

Introduction
Hypokalemia is a common electrolyte abnormality in clinical practice.1 Potassium is predominantly an intracellular cation with the intracellular to extracellular potassium ratio being crucial in maintaining the resting cell membrane potential. Extracellular potassium depletion increases this ratio, resulting in cell membrane hyperpolarization of most cells except Purkinje fibers. Purkinje fibers have paradoxical depolarization from sodium influx through K+ channel isoform 1 channels.2 Chronically low serum potassium levels <3.5 mEq/l have been associated with characteristic histopathological features of nonfatty degeneration of the convoluted tubules, varying from mild cytoplasmic vacuolization to extensive necrosis and sloughing of tubular cells.3, 4 Herein, we present an interesting renal biopsy case of acute on chronic kidney injury from hypokalemic nephropathy, review effects of low K on renal pathophysiology in experimental animal models and patient case series, and discuss association of low K with renal disease progression and mortality.
Case Presentation
A 49-year-old white woman presented to the renal clinic for evaluation of persistent low K. Her medical history is notable for biopsy-proven lymphocytic collagenous colitis treated with oral prednisone for 20 years with intermittent flares, rosuvastatin-associated myopathy, and numerous hospitalizations for myopathy secondary to low K beginning 7 years before her current presentation. Her medications include duloxetine, pregabalin, and trazodone. She denied use of any over-the-counter medications. She had no family history of renal disease or low K. Physical examination revealed a thin but well-developed woman of stated age, with a blood pressure of 123/84 mm Hg, a heart rate of 96 beats/min, and weight of 57 kg. Orthostatic vital signs revealed standing blood pressure of 107/75 mm Hg and heart rate of 94 beats/min. Skin turgor was normal. Her cardiopulmonary examination was benign with regular heart sounds, no murmurs, no lower extremity swelling, and clear lungs. Her abdominal examination demonstrated no evidence of tenderness or masses. She had no joint pain or swelling or skin rash, and her muscle bulk and strength were noted to be normal with appropriate gait and stance. Initial laboratory parameters in comparison to her previous available laboratory test results (Table 1) revealed an increase in serum creatinine levels from 1.7 to 2.1 mg% over 2 weeks. Dipstick urinalysis was notable for a specific gravity of 1.005 and a urine protein level of 19.8–49.6 mg/dl; microscopy showed 1 to 2 red blood cells per high power field and 1 to 3 white blood cells per high power field (present in previous urinalyses) with no cellular casts. A random urine protein to creatinine ratio was found to be elevated (1.2 and 1.5) on 2 occasions, which paralleled an increase in serum creatinine level. The urine potassium to creatinine ratio was 17.3 mEq/g, and the urine potassium level was 2.7 mmol/l. The urine potassium to creatinine ratio is usually <13 mEq/g in low K cases because of the transcellular shift of potassium.5 The higher urine potassium to creatinine ratio makes transcellular shift a less probable cause of her hypokalemia, and the low urine K level supports nonrenal losses of potassium in her stool. Her serum creatinine phosphokinase levels were 81 to 218 U/l (normal range) on multiple occasions when her serum potassium level was <2.5 mmol/l. Differential diagnoses of the progressive loss of renal clearance, female sex, age, proteinuria, and microscopic hematuria included glomerulonephritis, interstitial nephritis, and/or infiltrative diseases. Serological testing including antinuclear antibody, double-stranded DNA, antineutrophil cytoplasmic antibodies, hepatitis B, and hepatitis C was negative, and complement levels were not elevated. Urine protein electrophoresis showed abnormal bands in gamma and beta regions, but serum electrophoresis did not reveal monoclonal bands or spikes. Renal ultrasound revealed slightly hyperechoic cortices of both kidneys (size, 10.5 cm each) with a 7-mm nonobstructing stone found in the mid right kidney. Stone risk analysis revealed hypocitraturia (low citrate excretion, 39 mg/d), low potassium excretion (8 mEq/d), and low magnesium excretion (25 mg/d). The serum magnesium level was 2.3 mg/dl and phosphorus level 3.7 mg/dl (normal range) at the time of renal biopsy.
Table 1.
Chronology of serum laboratory parameters of the case
| Time of the laboratory test (month-year) | BUN level (mg/dl) | SCr level (mg/dl) | K level (mEq/l) |
|---|---|---|---|
| 01-2012 | 12.0 | 1.22 | 3.1 |
| 01-2015 | 19.3 | 1.76 | 3.1 |
| 02-2015 (biopsy) | 19.3 | 2.10 | 2.4 |
| 05-2015 | 12.4 | 1.72 | 3.7 |
BUN, blood urea nitrogen; K, serum potassium; Scr, serum creatinine.
