

Abstract
Introduction
There is no proven therapy to reverse or ameliorate fetal growth restriction (FGR). Sildenafil, a selective phosphodiesterase 5 (PDE5) inhibitor, has been reported to potentially play a therapeutic role in FGR, but this has not been established. Tadalafil is also a selective PDE5 inhibitor. We have demonstrated the efficacy of tadalafil against FGR along with short-term outcomes and the feasibility of tadalafil treatment. Based on the hypothesis that tadalafil will safely increase the likelihood of increased fetal growth in FGR, we designed this phase II study to prospectively evaluate the efficacy and safety of tadalafil against FGR.
Methods and analysis
This study is a multicentre, randomised controlled phase II trial. A total of 140 fetuses with FGR will be enrolled from medical centres in Japan. Fetuses will be randomised to receive either the conventional management for FGR or a once-daily treatment with 20 mg of tadalafil along with the conventional management until delivery. The primary endpoint is fetal growth velocity from the first day of the protocol-defined treatment to birth (g/day). To minimise bias in terms of fetal baseline conditions and timing of delivery, a fetal indication for delivery was established in this study. The investigator will evaluate fetal baseline conditions at enrolment and will decide the timing of delivery based on this fetal indication. Infants will be followed up for development until 1.5 years of age.
Ethics and dissemination
This study was approved by the Institutional Review Board of Mie University Hospital and each participating institution. Our findings will be widely disseminated through peer-reviewed publications.
Trial registration number
UMIN000023778.
Keywords: fetal growth restriction, phosphodiesterase 5 inhibitor, study protocol, tadalafil
Strengths and limitations of this study.
This is a multicentre, randomised controlled phase II trial to prospectively evaluate the efficacy and safety of tadalafil treatment in fetuses with fetal growth restriction (FGR), for which there is no proven therapy.
Participants include major medical centres providing treatment for fetuses with FGR according to the guidelines for obstetrical practice in Japan.
To minimise bias in terms of fetal baseline conditions and timing of delivery, a fetal indication for delivery was established in this study on the basis of the results from a multicentre survey in Japan.
The possible limitation is related to open-label trial features, in which enrolled participants receive either the conventional management for FGR according to the guidelines for obstetrical practice in Japan or a once-daily treatment with 20 mg of tadalafil in addition to the conventional management.
It is possible that small for gestational age (SGA)was included among the cases of FGR without Doppler abnormalities in this study.
Introduction
Fetal growth restriction (FGR) is a common complication of pregnancy that is associated with a variety of adverse perinatal outcomes. Although the main indication when treating FGR is to consider the appropriate termination of the pregnancy, fetal prematurity depending on gestational age (GA) is a serious problem, as shown by the multicentre survey of very low birthweight (VLBW) infants in Japan conducted by Kusuda et al.1 There is also no proven fetal therapy to reverse or ameliorate established FGR.2 To prevent FGR, nutritional and dietary supplementation, bed rest, and aspirin therapy have been investigated, but there is insufficient evidence for the routine indication of any of these treatments.3
Fetal growth is promoted by an adequate increase of uteroplacental perfusion. Vasodilation in the uteroplacental unit is probably due to the production and local release of nitric oxide (NO), which stimulates cyclic guanosine monophosphate (cGMP) production.4 cGMP is inactivated mainly by phosphodiesterase (PDE), and PDE5 exists mainly in vascular smooth muscle cells.
