoidance. Rats were trained for 10 days in platform-mediated avoidance (Fig. 1a). 20–40 min prior to extinction training (Day 11), we infused BDNF neutralizing antibodies (anti-BDNF) into IL to sequester extracellular BDNF, thereby preventing BDNF signaling [29, 31, 32]. As shown in Fig. 1b, anti-BDNF did not alter expression of avoidance, nor did it alter within-session extinction. Anti-BDNF infusions also did not alter locomotion (as bar-press activity) during the pretone period (S1A) or freezing to the tone (Figure S1B). The following day (Day 12), however, anti-BDNF infused rats showed elevated avoidance, as evidenced by a significant interaction in percent time spent on the platform (F(6,23) = 2.93, p = 0.010, η2 = 0.113). Post-hoc analysis revealed a significant effect in the first block of avoidance (p < 0.01). The following day (Day 13), there were no group differences. The effect of BDNF was specific to avoidance: there were no effects on freezing on Day 12 (S1B, Day 11–13, all p’s > 0.51). Furthermore, the effect on recall of extinction of avoidance was short-lived and seen only during the first extinction block. These results suggest that blocking BDNF signaling in IL during extinction training delays recall of extinction, but not its initial learning. The lack of effect on freezing by anti-BDNF differs from prior results with extinction of conditioned freezing [14].
Fig. 1.
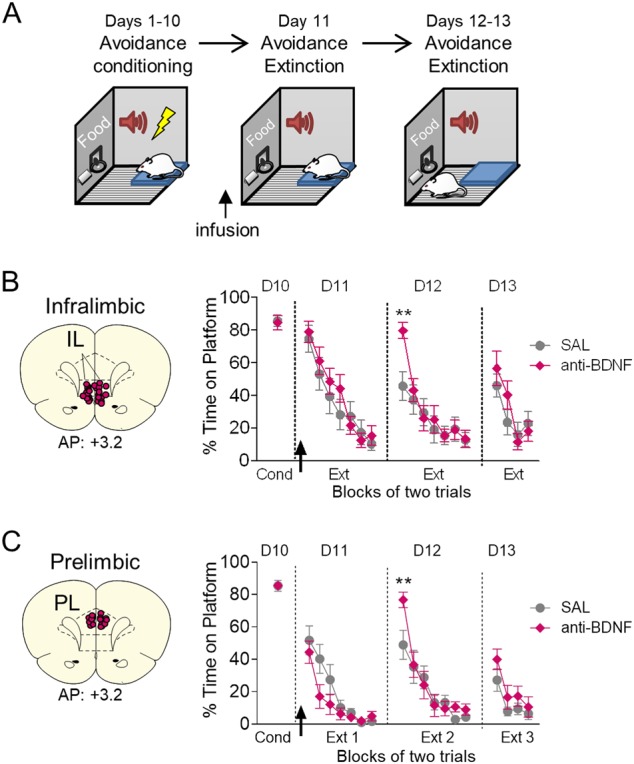
Blocking BDNF in IL or PL delays recall of extinction of avoidance. a Schematic of experimental design. b Left: Coronal drawings showing the location of the injector tips in infralimbic cortex (IL). Right: Infusion of BDNF binding antibody (anti-BDNF) into IL (arrow) prior to extinction of avoidance training delayed the recall of extinction the following day (Day 11, arrow; SAL n = 13; anti-BDNF n = 12). c Left: Coronal drawings showing the location of the injector tips in prelimbic cortex (PL). Right: Infusion of BDNF binding antibody (anti-BDNF) into PL (arrow) prior to extinction of avoidance training delayed the recall of extinction the following day (Day 11, arrow; SAL n = 15; anti-BDNF n = 13) (Right). Repeated-measures ANOVA followed by Tukey’s post hoc test. Data are shown as mean ± SEM in blocks of two trials. ** p < 0.01, interaction of treatment x trial
We next applied the same manipulation to PL (Fig. 1c). As with IL, infusion of anti-BDNF had no effect on within-session extinction (Day 11), avoidance or freezing to the tone (Fig. 1c and S1B), or bar pressing activity during the pretone period (S1A). However, on Day 12, anti-BDNF infused rats showed delayed recall of extinction, as evidenced by a significant treatment x trial interaction in percent time spent on the platform (F(6,26) = 2.53, p = 0.023, η2 = 0.089). Post-hoc analysis revealed a significant increase in the first block of avoidance (p < 0.01). On Day 13, there was no difference in the %time spent on the platform during the tone (Fig. 1c), nor were there any significant differences in freezing during the extinction sessions (S1B, all p’s > 0.19). Thus, similar to IL, blocking BDNF signaling in PL delays the recall of extinction of avoidance. This contrasts with extinction of conditioned freezing, in which neither BDNF signaling [14] nor activity in PL [7] are required.