Renal biopsy was done in February 2015 for worsening renal function with proteinuria and abnormal urine protein electrophoresis. Light microscopy revealed 17 of 33 glomeruli to be globally sclerotic. Nonsclerotic glomeruli were normocellular with segmentally increased mesangial matrix and no crescents. Moderate tubular atrophy with 40% cortical interstitial fibrosis and patchy lymphoplasmacytic infiltrates was present. Nonatrophic tubules were dilated with flattened epithelial cells, many of which contained irregular cytoplasmic vacuoles that were periodic acid–Schiff negative. Arterioles demonstrated moderate hyalinosis and intimal fibrosis of interlobular arteries. Immunofluorescence microscopy was negative for deposits. Electron microscopy showed no electron dense deposits and minimal podocyte foot process effacement, and the proximal and distal tubules contained many lysosomal vacuoles with a typical vacuolar membrane and electron lucent contents with or without various lipid materials (Figure 1, Figure 2, Figure 3, Figure 4).
Figure 1.
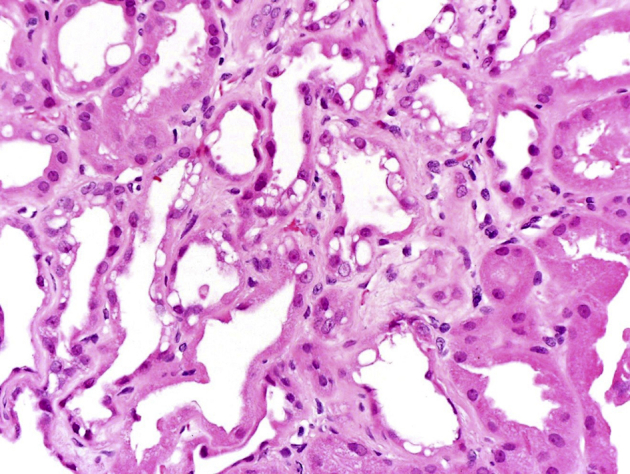
Light microscopy image of a kidney biopsy specimen in a patient with hypokalemia showing extensive vacuolization of the proximal tubules with acute tubular injury. Hematoxylin and eosin, original magnification ×200.
Figure 2.
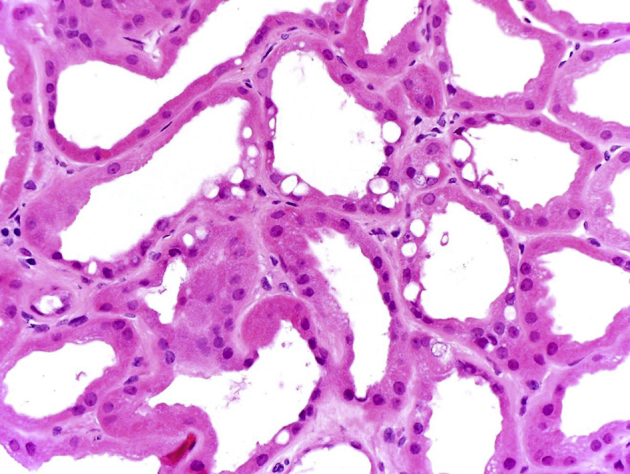
High-magnification image of a kidney biopsy specimen in the same patient showing irregular large intracytoplasmic vacuoles and tubular degeneration. Hematoxylin and eosin, original magnification ×400.
Figure 3.
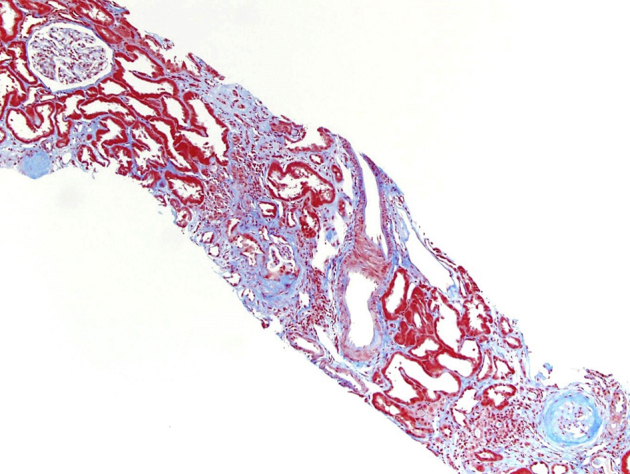
Light microscopy (trichrome-stained) image of a kidney biopsy specimen in the same patient showing sclerotic glomeruli and moderate interstitial fibrosis. Original magnification ×100.