It is now expected that PDE inhibitors could become therapeutic agents for FGR in light of the inhibitors’ artery dilation function, as confirmed in studies of erectile dysfunction and pulmonary hypertension.5 Sildenafil, a selective PDE5 inhibitor, has been shown to improve endothelial function in myometrial small arteries removed from women with pre-eclampsia and FGR.6 7 Although some reports have described an effect of sildenafil on maternal hypertension, the effectiveness of sildenafil for rats with FGR induced by L-NG-nitroarginine methyl ester (L-NAME) was not shown except in Baijnath et al.8–11 They reported that sildenafil improved the fetal growth of FGR induced by L-NAME.10 Their study showed the improvement of growth from 4 days postcoitum (dpc) to 8 dpc, but not from 8 dpc to 14 dpc. In mouse placenta, chorioallantoic attachment occurs at 8 dpc, and the mature circulatory pattern of maternal blood through the placenta is established by 10 dpc.12 13 In a clinical study, it was reported that sildenafil was associated with increased fetal abdominal circumference growth velocity in severe early-onset FGR, but the authors did not describe the fetal growth velocity or birth weight.14 The Sildenafil for Severe Fetal Growth Restriction (STRIDER) UK group recently reported obtaining no evidence of a beneficial effect of sildenafil on survival or short-term neonatal outcomes.15
Tadalafil is another selective PDE5 inhibitor with a longer half-life and a more rapid onset of action compared with sildenafil.5 There are several reports showing the safety of tadalafil treatment for pregnant women.16–18 Regarding the plasma concentration and the bioavailability of the drug, tadalafil is less susceptible to the intake of a high-fat meal and less influenced by sex compared with sildenafil.19 20 Our animal experiments demonstrated that tadalafil treatment dilates the maternal blood sinuses in the placenta, which leads to increased placental growth factor (PlGF) production and contributes to the facilitation of fetal growth.21 In those animal experiments, we can safely presume that tadalafil treatment contributed to the facilitation of fetal growth via mechanisms associated with NO signalling, because the tadalafil treatment was initiated after blood spaces in the placenta were narrowed by the L-NAME treatment and an elevated urinary excretion of cGMP was observed.
We also retrospectively analysed the cases of 11 Japanese singleton pregnant women showing FGR who received tadalafil along with conventional management for FGR at Mie University Hospital from July 2015 to February 2016 (the tadalafil group).22 The women were matched for maternal age, parity and GA, and we also estimated fetal weight at enrolment among 14 singleton pregnant women who had received only the conventional management for FGR in 2014 (the conventional management group). The conventional management for FGR was performed according to the guidelines for obstetric practice in Japan.23 This retrospective study showed that both fetal growth velocity from enrolment to birth and birth weight were significantly higher in the tadalafil group than in the conventional management group. Moreover, the prevalence of respiratory distress syndrome (RDS) was significantly lower in the tadalafil group compared with the conventional management group.
As the next step, a phase I trial was conducted to confirm the safety of tadalafil administration for FGR.24 No severe adverse event was seen following the initiation of a daily tadalafil dose of 10 mg, 20 mg or 40 mg except for one intrauterine fetal death case. That case was immediately reviewed by the Safety Evaluation Committee, which concluded that the intrauterine fetal death was due to velamentous insertion of the umbilical cord.25 We concluded that tadalafil treatment was feasible in pregnant women showing FGR.24
Based on the above data, we hypothesised that tadalafil therapy can safely increase the likelihood of increased fetal growth in fetuses with FGR, and we designed the present multicentre, randomised controlled phase II study to further examine the efficacy and safety of fetal therapy with tadalafil (A multicenter phase II trial of the efficacy and safety of tadalafil in fetus with early-onset growth restriction: TADAFER II). This study will prospectively evaluate the safety and efficacy of tadalafil against FGR with the participation of major medical centres providing treatment for fetuses with FGR according to the guidelines for obstetrical practice in Japan.
Methods
Study design
This study is a multicentre, randomised controlled phase II trial.
Study period
The planned study period is from the date of ethics approval to February 2021. The patient registration period will last until December 2018 starting from the date of ethics approval. The data collection and follow-up will be performed until 1.5 years after birth of the children registered under this study. Data collected by the end of the neonatal evaluation period will be subjected to statistical analysis.
Patient selection
The inclusion criteria are as follows: (1) pregnant women ≥20 years; (2) estimated fetal weight (EFW) should be less than 1.5 SD of the mean EFW for GA; (3) GA should be between 20+0 and 33+6 weeks; (4) the expected date of confinement is determined using the criteria in the guidelines for obstetrical practice in Japan (2014); (5) only singleton pregnant patients should be selected; and (6) signed written informed consent should be obtained from the patients.