Extinction of avoidance increases BDNF expression in the vHPC, but not in the basal amygdala
The results above suggest that BDNF released into IL and PL is necessary for extinction of avoidance. There are various potential BDNFergic inputs to the prefrontal cortex, including the vHPC, BA, MD, and/or IL and PL themselves. Therefore, we sought to identify the areas that exhibit extinction-induced increases in neuronal BDNF expression. Following avoidance conditioning, rats were exposed to either 20 extinction tones (Extinction group) or 2 extinction tones (No Extinction group, Fig. 2a) in a single session. Both groups remained in the test chambers for 71 min with access to food on a VI-30 reward schedule. Following the extinction session, animals were placed in their home cages for an additional 2 h before being sacrificed for immunohistochemistry [14]. Neuronal BDNF was assessed for each structure by measuring the percent overlap between BDNF expression and the neuronal marker NeuN (Fig. 2b [14, 33]).
Fig. 2.

Avoidance extinction increases BDNF expression in ventral hippocampal neurons, but not in amygdala or PFC neurons. a Training in extinction of avoidance for the extinction group (20 tones) and the no extinction group (2 tones). Blocks of two trials shown. b Representative micrographs showing labeling of NeuN (neurons, green), BDNF (red), and NeuN/BDNF overlap (yellow) in mediodorsal thalamus (MD). c BDNF/NeuN co-expression in the prelimbic cortex (PL), infralimbic cortex (IL), ventral hippocampus (vHPC), MD, and basal amygdala (BA), as indicated by the % area of overlap between area of BDNF label and NeuN. Extinction training significantly increased levels of neuronal BDNF in the CA1 subregion of vHPC and MD, but not in PFC or BA. Unpaired Student’s t-test between Extinction and No Extinction groups (Extinction n = 8; No Extinctions n = 5). d Red dashed lines represent dissected vmPFC region for Western Blot analysis (Upper left). Quantification of total BDNF levels and pTrkB levels respectively in vmPFC as normalized to GAPDH levels (Volume (INT/GAPDH INT)) in vmPFC between Extinction and No Extinction groups (Lower left). Red dashed lines represent dissected vHPC region (Upper right). Quantification of total BDNF levels and pTrkB levels respectively in vHPC as normalized to GAPDH levels (Volume (INT/GAPDH INT)) in vHPC between Extinction and No Extinction groups (Lower right). Unpaired Student’s t-test between Extinction and No Extinction groups (Extinction n = 4; No Extinctions n = 6). ** p < 0.01. * p < 0.05
As shown in Fig. 2c, there were no differences in BDNF levels between Extinction and No Extinction groups in IL, PL, or BA (all p’s > 0.831). However, extinction training increased neuronal BDNF in the vHPC (t(11) = 3.640, p = 0.039, Cohen’s D = 2.182) and MD (t(11) = 2.215, p = 0.049, Cohen’s D = 1.297). Thus, similar to extinction of conditioned freezing [14], extinction of avoidance increases BDNF colocalization with NeuN in vHPC, but not of IL or PL. In contrast to extinction of freezing [14], avoidance extinction failed to increase BDNF colocalization in the BA. No extinction controls exhibited virtually no avoidance behavior in the absence of tones (Supplementary Figure 2), ruling out extinction of contextual cues associated with avoidance.
We observed that infusing anti-BDNF into IL or PL delayed recall of extinction, however, extinction did not increase the number of BDNF expressing cells in IL or PL. This could indicate that extinction caused release of BDNF from inputs to prefrontal cortex, rather than release from local neurons. Consistent with this hypothesis, Western Blot analysis (Fig. 2d) showed that avoidance extinction increased both BDNF and phosphorylated BDNF receptor (pTrkB) levels in vmPFC (t(8) = 3.378, p < 0.001, Cohen’s D = 4.719 and (t(8) = 2.479, p = 0.0382, Cohen’s D = 1.596 respectively), whereas only BDNF levels were increased in vHPC (t(8) = 2.601, p = 0.032, Cohen’s D = 1.492). The observed increase in vmPFC BDNF levels with Western Blots, but not immunohistochemistry, suggests that the extinction-related BDNF in vmPFC resides within terminals or extracellular space. The increase in pTrkB in vmPFC further supports synaptic release of BDNF in vmPFC from vHPC inputs in extinction of avoidance.
Avoidance extinction increases BDNF expression in vHPC neurons projecting to IL and PL
We next investigated whether neurons expressing extinction-related BDNF in vHPC and MD project to PL or IL, by combining immunohistochemistry with retrograde tracers injected into either IL or PL (Fig. 3a). Rats injected with the retrograde tracer Alexa 555 conjugated to cholera subunit B (CTB) were trained in avoidance (10 days). On Day 11, rats were exposed to either 20 extinction tones (Extinction group) or 2 extinction tones (No Extinction group) in a single session. Both groups remained in the test chambers for 71 min with access to food pellets delivered on a VI-30 reward schedule. Following extinction training, rats were returned to their home cages and sacrificed 2 h later for immunohistochemistry. For each structure, we measured the percentage of overlap of expression of BDNF and CTB [34]. Examples of double-labeled neurons in vHPC and MD are shown in Fig. 3b.