Figure 4.
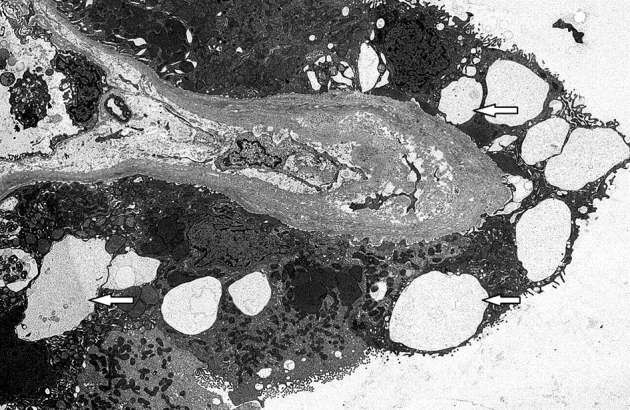
Electron microscopy image of a kidney biopsy specimen in the same patient showing lysosomal vacuoles (arrows) with a typical vacuolar membrane and electron lucent contents in the proximal and distal tubules. Original magnification ×4000.
Clinical Follow-up
The patient was treated with oral potassium chloride 60 mEq twice daily until diarrhea resolved. Evaluation of the patient’s acute diarrhea led to a repeat biopsy of the terminal ileum and colon, reconfirming the presence of lymphocytic collagenous colitis in March 2015. Oral budesonide twice daily (dose range, 3–9 mg) resulted in remission of her colitis and improvement in low K. Sodium bicarbonate was added to correct mild metabolic acidosis during her acute diarrheal flares. The patient achieved clinical remission with a serum potassium level of 4.3 mEq/l, a serum creatinine level of 1.68 mg/dl, and a spot urine protein to creatinine ratio of 0.3. Persistent microscopic hematuria (7 red blood cells per high power field) is presently attributed to her renal lithiasis and negative urologic evaluation.
Discussion
Uncomplicated potassium depletion over 2 to 6 weeks in rats produces a spectrum of lesions in the convoluted and collecting tubular epithelium, including cloudy swelling, extensive vacuolization, fatty and calcific degeneration, thickening and fibrillation of the basement membrane of the thin ascending limb of loop of Henle, necrosis, and atrophy.6 “Vacuolar degeneration” in the kidneys of deceased patients with chronic dysentery, first described in 1919 by Jaffe and Sternberg, was attributed to intestinal losses of unspecified “nutritional substances”.7 Perkins et al. recognized this entity in 1950 in humans.8 Conn and Johnson coined the term “kaliopenic nephropathy” for renal lesions with tubular vacuolization in association with long-standing potassium depletion of various etiologies.9 “Hypokalemic nephropathy” was coined by Cremer and Bock in the late 1970s when they recognized similar histological findings of vacuolar lesions with chronic interstitial fibrosis in hypokalemic rat renal biopsies as well as human renal biopsies and autopsies.10 Human renal biopsies by light microscopy revealed large empty vacuoles that did not stain for fat or glycogen. Approximately two-thirds of the changes were found in the proximal tubules and one-third in the distal tubules. Electron microscopy showed extracellular vacuoles in the tubular basal membrane11 and various sized vesicles in the intracellular space surrounded by the basement membrane. Disappearance of both the vacuolar and the nonspecific degenerative tubular changes after repletion of the potassium deficit in 2 patients led support to the hypothesis that both lesions were in fact related to potassium deficiency.3
Cytoplasmic vacuolar changes in the tubular epithelium can be seen in various conditions such as treatment with hypertonic solutions, acute calcineurin inhibitor toxicity, and sodium ethylenediamine tetraacetate poisoning as described below. The hydropic changes consist of a fine diffuse vacuolar clearing of the cytoplasm of the proximal tubular epithelium in patients treated with hypertonic solutions (sucrose, mannitol, high-molecular-weight dextrans, radiopaque contrast material, or i.v. Ig),12 whereas isometric vacuolization reflects distended lysosomes and dilated endoplasmic reticulum in patients with acute calcineurin inhibitor toxicity.13 Ethylenediamine tetra-acetic acid poisoning shows the cytoplasm of renal tubules filled with fine, small, clear vacuoles best seen at the base of the proximal tubular epithelium with brush border usually well preserved.14 The vacuoles in hypokalemic nephropathy in comparison to these conditions are larger, irregular, and more coarse and do not stain for fat or glycogen.15 Electron microscopy showed extracellular vacuoles in the basal compartment of the tubules11 and variously sized vesicles in the intracellular space.16