The exclusion criteria are as follows: (1) a result from the antepartum fetal tests, done at enrolment, which indicates that delivery should be attempted*; (2) history of allergy to tadalafil; (3) concurrent medications that interact adversely with tadalafil; (4) contraindication of tadalafil treatment due to renal disease; (5) contraindication of tadalafil treatment due to liver disease; (6) contraindication of tadalafil treatment due to uncontrolled arrhythmia, hypertension (blood pressure (BP) >170/100 mm Hg) and hypotension (BP <80/40 mm Hg); (7) fetus with suspected chromosomal disorder and/or multiple congenital anomalies; (8) contraindication of tadalafil treatment due to retinitis pigmentosa, coagulation defect, active gastric and/or intestinal ulcer, or venous obstructive disease; and (9) investigators’ decision that the entry is inappropriate**.
*To minimise bias in terms of fetal baseline condition at enrolment, a fetal indication for delivery was established on the basis of the results from the multicentre survey of VLBW infants in Japan using a network database, in which the 82 level III perinatal centres were registered. The survey data included infant survival rate in the neonatal intensive care unit (NICU), categorised by birth weight and gestational week at birth (figure 1).1 The infant survival rate data acquired from the survey were preprocessed with the moving average method and divided into three groups. The first group was defined as ‘Zone 1’, where the infant survival rate in the NICU was less than 60%. The second group was defined as ‘Zone 2’, where the infant survival rate in the NICU ranged from 60% to 95%. The third group was defined as ‘Zone 3’, where the infant survival rate in the NICU was 95% or higher. All patients in our study will undergo antepartum fetal tests consisting of the evaluation of fetal well-being by ultrasonography, including Doppler imaging of umbilical arterial blood flow, non-stress test, contraction stress test and biophysical profile scoring depending on GA, to evaluate possible pregnancy termination by the investigator at enrolment (table 1).23 25 26
Figure 1.

Infant survival rate in the neonatal intensive care unit (NICU) categorised by birth weight and gestational week at birth (%). This figure is established on the basis of the results from the multicentre survey of very low birthweight infants in Japan using a network database. The survey data included infant survival rates in the NICU, categorised by birth weight and gestational week at birth.1 The infant survival rate data acquired from the survey were preprocessed with the moving average method and divided into three groups. The first group was defined as ‘Zone 1’, where the infant survival rate in the NICU was less than 60% (highlighted by the red background). The second group was defined as ‘Zone 2’, where the infant survival rate in the NICU ranged from 60% to 95% (highlighted by the yellow background). The third group was defined as ‘Zone 3’, where the infant survival rate in the NICU was 95% or higher (highlighted by the blue background).
Table 1.
A fetal indication for delivery in the tadalafil for fetus with early-onset growth restriction (TADAFER) II study23 25 26
| Infant survival rate in the neonatal intensive care unit (NICU) (see figure 1) | |
| Zone 1 | Decide timing of delivery depending on available therapeutic measures at the NICU in each institute. |
| Zone 2 | Consider delivery if at least one of three findings is made, but place a high priority on the determination by the investigators.
|
| Zone 3 | Consider delivery if at least one of the following five findings is made, but place a high priority on the determination by the investigators.
|
**Regarding exclusion criterion 9 (the investigator decides that entry is inappropriate), this study excludes mothers with mental or psychiatric problems, since poor judgement capabilities that are often associated with such conditions may not be compatible with inclusion criterion 6.
Registration
The study protocol defines all the procedures and schedules that the investigator must abide by to complete this clinical study, including patient selection and registration, fetal treatment of FGR, and follow-up (figure 2). Patients who satisfy all inclusion criteria and do not meet any of the exclusion criteria will be eligible for inclusion in the study. Individual study sites will be responsible for guiding potential participants through the informed consent process, including patients who have been referred to them for treatment purposes. The investigator will enter an eligible patient’s information into the eligibility confirmation form on the website of this clinical trial (the Clinical Trial Data Management System: Japanese-only website). The data management system will check the content of the form before registering the patient. For patients who meet all inclusion criteria without violating any of the exclusion criteria listed above, the data management system will register and allocate them to the two arms in an allocation ratio of 1:1, one group receiving the conventional management of FGR according to the guidelines for obstetrical practice in Japan23 and the other receiving once-daily treatment with 20 mg tadalafil added to conventional management after adjustment for study sites and GA (<28 or ≥28 weeks of gestation). The investigators are blinded to the allocation algorithm. Enrolled participants will receive fetal therapy within 7 days of registration. The investigator will enter patients’ data into the case report form on the website of this clinical trial (the Clinical Trial Data Management System).