Fig. 3.

Avoidance extinction increases BDNF expression in vHPC neurons projecting to IL and PL. a Left: Coronal drawings showing the minimum (dark orange) and maximum (light orange) spread of infusions of retrograde tracer cholera toxin-B (CTB) in IL (left) and PL (right). Right: Training in extinction of avoidance for the extinction group (20 tones) and the no extinction group (2 tones). b Representative micrographs showing labeling by CTB (red), BDNF (green), and CTB/BDNF overlap (yellow) in CA1 of vHPC (upper) and mediodorsal thalamus (lower). c (Upper) BDNF expression in neurons in vHPC, MD, and BA that project to IL, in Extinction and No extinction groups, as measured by percent of overlap between image area of BDNF and CTB labels. (Lower) Similar analysis for projections to PL. Unpaired Student’s t-test between Extinction and No Extinction groups (n = 5 per group). *p < 0.05
As shown in Fig. 3c, extinction augmented the proportion of BDNF expressing neurons projecting to IL in vHPC (t(8) = 4.129, p = 0.0033 3, Cohen’s D = 2.610), but not in MD (t(8) = 0.4518, p = 0.6634) or BA (t(8) = 0.3746, p = 0.7177). Similarly for PL projections, extinction augmented the proportion of BDNF expressing neurons in vHPC (t(8) = 2.448, p = 0.0401, Cohen’s D = 1.548), but not in MD (t(8) = 0.1667, p = 0.8718) or BA (t(8) = 0.9826, p = 0.3546; Fig. 3c). This extinction-induced increase in the proportion of BDNF-expressing projection neurons was not due to group differences in the number of CTB+ neurons analyzed (71.80 and 73.725 for the IL projection, and 43.1 and 44.2 for the PL projection for the Extinction and no Extinction groups respectively). Nor was there any context-induced avoidance behavior (S2). Thus, extinction increases the expression of BDNF in vHPC neurons projecting to both IL and PL. In contrast, extinction-related neurons in MD do not project to PL or IL.
Reducing BDNF production in vHPC neurons impairs extinction of avoidance
Our results are consistent with the idea that the release of BDNF from vHPC inputs to IL and PL is necessary for extinction of avoidance. However, because this link is correlative, it’s still possible that extinction-related BDNF originates in other structures. Therefore, to determine the necessity of BDNF in vHPC, we attempted to reduce BDNF production in vHPC by infusing a lentivirus expressing the CRISPR/Cas9 system [35] targeting the bdnf gene (RatBDNF_0_18_CGG, obtained from Sigma-Aldrich). The CRISPR/Cas9 system introduces double strand breaks in the gene of interest. The attempted repair by the error prone non-homologous end-joining pathway leads to the introduction of errors that prevent transcription, effectively knocking out the gene [35]. Because this technique had never been used for behavioral studies of BDNF, we first tested the system in neuroblastoma cells to confirm that CRISPR/Cas9 could reduce BDNF production. Because BDNF is cleaved from its precursor proBDNF, knocking out bdnf will result in the loss of both proBDNF and BDNF. Accordingly, we observed that the CRISPR/Cas9 system reduced BDNF production in neuroblastoma cells, as confirmed by the reduction in proBDNF and BDNF bands in western blots (Fig. 4a).
Fig. 4.

Reducing BDNF production in vHPC neurons impairs extinction of avoidance. a Western blot showing bands for proBDNF (32 kDa), BDNF (14 kDa), and GAPDH (37 kDa) of neuroblastoma cells belonging to non-target control and bdnf knockdown group. b Schematic of experimental design for the use of CRISPR/Cas9 system to determine if BDNF production is necessary for extinction of avoidance. c Extinction training for the group with CRISPR/Cas9 system targeting the bdnf gene (bdnf knockdown) and non-target controls (bdnf knockdown, n = 6; non-target controls, n = 8) following injection of lentiviri expressing the CRISPR/Cas9 system (arrow). 2 h following the extinction session, hippocampi were extracted for western blots. d Left: Sample western blot showing bands for proBDNF (32 kDa), BDNF (14 kDa), and GAPDH (37 kDa) of hippocampi belonging to non-target control and bdnf knockdown rats. Right: Quantification of total BDNF levels normalized to GAPDH levels (Volume (total BDNF INT/GAPDH INT)) in non-target controls and bdnf knockdown (non-target controls, n = 8; bdnf knockdown, n = 6). e Micrograph and coronal drawing showing the minimum (dark orange) and maximum (light orange) spread of lentivirus expression in vHPC as revealed by GFP. f Reducing BDNF production with the CRISPR/Cas9 system prior to extinction of avoidance impaired recall of extinction the following day (Day 26; non-targeting n = 11; bdnf targeting n = 12). Inset: Freezing to the first 2 extinction tones on Day 26. Unpaired Student’s t-test or repeated-measures ANOVA followed by Tukey post hoc test. Data are shown as mean ± SEM in blocks of two trials. *p < 0.05, treatment group