On the basis of both animal and human studies, pathogenesis of hypokalemic nephropathy is presumed to be due to renal vasoconstriction, reduced medullary blood flow, and impaired renal angiogenesis. There is evidence of progressive capillary loss, reduced endothelial cell proliferation, and loss of vascular endothelial growth factor expression.17 In addition, there is increased renal ammonia genesis18 activating the alternative complement pathway and increased profibrotic cytokine expression (insulin-like growth factor-1 and transforming growth factor). Animal studies in hypokalemic rats suggest an imbalance of intrarenal vasoactive mediators from increased angiotensin-converting enzyme expression by interstitial cells at the site of damage and by the proximal tubule brush border. This leads to increased levels of intrarenal and cortical angiotensin II as well as endothelin 1 and decreased levels of urinary nitrites and kallikrein.19
Functional effects of low K on the kidney include a vasopressin-resistant urinary concentrating defect mediated by rapid reversible aquaporin 2 channel defects in the collecting duct. Decreased numbers of apical Na-K-Cl cotransporters and renal outer medullary potassium channels in the thick ascending loop of Henle leads to decreased sodium reabsorption and abnormalities in the countercurrent mechanism.20 The most common urinary abnormality is a low fixed urine specific gravity (1.003–1.005). This was evident in our patient who presented with a persistent low specific gravity of 1.005 despite ongoing diarrhea. Sodium chloride retention can occur from upregulation of angiotensin II, angiotensin 1 receptor, and sodium-hydrogen exchanger 3 channels in the proximal tubule.21 Our patient did not have evidence for salt and water retention on physical examination. Low K can also lead to hypocitraturia by causing intracellular acidosis and a decrease in tubular pH, both of which lead to increased citrate uptake and metabolism by proximal tubular cells,22 thus increasing risk of stone formation as found in our patient. Persistent tubular proteinuria in the form of minimal albuminuria is another clinical feature of low K. Urinalysis can reveal low-grade pyuria and microscopic hematuria, with active urine sediment being uncommon. Low K has also been associated with hypophosphatemia and/or increased urinary phosphate excretion. In rat studies, low K causes inhibition of Na/Pi cotransport activity by posttranslational mechanisms including alterations in glucosylceramide content and membrane lipid dynamics.23 Patients with prolonged low K can present with decreased renal clearance from chronic interstitial fibrosis and vascular changes as found in our case.
A PubMed literature review was performed using terms hypokalemia and nephropathy that produced 19 articles on adults over the past 60 years. Cases summarized in Table 224, 25, 26, 27, 28, 29, 30, 31, 32 exclude 5 non-English language articles that could not be accessed. Reported cases present with various diagnoses including diarrheal diseases (ulcerative colitis, regional enteritis, cholera, bacillary dysentery, sprue syndrome, Whipple disease, malabsorption syndrome, and laxative abuse), hyperadrenalism (primary aldosteronism and cortisone treatment), renal disease with renal potassium wasting (Fanconi syndrome, renal tubular acidosis, cystinosis, and diuretic abuse), and anorexia nervosa. Common clinical findings include polyuria, low urine specific gravity, and tubular proteinuria with inactive urinary sediment like the findings in our patient. The duration of low K levels ranged from 18 days to 16 years, affecting men and women equally; ages ranged from 18 to 68 years; serum potassium levels ranged from 0.5 to 3.2 mEq/l in noncholera cases, but from 3.6 to 5.6 mEq/l in cholera cases with oligoanuric acute kidney injury. It is interesting to note that vacuolar changes were present despite normal serum potassium levels in patients with cholera, raising the possibility that total body potassium depletion and intracellular K depletion are probably more important than serum potassium depletion in causing these vacuolar lesions.
Table 2.