Figure 2.
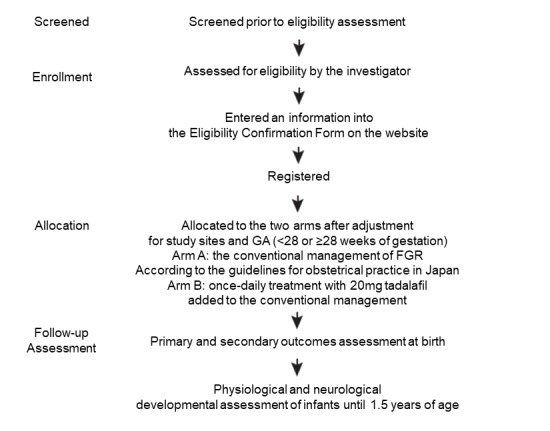
Summary of the study design. FGR, fetal growth restriction; GA, gestational age.
Fetal treatment protocol
The investigator will provide the fetal therapy as described below.
Arm A
The conventional management of FGR according to the guidelines for obstetrical practice in Japan will be followed.23 Briefly, the conventional management of FGR consists of evaluation of fetal well-being on ultrasonography, including Doppler imaging of umbilical arterial blood flow, non-stress test, contraction stress test and biophysical profile scoring depending on GA to evaluate possible pregnancy termination.
Arm B
Patients will be provided a once-daily treatment with 20 mg tadalafil added to the conventional management until delivery.
The investigators are blinded to the allocation algorithm. Enrolled participants will receive fetal therapy within 7 days of registration.
Endpoints
Primary endpoint
The primary endpoint is fetal growth velocity from the first day of the protocol-defined treatment to birth (g/day), and is calculated using the following formula:
Rationale for the primary endpoint
Our primary hypothesis is that tadalafil therapy will increase the likelihood of increased fetal growth velocity in fetuses with FGR. Taking into account the results of our retrospective study demonstrating that tadalafil treatment increased fetal growth velocity (g/day), which was the primary outcome of the retrospective study, and decreased the incidence of RDS, an improvement in fetal growth velocity from the first day of the protocol-defined treatment to birth (g/day) is an important indicator of the therapeutic benefits for fetuses with FGR.22 The cases of fetal death were included in the analysis of primary endpoint.
Secondary endpoints
Completion rate of the treatment regimen
Completion rate of the treatment regimen is defined as the percentage of enrolled patients who receive the protocol-defined treatment for more than 7 days.
Efficacy endpoints
1. Estimated fetal weight (g).
Estimated fetal weight (EFW) is calculated using the following formula27:
2. Fetal growth velocity in the 2 weeks after the protocol-defined treatment and in the 2 weeks after 1 week of the protocol-defined treatment (g/day).
Fetal growth velocity in the 2 weeks after the protocol-defined treatment (g/day) is calculated using the following formula:
Fetal growth velocity in the 2 weeks after 1 week of the protocol-defined treatment (g/day) is calculated using the following formula:
3. Fetal growth rate in the 2 weeks after the protocol-defined treatment and from the first day of the protocol-defined treatment to birth (%/day).
Fetal growth rate in the 2 weeks after the protocol-defined treatment (%/day) is calculated using the following formula:
Fetal growth rate in the 2 weeks after the protocol-defined treatment (%/day)
Fetal growth rate from the first day of the protocol-defined treatment to birth (%/day) is calculated using the following formula:
Fetal growth rate from the first day of the protocol-defined treatment to birth (%/day)
4. Fetal head circumference (cm).
The fetal head circumference was measured at the plane of the third ventricle with the thalamus in the central portion and the cavum septi pellucidi visible in the anterior portion.