We next tested if targeting the bdnf gene in the vHPC in vivo reduces extinction levels of BDNF. Rats were conditioned for 10 days, and on Day 11 were infused in vHPC with lentiviri expressing the CRISPR/Cas9 system targeting bdnf (Fig. 4b). This was followed by 14 days of recovery to (1) fully express the virus, (2) knockdown bdnf, and (3) allow for degradation of already existing BDNF. On Day 25, rats were presented with 20 extinction tones (Fig. 4c). Extinction was somewhat slower in rats with bdnf knockdown, but this difference was not statistically significant (F(1,12) = 2.23, p = 0.161, η2 = 0.157). 2 h after training, vHPC was dissected and prepared for western blots. CRISPR/Cas9 significantly reduced BDNF expression in the vHPC (Fig. 4d) (t(4) = 2.891, p = 0.014, Cohen’s D = 1.642). In a separate set of animals, we determined whether knocking down BDNF in the vHPC would impair extinction of avoidance. Fig. 4e (left) shows the extent of lentiviral expression in the ventral hippocampus as indicated by expression of GFP. As shown in Fig. 4f, rats with bdnf knockdown showed a significant difference in within-session extinction of avoidance on day 25 (main effect of group, F(1,21) = 4.44, p = 0.047, η2 = 0.174). The following day, rats with bdnf knockdown showed significantly higher avoidance during recall (day 26, F(1,21) = 6.41, p = 0.019, η2 = 0.234). Post-hoc analysis on day 26 revealed a significant effect in the first and fourth blocks of avoidance (p < 0.05). This effect was specific to avoidance, as we did not observe any effect on freezing (Fig. 4f inset). Altogether, our findings suggest that BDNF released by vHPC into both PL and IL is required for the extinction of avoidance.
Discussion
In the present study, we demonstrated that recall of extinction of platform-mediated avoidance relies on BDNF in both IL and PL subdivisions of the medial prefrontal cortex. We also showed that extinction of avoidance increases BDNF expression in vHPC and MD neurons, but not in BA neurons, and this increase was observed in vHPC neurons projecting to IL and PL. Moreover, we showed that interfering with BDNF production within the vHPC impairs the extinction of avoidance.
BDNF packaging and trafficking is a highly regulated process. BDNF is stored at dense core vesicles at synaptic terminals [36, 37] ready to be released into the synapse by high frequency firing. The BDNF increases we observed may be required to replenish terminal BDNF supply following release. BDNF is transported at an average rate of 1.18 µm/s [38], which is sufficiently fast to transport BDNF from vHPC to vmPFC within the 2 hour time period we analyzed. BDNF activates TrkB receptors which in turn activate molecular processes required for plasticity [32, 39, 40]. Consistent with the idea of increased BDNF release from terminals in vmPFC, we observed extinction-induced activation of the TrkB receptor in vmPFC, but not in vHPC.
Our findings with anti-BDNF in IL are consistent with the observation that pharmacological inactivation of IL impairs extinction of avoidance in this task [18]. Inactivation of IL also increased freezing during avoidance [18], suggesting that IL may enable avoidance behavior by reducing freezing [41]. In the present study, however, reducing BDNF in IL delayed recall of extinction of avoidance without increasing freezing, consistent with an impairment in extinction-associated plasticity rather than a reduction in IL activity. The effect was seen only in the first trial block, suggesting that BDNF-mediated plasticity in IL facilitates initial recall of extinction learning. Perhaps a shorter or weaker extinction protocol may reveal a more marked extinction deficit by making recall of extinction more challenging.
The observed effects on extinction recall are only present during the second extinction session following sequestration of extracellular BDNF in IL or PL, or by knocking down BDNF production in the vHPC. In the case of BDNF sequestration in IL or PL, the animals are able to learn normally during the second extinction session because they are drug-free. In the case of the CRISPR-Cas9 experiment, the animals are able to compensate for the learning deficit because there is likely still some BDNF available for learning, as not all the vHPC neurons express the CRISPR-Cas9 (transfection is not 100%).
In contrast to previous findings showing that PL is not necessary for extinction of conditioned freezing [7, 14, 42], we found here that BDNF mediated plasticity in PL is necessary for recall of extinction of avoidance the following day, thereby adding to PL’s reported role in avoidance expression ([18, 43] but see ref. [41]). It is unlikely that the effect of anti-BDNF in PL is due to an unintended diffusion into neighboring IL, because infusing the same volume of anti-BDNF into PL or IL had differing effects on extinction of conditioned freezing [14]. Consistent with our PL findings, it was recently reported that pre-training lesions of PL impair extinction of lever-press active avoidance [43], and extinction of avoidance in our task activates PL neurons projecting to ventral striatum (Martínez-Rivera et al., 2018 in press) [44].