Summary of hypokalemic nephropathy cases found in the literature search
| Study | Age (yr) | M/F | K level (mEq/l) | Etiology | Polyuria | Albuminuria | Maximum SG | Renal function: BUN/Cr/CrCl | Kidney abnormality |
|---|---|---|---|---|---|---|---|---|---|
| Watson and Katz25 | 44 | F | 1.9–2.3 | Excessive 9α-fluorohydrocortisone to adrenalectomized patients, causing hypokalemia for 1 wk | + | − | 1.010 | BUN 16 mg% | ND |
| Benyajati et al.26 | 33 | M | 3.6–5.6 | Acute diarrhea due to cholera for 7 h | − | ND | ND | BUN 46–264 mg% | Autopsy: “fine to occasionally large vacuolization in the cytoplasm” and severe ischemic ATN |
| 24 | M | 4.1–7.1 | Acute cholera diarrhea for 10 h | Anuric | ND | ND | BUN 42–133 mg% | No vacuolization and ischemic ATN | |
| 56 | M | 3.7–4.3 | Acute cholera diarrhea for 24 h | Anuric | ND | ND | BUN 57–200 mg% | Autopsy: “severe vacuolar degeneration most marked in the convoluted tubular cells” and mild ATN | |
| 53 | M | ND | Acute cholera diarrhea for 6 h and opium addiction for 30 yr | Anuric | + | ND | BUN 50–100 mg% | Autopsy: “severe degree of vacuolar degeneration” | |
| 18 | M | Normal | Cholera diarrhea for 10 h | Anuric | + | ND | BUN 44–266 mg% | ND | |
| Galloway et al.27 | 57 | M | 2.2 | Diarrhea: tropical sprue for 3 wk | ND | + | 1.015 | BUN 23 mg% | ND |
| 41 | F | 0.5 | Vomiting for 3 wk | ND | + | Not obtained | BUN 66 mg% | Autopsy: “tubular vacuolization” | |
| 30 | F | 3.1 | Vomiting for 6 wk, struma ovarii | ND | + | 1.010 | BUN 104 mg% | Autopsy: “diffuse degeneration of the tubules with swelling” | |
| Cameron et al.28 | 54 | F | 2–2.3 | Diarrhea for 13 yr due to functioning ganglioneuroblastoma | ND | ND | 1.004–1.022 | CrCl 66 l/d | ND |
| Wagner et al.29 | 38 | F | 2–3.3 | Abuse of diuretics for 10 yr | ND | ND | 1.010–1.014 | Cr 1.7–2.0 mg% | “Focal vacuolization of the proximal tubular epithelium” |
| Buridi et al.30 | 53 | F | 1.3 | Chronic osmotic diarrhea due to the consumption of high fructose corn syrup drink (Big Red, Louisville, KY) | + | ND | 1.005 | Cr 13 mg% | Biopsy not done |
| Ishikawa et al.31 | 29 | F | 1.7 | Due to anorexia nervosa for 10 yr, purging and licorice ingestion (30 tablets/d for 4 mo) | + | ND | 1.005 | Cr 1.5 mg% | “Severely damaged tubular cells with intense vacuolar formations” |
| Yasuhara et al.32 | 26 | F | 2 | 8-y h/o binge/purge anorexia nervosa | ND | ND | ND | BUN 10.3 mg% Cr 0.9 mg% CrCl 58 l/d |
Initial biopsy: “juxtaglomerular hyperplasia” Autopsy: “interstitial nephritis and proximal tubular swelling” |
| Bock et al.24 | 19–68 | 4 M, 17 F | 2.0–3.2 | 2–16 yr h/o hypokalemia Anorexia nervosa with drug abuse (laxatives/diuretics) or 1°/2° aldosteronism |
ND | Yes | ND | CrCl <90 ml/min per 1.73 m2 in 14 of 21 patients | “Hydropic degeneration, atrophy, destruction, and regeneration of tubular cells most striking in the proximal tubule” |
+, present; −, absent; 1°, primary; 2°, secondary; ATN, acute tubular necrosis; BUN, blood urea nitrogen; Cr, creatinine; CrCl, creatinine clearance; F, female; h/o, history of; K, serum potassium; M, male; ND, no data; SCr, serum creatinine; SG, specific gravity.