PL is thought to be critical for strategy switching and behavioral flexibility [45, 46]. PL neurons signal shifts in strategy, and PL is necessary for behavioral shifts in response to rule changes [47–50]. In the platform-mediated avoidance task, extinction of avoidance can be seen as a rule change requiring a strategy shift in decision making from tone-triggered “gain of safety/loss of food”, to tone-triggered “loss of safety/gain of food”. Thus, unlike extinction of freezing, BDNF signaling in PL may be necessary for the plasticity associated with the switching of strategies in extinction of avoidance. PL BDNF-mediated plasticity may also enhance signaling in PL neurons projecting to IL [51], further facilitating extinction learning.
In BA, BDNF is required for the extinction of conditioned freezing [12, 52], and extinction training increases BDNF expression in BA [12, 14]. In contrast, we observed that extinction of avoidance did not increase BDNF expression in BA. Thus, unlike extinction of conditioned freezing, extinction of avoidance does not require BDNF production within the amygdala. The amygdala may receive BDNF from other inputs to mediate extinction of avoidance, or may not be involved in this type of extinction. Further experiments are needed to determine the role of BA BDNF in extinction of avoidance.
Neuronal activity patterns in MD have been associated with successful extinction of conditioned freezing. Tonic activity patterns in MD are associated with extinction success, and burst activity patterns are associated with extinction failure [53]. Furthermore, MD has been implicated in behavioral flexibility and strategy shifting [54–59]. MD could therefore be involved in the reward/avoidance strategy switching inherent in the extinction phase of our task. We observed extinction-related increases in BDNF expression in MD, but not in MD neurons that project to IL or PL. If BDNF in MD is necessary for extinction of avoidance, it is likely being released at sites other than mPFC, such as the orbital cortex, agranular insular cortex, and dorsal anterior cingulate cortex [60–62]. In fact, the central zone of MD, where we observed increased BDNF, projects to both the ventral anterior insular cortex and lateral orbital cortex [62].
Unlike in BA, BDNF in the vHPC was increased by extinction of avoidance and reducing BDNF production in vHPC impairs the recall of extinction of avoidance. Prior work has shown that BDNF in vHPC is required for the extinction of conditioned freezing [13, 63]. Furthermore, infusion of BDNF in vHPC induces extinction of freezing in the absence of training [29] and increases IL firing rate [14]. The projection from vHPC to medial prefrontal cortex is robust [64], and we observed that extinction of avoidance increased BDNF in vHPC neurons that project to PL and IL. The vHPC is required for extinction of conditioned freezing [7], and the projection from vHPC to PL has been shown to gate context-dependent expression of conditioned fear [65–67] and anxiety-like behavior [68]. During extinction of avoidance, hippocampal BDNF may be mediating plasticity required for setting the conditions under which avoidance is no longer advantageous. Our cumulative data strongly suggest that plasticity occurs in the hippocampal-prefrontal pathway to mediate behavioral change in extinction. One caveat, however, is that we did not directly test this connection. It is possible that some effects of knocking down hippocampal BDNF may have been mediated by projections to the BA. Further experiments are required to fully test these possibilities.
In PTSD, the pathway from the hippocampus to prefrontal cortex is deficient [69, 70], and PTSD symptoms are correlated with decreased connectivity in the hippocampal-vmPFC pathway [69, 70]. There is also a correlation between the activation of fear-associated structures and the severity of avoidance symptoms in PTSD [71]. This suggests that elevated fear may promote persistent avoidance, an idea supported by recent studies in rodents [24, 26]. Persistent avoidance leads to psychosocial dysfunction and a reduction in the opportunities for spontaneously-occurring extinction of the traumatic memory [15]. Our results suggest that a disrupted hippocampal-prefrontal pathway may be a common substrate for both excessive fear responses and persistent avoidance observed in PTSD. Increasing levels of BDNF in the hippocampus or PFC/ACC may repair or compensate for the structural deficits associated with PTSD, and increase plasticity required for extinction -based therapies that target fear and avoidance.
Electronic supplementary material
Acknowledgements
The authors thank Kelvin Quiñones-Laracuente, Hector Bravo-Rivera, Maria M Diehl, Pablo A. Pagán Rivera, and Jose Colom for their help with behavioral experiments. We also thank Zarkalys Quintero and Carlos Rodriguez for technical support.
Funding
This study was supported by NIH grants R36-MH106332 to LER-V and R37-MH058883 and P50-MH106435 to GJQ, and a grant from the University of Puerto Rico President’s Office to GJQ, NIGMS SC1GM113691 to LMM, and NIMH U54MD007600 (UPR-MSC RCMI Translational Proteomics Center).
Competing interests
The authors declare that they have no conflict of interest.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41386-018-0176-8).
References
- 1.Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, et al. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol Psychiatry. 2009;66:1075–82. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shvil E, Sullivan GM, Schafer S, Markowitz JC, Campeas M, Wager TD, et al. Sex differences in extinction recall in posttraumatic stress disorder: a pilot fMRI study. Neurobiol Learn Mem. 2014;113:101–8. doi: 10.1016/j.nlm.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zuj DV, Palmer MA, Lommen MJ, Felmingham KL. The centrality of fear extinction in linking risk factors to PTSD: a narrative review. Neurosci Biobehav Rev. 2016;69:15–35. doi: 10.1016/j.neubiorev.2016.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Falls WA, Miserendino MJ, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–63. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herry C, Ciocchi S, Senn V, Demmou L, Muller C, Luthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–6. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- 6.Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Luthi A. Neuronal circuits of fear extinction. Eur J Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- 7.Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–38. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sotres-Bayon F, Diaz-Mataix L, Bush DE, LeDoux JE. Dissociable roles for the ventromedial prefrontal cortex and amygdala in fear extinction: NR2B contribution. Cereb Cortex. 2009;19:474–82. doi: 10.1093/cercor/bhn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry. 2011;168:163–72. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Felmingham KL, Dobson-Stone C, Schofield PR, Quirk GJ, Bryant RA. The brain-derived neurotrophic factor Val66Met polymorphism predicts response to exposure therapy in posttraumatic stress disorder. Biol Psychiatry. 2013;73:1059–63. doi: 10.1016/j.biopsych.2012.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science. 2010;327:863–6. doi: 10.1126/science.1181886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chhatwal JP, Stanek-Rattiner L, Davis M, Ressler KJ. Amygdala BDNF signaling is required for consolidation but not encoding of extinction. Nat Neurosci. 2006;9:870–2. doi: 10.1038/nn1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heldt SA, Stanek L, Chhatwal JP, Ressler KJ. Hippocampus-specific deletion of BDNF in adult mice impairs spatial memory and extinction of aversive memories. Mol Psychiatry. 2007;12:656–70. doi: 10.1038/sj.mp.4001957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosas-Vidal LE, Do-Monte FH, Sotres-Bayon F, Quirk GJ. Hippocampal-prefrontal BDNF and memory for fear extinction. Neuropsychopharmacology. 2014;39:2161–9. doi: 10.1038/npp.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foa EB. Social anxiety disorder treatments: psychosocial therapies. J Clin Psychiatry. 2006;67:27–30. [PubMed] [Google Scholar]
- 16.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Washington DC: 2013.
- 17.Aupperle RL, Melrose AJ, Stein MB, Paulus MP. Executive function and PTSD: disengaging from trauma. Neuropharmacology. 2012;62:686–94. doi: 10.1016/j.neuropharm.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bravo-Rivera C, Roman-Ortiz C, Brignoni-Perez E, Sotres-Bayon F, Quirk GJ. Neural structures mediating expression and extinction of platform-mediated avoidance. J Neurosci. 2014;34:9736–42. doi: 10.1523/JNEUROSCI.0191-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mowrer OH, Lamoreaux RR. Fear as an intervening variable in avoidance conditioning. J Comp Psychol. 1946;39:29–50. doi: 10.1037/h0060150. [DOI] [PubMed] [Google Scholar]
- 20.Overmier JB, Brackbill RM. On the independence of stimulus evocation of fear and fear evocation of responses. Behav Res Ther. 1977;15:51–6. doi: 10.1016/0005-7967(77)90087-0. [DOI] [PubMed] [Google Scholar]
- 21.Burgos-Robles A, Vidal-Gonzalez I, Quirk GJ. Sustained conditioned responses in prelimbic prefrontal neurons are correlated with fear expression and extinction failure. J Neurosci. 2009;29:8474–82. doi: 10.1523/JNEUROSCI.0378-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diehl MM, Bravo-Rivera C, Rodriguez-Romaguera J, Pagan-Rivera PA, Burgos-Robles A, Roman-Ortiz C et al. Active avoidance requires inhibitory signaling in the rodent prelimbic prefrontal cortex. Elife 2018;7:e34657. [DOI] [PMC free article] [PubMed]
- 23.Ilango A, Shumake J, Wetzel W, Ohl FW. Contribution of emotional and motivational neurocircuitry to cue-signaled active avoidance learning. Front Behav Neurosci. 2014;8:372. doi: 10.3389/fnbeh.2014.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bravo-Rivera C, Roman-Ortiz C, Montesinos-Cartagena M, Quirk GJ. Persistent active avoidance correlates with activity in prelimbic cortex and ventral striatum. Front Behav Neurosci. 2015;9:184. doi: 10.3389/fnbeh.2015.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–31. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez-Romaguera J, Greenberg BD, Rasmussen SA, Quirk GJ. An avoidance-based rodent model of exposure with response prevention therapy for obsessive-compulsive disorder. Biol Psychiatry. 2016;80:534–40. [DOI] [PMC free article] [PubMed]