In a case series of 21 patients by Cremer and Bock, 13 of whom had renal biopsies, intraglomerular lesions of mesangial hyalinosis and mesangial sclerosis along with basement membrane thickening with arteriosclerosis were observed irrespective of an underlying history of hypertension.10 This is like biopsy findings in our patient. Upon carefully looking at biopsy differences in a different case series of 14 patients by Bock et al., 7 patients with malnutrition and/or abuse of laxatives and/or diuretics (group A) compared with 7 patients with primary or secondary aldosteronism (group B), a hyperplastic juxtaglomerular apparatus was seen in those in group A; however, a hypoplastic juxtaglomerular apparatus was found in those in group B. Biopsies in both groups A and B showed diffuse chronic interstitial nephritis.24 Most renal biopsies in a study by Riemenschneider and Bohle (mean duration of low K levels, 10 years) showed fibrosis and lymphocytic infiltration in the renal interstitium as seen in our patient.15
Because low K can lead to pathologic abnormalities in the renal tubular cells and fibrosis of the renal interstitium, particularly with the presence of chronically low K levels, the possibility of chronic kidney disease (CKD) progression in patients with low normal or intermittent low K levels can be entertained. Five observational studies have evaluated the effects of potassium on mortality and end-stage renal disease (ESRD).33, 34, 35, 36, 37 A prospective observational study in 2010 of 820 patients with CKD followed for a mean duration of 2.6 years showed a hazard ratio (HR) for mortality of 2.40 for a serum K level of 3.4 mEq/l, 1.65 for a serum K level of 3.8 mEq/l, 1.34 for a serum K level of 6 mEq/l in comparison to the reference serum K level of 5.0 mEq/l. This study also showed for the first time that a serum K level of ≤4.0 mmol/l was significantly associated with an increased risk of ESRD with an HR of 1.87, which reduced to 1.56 after adjustment for albumin.33 A subsequent retrospective study in 2012 of 1227 male patients with CKD also showed that low K was associated with higher mortality and faster CKD progression independent of race (a lower potassium level of 1 mEq/l was associated with an adjusted difference in slopes of an estimated glomerular filtration rate of −0.13 ml/min per 1.73 m2 per yr).34 A retrospective evaluation of 36,359 patients with CKD in 2015 confirmed the U-shaped mortality curve with an HR of 1.95 for a serum K level of <3.5 mEq/l but refuted the association of low K with ESRD (HR, 0.97).35 However, a larger 2016 retrospective study of 55,266 patients with CKD reconfirmed higher rates of death (HR, 1.06 for a serum K level of 3.5–3.9 mEq/l; HR, 3.05 for a serum K level of <3.5 mEq/l), major adverse cardiovascular events (HR, 1.89 with a serum K level of <3.5 mEq/l; HR, 1.27 with a serum K level of 3.5–3.9 mEq/l), and hospitalization associated with low K.36 A more recent retrospective study of 9651 community dwellers from the Multi-Ethnic Study of Atherosclerosis and the Cardiovascular Health Study with a median follow-up duration of 10.5 years showed a J-shaped curve for outcomes of mortality, cardiovascular disease death, non–cardiovascular disease death, and serum K. The group of patients with a serum K level of <3.5 mEq/l had a nonsignificant HR of 1.13 for mortality.37 Taken together, mild low K (a serum K level of ≤4) is associated with higher mortality risk in CKD stages 3 to 5 but not in CKD stages 1 and 2, although specific reasons are yet to be investigated. The risk of ESRD progression with low K can be debated, given discrepant findings from a prospective observational study and a large retrospective observational study.35, 37
In summary, hypokalemic nephropathy is a potential complication of subacute and chronic low K (Table 3). Biopsy findings include intracytoplasmic vacuoles in tubular cells, chronic inflammation, and interstitial fibrosis. Pathogenesis is mediated via an imbalance in vasoactive mediators, leading to vasoconstriction and medullary ischemia. Neither the serum potassium level nor the duration of low K level that leads to hypokalemic nephropathy is clearly known. Common clinical findings include low urine specific gravity, polyuria, tubular proteinuria, and inactive urinary sediment. Although the risk of CKD and ESRD with untreated hypokalemic nephropathy seems logical, it has yet to be unequivocally confirmed. Given that large numbers of patients are treated with diuretics for various disease entities with the risk of subacute and chronic low K levels, future prospective studies are needed to identify the risk of low K and CKD progression and to identify the effect of timely treatment of low K on the reversal of renal damage.
Table 3.
Teaching points
|
|
|
|
|
Disclosure
All the authors declared no competing interests. We would like to acknowledge Ms. Jennifer Blankenship, the librarian, for her support in literature search. This case report is the result of work supported with resources and the use of facilities at the Salem VA Medical Center, Salem, VA. It is unfunded. The contents do not represent the views of the US Department of Veterans Affairs or the United States Government.
Contributor Information
Hima Bindu Yalamanchili, Email: Himabindu.yalamanchili@va.gov.
Devasmita Choudhury, Email: devasmita.dev@va.gov.