- 27.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4th edn. San Diego: Academic Press; 1998. [Google Scholar]
- 28.Rattiner LM, Davis M, French CT, Ressler KJ. Brain-derived neurotrophic factor and tyrosine kinase receptor B involvement in amygdala-dependent fear conditioning. J Neurosci. 2004;24:4796–806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–90. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Do-Monte FH, Rodriguez-Romaguera J, Rosas-Vidal LE, Quirk GJ. Deep brain stimulation of the ventral striatum increases BDNF in the fear extinction circuit. Front Behav Neurosci. 2013;7:102. doi: 10.3389/fnbeh.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alonso M, Vianna MR, Izquierdo I, Medina JH. Signaling mechanisms mediating BDNF modulation of memory formation in vivo in the hippocampus. Cell Mol Neurobiol. 2002;22:663–74. doi: 10.1023/A:1021848706159. [DOI] [PubMed] [Google Scholar]
- 32.Slipczuk L, Bekinschtein P, Katche C, Cammarota M, Izquierdo I, Medina JH. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS ONE. 2009;4:e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–11. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 34.Penzo MA, Robert V, Tucciarone J, De Bundel D, Wang M, Van Aelst L, et al. The paraventricular thalamus controls a central amygdala fear circuit. Nature. 2015;519:455–9. doi: 10.1038/nature13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 36.Lessmann V, Brigadski T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: an update. Neurosci Res. 2009;65:11–22. doi: 10.1016/j.neures.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Song M, Martinowich K, Lee FS. BDNF at the synapse: why location matters. Mol Psychiatry. 2017;22:1370–5. doi: 10.1038/mp.2017.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao X, Chen XQ, Han E, Hu Y, Paik P, Ding Z, et al. TRiC subunits enhance BDNF axonal transport and rescue striatal atrophy in Huntington’s disease. Proc Natl Acad Sci USA. 2016;113:E5655–64. doi: 10.1073/pnas.1603020113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Musumeci G, Minichiello L. BDNF-TrkB signalling in fear learning: from genetics to neural networks. Rev Neurosci. 2011;22:303–15. doi: 10.1515/rns.2011.031. [DOI] [PubMed] [Google Scholar]
- 40.Black IB. Trophic regulation of synaptic plasticity. J Neurobiol. 1999;41:108–18. doi: 10.1002/(SICI)1097-4695(199910)41:1<108::AID-NEU14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 41.Moscarello JM, LeDoux JE. Active avoidance learning requires prefrontal suppression of amygdala-mediated defensive reactions. J Neurosci. 2013;33:3815–23. doi: 10.1523/JNEUROSCI.2596-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang CH, Berke JD, Maren S. Single-unit activity in the medial prefrontal cortex during immediate and delayed extinction of fear in rats. PLoS ONE. 2010;5:e11971. doi: 10.1371/journal.pone.0011971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fragale JE, Khariv V, Gregor DM, Smith IM, Jiao X, Elkabes S, et al. Dysfunction in amygdala-prefrontal plasticity and extinction-resistant avoidance: a model for anxiety disorder vulnerability. Exp Neurol. 2016;275:59–68. doi: 10.1016/j.expneurol.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martínez-Rivera FJ, Bravo-Rivera C, Velázquez-Díaz CD, Montesinos-Cartagena M, Quirk GJ. Prefrontal circuits signaling retrieval and extinction of active avoidance. Psychopharmacology (in press). [DOI] [PMC free article] [PubMed]
- 45.Dalley JW, Cardinal RN, Robbins TW. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev. 2004;28:771–84. doi: 10.1016/j.neubiorev.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 46.Kesner RP, Churchwell JC. An analysis of rat prefrontal cortex in mediating executive function. Neurobiol Learn Mem. 2011;96:417–31. doi: 10.1016/j.nlm.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 47.Durstewitz D, Vittoz NM, Floresco SB, Seamans JK. Abrupt transitions between prefrontal neural ensemble states accompany behavioral transitions during rule learning. Neuron. 2010;66:438–48. doi: 10.1016/j.neuron.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 48.Floresco SB, Block AE, Tse MT. Inactivation of the medial prefrontal cortex of the rat impairs strategy set-shifting, but not reversal learning, using a novel, automated procedure. Behav Brain Res. 2008;190:85–96. doi: 10.1016/j.bbr.2008.02.008. [DOI] [PubMed] [Google Scholar]
- 49.Oualian C, Gisquet-Verrier P. The differential involvement of the prelimbic and infralimbic cortices in response conflict affects behavioral flexibility in rats trained in a new automated strategy-switching task. Learn Mem. 2010;17:654–68. doi: 10.1101/lm.1858010. [DOI] [PubMed] [Google Scholar]
- 50.Rich EL, Shapiro M. Rat prefrontal cortical neurons selectively code strategy switches. J Neurosci. 2009;29:7208–19. doi: 10.1523/JNEUROSCI.6068-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marek R, Xu L, Sullivan RKP, Sah P. Excitatory connections between the prelimbic and infralimbic medial prefrontal cortex show a role for the prelimbic cortex in fear extinction. Nat Neurosci. 2018;21:654–8. doi: 10.1038/s41593-018-0137-x. [DOI] [PubMed] [Google Scholar]
- 52.Heldt SA, Zimmermann K, Parker K, Gaval M, Ressler KJ. Bdnf deletion or TrkB impairment in amygdala inhibits both appetitive and aversive learning. J Neurosci. 2014;34:2444–50. doi: 10.1523/JNEUROSCI.4085-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee S, Ahmed T, Kim H, Choi S, Kim DS, Kim SJ, et al. Bidirectional modulation of fear extinction by mediodorsal thalamic firing in mice. Nat Neurosci. 2012;15:308–14. doi: 10.1038/nn.2999. [DOI] [PubMed] [Google Scholar]
- 54.Hunt PR, Aggleton JP. Neurotoxic lesions of the dorsomedial thalamus impair the acquisition but not the performance of delayed matching to place by rats: a deficit in shifting response rules. J Neurosci. 1998;18:10045–52. doi: 10.1523/JNEUROSCI.18-23-10045.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitchell AS. The mediodorsal thalamus as a higher order thalamic relay nucleus important for learning and decision-making. Neurosci Biobehav Rev. 2015;54:76–88. doi: 10.1016/j.neubiorev.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Mitchell AS, Chakraborty S. What does the mediodorsal thalamus do? Front Syst Neurosci. 2013;7:37. doi: 10.3389/fnsys.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ostlund SB, Balleine BW. Differential involvement of the basolateral amygdala and mediodorsal thalamus in instrumental action selection. J Neurosci. 2008;28:4398–405. doi: 10.1523/JNEUROSCI.5472-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parnaudeau S, Taylor K, Bolkan SS, Ward RD, Balsam PD, Kellendonk C. Mediodorsal thalamus hypofunction impairs flexible goal-directed behavior. Biol Psychiatry. 2015;77:445–53. doi: 10.1016/j.biopsych.2014.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bradfield LA, Hart G, Balleine BW. The role of the anterior, mediodorsal, and parafascicular thalamus in instrumental conditioning. Front Syst Neurosci. 2013;7:51. doi: 10.3389/fnsys.2013.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krettek JE, Price JL. The cortical projections of the mediodorsal nucleus and adjacent thalamic nuclei in the rat. J Comp Neurol. 1977;171:157–91. doi: 10.1002/cne.901710204. [DOI] [PubMed] [Google Scholar]
- 61.Matyas F, Lee J, Shin HS, Acsady L. The fear circuit of the mouse forebrain: connections between the mediodorsal thalamus, frontal cortices and basolateral amygdala. Eur J Neurosci. 2014;39:1810–23. doi: 10.1111/ejn.12610. [DOI] [PubMed] [Google Scholar]
- 62.Ray JP, Price JL. The organization of the thalamocortical connections of the mediodorsal thalamic nucleus in the rat, related to the ventral forebrain-prefrontal cortex topography. J Comp Neurol. 1992;323:167–97. doi: 10.1002/cne.903230204. [DOI] [PubMed] [Google Scholar]
- 63.Sakata K, Martinowich K, Woo NH, Schloesser RJ, Jimenez DV, Ji Y et al. Role of activity-dependent BDNF expression in hippocampal-prefrontal cortical regulation of behavioral perseverance. Proc Natl Acad Sci USA. 2013;110:15103-08 [DOI] [PMC free article] [PubMed]
- 64.Hoover WB, Vertes RP. Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct Funct. 2007;212:149–79. doi: 10.1007/s00429-007-0150-4. [DOI] [PubMed] [Google Scholar]
- 65.Hugues S, Garcia R. Reorganization of learning-associated prefrontal synaptic plasticity between the recall of recent and remote fear extinction memory. Learn Mem. 2007;14:520–4. doi: 10.1101/lm.625407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin J, Maren S. Fear renewal preferentially activates ventral hippocampal neurons projecting to both amygdala and prefrontal cortex in rats. Sci Rep. 2015;5:8388. doi: 10.1038/srep08388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sotres-Bayon F, Sierra-Mercado D, Pardilla-Delgado E, Quirk GJ. Gating of fear in prelimbic cortex by hippocampal and amygdala inputs. Neuron. 2012;76:804–12. doi: 10.1016/j.neuron.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Padilla-Coreano N, Bolkan SS, Pierce GM, Blackman DR, Hardin WD, Garcia-Garcia AL, et al. Direct ventral hippocampal-prefrontal input is required for anxiety-related neural activity and behavior. Neuron. 2016;89:857–66. doi: 10.1016/j.neuron.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Admon R, Leykin D, Lubin G, Engert V, Andrews J, Pruessner J et al. Stress-induced reduction in hippocampal volume and connectivity with the ventromedial prefrontal cortex are related to maladaptive responses to stressful military service. Hum Brain Mapp. (2013);34:2808-16. [DOI] [PMC free article] [PubMed]
- 70.Godsil BP, Kiss JP, Spedding M, Jay TM. The hippocampal-prefrontal pathway: the weak link in psychiatric disorders? Eur Neuropsychopharmacol. 2013;23:1165–81. doi: 10.1016/j.euroneuro.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 71.Sripada RK, Garfinkel SN, Liberzon I. Avoidant symptoms in PTSD predict fear circuit activation during multimodal fear extinction. Front Hum Neurosci. 2013;7:672. doi: 10.3389/fnhum.2013.00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.