References
- 1.Eliacik E., Yildirim T., Sahin U. Potassium abnormalities in current clinical practice: frequency, causes, severity and management. Med Princ Pract. 2015;24:271–275. doi: 10.1159/000376580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Christe G. Effects of low [K+]o on the electrical activity of human cardiac ventricular and Purkinje cells. Cardiovasc Res. 1983;17:243–250. doi: 10.1093/cvr/17.4.243. [DOI] [PubMed] [Google Scholar]
- 3.Relman A.S., Schwartz W.B. The nephropathy of potassium depletion: a clinical and pathological entity. N Engl J Med. 1956;255:195–203. doi: 10.1056/NEJM195608022550501. [DOI] [PubMed] [Google Scholar]
- 4.Kulka J.P., Pearson C.M., Robins S.L. A distinctive vacuolar nephropathy associated with intestinal disease. Am J Pathol. 1950;26:349–377. [PMC free article] [PubMed] [Google Scholar]
- 5.Lin S.H., Lin Y.F., Chen D.T. Laboratory tests to determine the cause of hypokalemia and paralysis. Arch Intern Med. 2004;164:1561–1566. doi: 10.1001/archinte.164.14.1561. [DOI] [PubMed] [Google Scholar]
- 6.MacDonald M.K., Sabour M.S., Lambie A.T., Robson J.S. The nephropathy of experimental potassium deficiency: an electron microscopic study. Q J Exp Physiol Cogn Med Sci. 1962;47:262–272. doi: 10.1113/expphysiol.1962.sp001606. [DOI] [PubMed] [Google Scholar]
- 7.Jaffé R.H., Sternberg H. Über die vakuoläre Nierendegeneration bei chronischer Ruhr [About Vakuolare kidney degeneration in the case of chronic dysentery] Virchows Arch Path Anat. 1920;227:313–319. [Google Scholar]
- 8.Perkins J.G., Petersen A.B., Riley J.A. Renal and cardiac lesions in potassium deficiency due to chronic diarrhea. Am J Med. 1950;8:115–123. doi: 10.1016/0002-9343(50)90348-2. [DOI] [PubMed] [Google Scholar]
- 9.Conn J.W., Johnson R.D. Kaliopenic nephropathy. Am J Clin Nutr. 1956;4:523. doi: 10.1093/ajcn/4.5.523. [DOI] [PubMed] [Google Scholar]
- 10.Cremer W., Bock K.D. Symptoms and course of chronic hypokalemic nephropathy in man. Clin Nephrol. 1977;7:112–119. [PubMed] [Google Scholar]
- 11.Biava C.G., Dyrda I., Genest I., Bencosme S.A. Kaliopenic nephropathy: a correlated light and electron microscopic study. Lab Invest. 1963;12:443–453. [PubMed] [Google Scholar]
- 12.Dickenmann M., Oettl T., Mihatsch M.J. Osmotic nephrosis: acute kidney injury with accumulation of proximal tubular lysosomes due to administration of exogenous solutes. Am J Kidney Dis. 2008;51:491–503. doi: 10.1053/j.ajkd.2007.10.044. [DOI] [PubMed] [Google Scholar]
- 13.Liptak P., Ivanyi B. Primer: histopathology of calcineurin-inhibitor toxicity in renal allografts. Nat Clin Pract Nephrol. 2006;2:398–404. doi: 10.1038/ncpneph0225. [DOI] [PubMed] [Google Scholar]
- 14.Dudley H.R., Ritchie A.C., Schilling A., Baker W.H. Pathologic changes associated with the use of sodium ethylene diamine tetra-acetate in the treatment of hypercalcemia: report of two cases with autopsy findings. N Engl J Med. 1955;252:331–337. doi: 10.1056/NEJM195503032520901. [DOI] [PubMed] [Google Scholar]
- 15.Riemenschneider T., Bohle A. Morphologic aspects of low-potassium and low-sodium nephropathy. Clin Nephrol. 1983;19:271–279. [PubMed] [Google Scholar]
- 16.Cremer W., Blumcke S. Zur Morphologie der kaliopenischen Nephropathie beim Menschen [To morphology of the kaliopenic nephropathy in people] Nieren u Hochdruckkrankh. 1977;2:71–78. [Google Scholar]
- 17.Reungjui S., Roncal C.A., Sato W. Hypokalemic nephropathy is associated with impaired angiogenesis. J Am Soc Nephrol. 2008;19:125–134. doi: 10.1681/ASN.2007030261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tizianello A., Garibotto G., Robaudo C. Renal ammoniagenesis in humans with chronic potassium depletion. Kidney Int. 1991;40:772–778. doi: 10.1038/ki.1991.274. [DOI] [PubMed] [Google Scholar]
- 19.Suga S., Yasui N., Yoshihara F. Endothelin A receptor blockade and endothelin B receptor blockade improve hypokalemic nephropathy by different mechanisms. J Am Soc Nephrol. 2003;14:397–406. doi: 10.1097/01.asn.0000046062.85721.ac. [DOI] [PubMed] [Google Scholar]
- 20.Khositseth S., Uawithya P., Somparn P. Autophagic degradation of aquaporin-2 is an early event in hypokalemia-induced nephrogenic diabetes insipidus. Sci Rep. 2015;5:18311. doi: 10.1038/srep18311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suga S., Phillips M.I., Ray P.E. Hypokalemia induces renal injury and alterations in vasoactive mediators that favor salt sensitivity. Am J Physiol Renal Physiol. 2001;281:F620–F629. doi: 10.1152/ajprenal.2001.281.4.F620. [DOI] [PubMed] [Google Scholar]
- 22.Zuckerman J.M., Assimos D.G. Hypocitraturia: pathophysiology and medical management. Rev Urol. 2009;11:134–144. [PMC free article] [PubMed] [Google Scholar]
- 23.Zajicek H.K., Wang H., Puttaparthi K. Glycosphingolipids modulate renal phosphate transport in potassium deficiency. Kidney Int. 2001;60:694–704. doi: 10.1046/j.1523-1755.2001.060002694.x. [DOI] [PubMed] [Google Scholar]
- 24.Bock K.D., Cremer W., Werner U. Chronic hypokalemic nephropathy: a clinical study. Klin Wochenschr. 1978;56(Suppl 1):91–96. doi: 10.1007/BF01477459. [DOI] [PubMed] [Google Scholar]
- 25.Watson J.F., Katz F.H. Hypokalemic nephropathy in an adrenalectomized patient. Am J Med. 1959;27:844–848. doi: 10.1016/0002-9343(59)90203-7. [DOI] [PubMed] [Google Scholar]
- 26.Benyajati C., Keoplug M., Beisel W. Acute renal failure in Asiatic cholera: clinicopathologic correlations with acute tubular necrosis and hypokalemic nephropathy. Ann Intern Med. 1960;52:960–975. doi: 10.7326/0003-4819-52-5-960. [DOI] [PubMed] [Google Scholar]
- 27.Galloway N.C., Patterson R.M., Thorpe Ray C. Hypokalemic nephropathy of vomiting. South Med J. 1964;57:1303–1308. doi: 10.1097/00007611-196411000-00009. [DOI] [PubMed] [Google Scholar]
- 28.Cameron D.G., Warner H.A., Szabo A.J. Chronic diarrhea in an adult with hypokalemic nephropathy and osteomalacia due to a functioning ganglioneuroblastoma. Trans Am Clin Climatol Assoc. 1967;78:205–217. [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner P.K., Pippig L., Thoenes W. [Histopathology of the kidney in pseudo-Bartter’s syndrome induced by chronic abuse of diuretics (author’s transl)] Klin Wochenschr. 1979;57:135–142. doi: 10.1007/BF01476054. [in German] [DOI] [PubMed] [Google Scholar]
- 30.Buridi A., Corman L., Redinger R. Hypokalemic nephropathy and nephrogenic diabetes insipidus due to excessive consumption of a soft drink. South Med J. 1998;91:1079–1082. doi: 10.1097/00007611-199811000-00021. [DOI] [PubMed] [Google Scholar]
- 31.Ishikawa S., Kato M., Tokuda T. Licorice-induced hypokalemic myopathy and hypokalemic renal tubular damage in anorexia nervosa. Int J Eat Disord. 1999;26:111–114. doi: 10.1002/(sici)1098-108x(199907)26:1<111::aid-eat16>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 32.Yasuhara D., Naruo T., Taguchi S. “End-stage kidney” in longstanding bulimia nervosa. Int J Eat Disord. 2005;38:383–385. doi: 10.1002/eat.20198. [DOI] [PubMed] [Google Scholar]
- 33.Korgaonkar S., Tilea A., Gillespie B.W. Serum potassium and outcomes in CKD: insights from the RRI-CKD cohort study. Clin J Am Soc Nephrol. 2010;5:762–769. doi: 10.2215/CJN.05850809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hayes J., Kalantar-Zadeh K., Lu J.L. Association of hypo- and hyperkalemia with disease progression and mortality in males with chronic kidney disease: the role of race. Nephron Clin Pract. 2012;120:c8–c16. doi: 10.1159/000329511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakhoul G.N., Huang H., Arrigain S. Serum potassium, end-stage renal disease and mortality in chronic kidney disease. Am J Nephrol. 2015;41:456–463. doi: 10.1159/000437151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luo J., Brunelli S., Jensen D.E., Yang A. Association between serum potassium and outcomes in patients with reduced kidney function. Clin J Am Soc Nephrol. 2016;11:90–100. doi: 10.2215/CJN.01730215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hughes-Austin J.M., Rifkin D., Beben T. The relation of serum potassium concentration with cardiovascular events and mortality in community-living individuals. Clin J Am Soc Nephrol. 2017;12:245–252. doi: 10.2215/CJN.06290616. [DOI] [PMC free article] [PubMed] [Google Scholar]