5. Doppler imaging of umbilical arterial blood flow.
Umbilical arterial blood flow was examined by Doppler ultrasound according to the Society for Maternal-Fetal Medicine Clinical Guidelines.28
6. Deepest amniotic fluid pocket (cm).
The deepest amniotic fluid pocket was measured by transabdominal ultrasonography.
Fetal head circumference, deepest amniotic fluid pocket and Doppler imaging of umbilical arterial blood flow are evaluated according to the flow chart as shown in table 2:
Table 2.
Fetal head circumference, deepest amniotic fluid pocket and Doppler imaging of umbilical arterial blood flow evaluation flow chart
| Visit 1 | Visit 2 | Visit 3 | Visit 4 | Visit 5 | Every 2 weeks before 36 weeks of GA after visit 5 | Every 1 week at or after 37 weeks of GA | |
| Day of enrolment | 1 week after the enrolment | 2 weeks after the enrolment | 3 weeks after the enrolment | 4 weeks after the enrolment | |||
| Fetal head circumference | ● | ● | ● | ● | ● | ● | ● |
| Deepest amniotic fluid pocket | ● | ● | ● | ● | ● | ● | ● |
| Doppler imaging of umbilical arterial blood flow | ● | ● | ● | ● | ● | ● | ● |
GA, gestational age.
7. Prolongation of GA at birth (days).
Prolongation of GA at birth is defined as days from the first day of the protocol-defined treatment to birth.
8. Birth weight (g).
Birth weight is defined as the weight of the infant at birth.
9. GA at birth.
GA at birth is defined as the gestational age at birth.
10. Apgar score.
The Apgar score consists of an evaluation of five factors: heart rate, respiratory effort, muscle tone, responsiveness, and colour at 1 min and 5 min after birth.
11. Umbilical artery pH and base excess values.
Umbilical artery pH and base excess are measured at delivery.
12. Incidence rate of pre-eclampsia.
Incidence rate of pre-eclampsia is defined as the percentage of enrolled patients who develop pre-eclampsia after the protocol-defined treatment.
13. Paediatric developmental assessment until 1.5 years of age.
Paediatric developmental assessment includes physiological and neurological developmental assessment, and infant complications including cerebral palsy, epilepsy and death. In the neurodevelopment test in this study, the Kyoto Scale of Psychological Development 2001 was used. Evaluation of neurodevelopment was performed by a paediatric neurologist.
Safety endpoints
1. Incidence rate of obstetric complications.
Incidence rate of obstetric complications including hypertensive disorders of pregnancy (HDP) is defined as the percentage of enrolled patients who develop obstetric complications after the protocol-defined treatment.
2. Perinatal mortality.
Perinatal mortality is defined to include stillbirths (occurring after 22 weeks of gestation) and neonatal deaths (occurring up to 7 days after birth).
3. Neonatal mortality.
Neonatal mortality is defined as neonatal deaths occurring up to 28 days after birth.
Stopping criteria
The investigator must discontinue the protocol-defined treatment when certain events prevent continuation of the protocol treatment. These events include the following:
The mother has withdrawn her consent for participation in this study.
- Certain events prevent continuation of the protocol treatment, which include the following:
- A serious adverse drug reaction to tadalafil has developed.
- The investigator’s decision to prioritise other management including termination of the pregnancy instead of continuation of the protocol-defined treatment.
- The investigator’s decision that it is inappropriate to continue with the protocol treatment.
- The mother’s poor compliance or discontinuation of the protocol treatment.
Criteria for delivery
In this study, to minimise bias in terms of the timing of delivery, a fetal indication for delivery is established on the basis of the results from the multicentre survey of VLBW infants in Japan using a network database (figure 1 and table 1). After registration, all patients will receive the conventional management of FGR according to the guidelines for obstetrical practice in Japan regardless of the treatment arm.23 Briefly, the conventional management of FGR consists of the evaluation of fetal well-being on ultrasonography, Doppler imaging of umbilical arterial blood flow, non-stress test, contraction stress test and biophysical profile scoring depending on GA to evaluate possible pregnancy termination. The investigator will evaluate the fetal condition and decide timing of delivery referring to table 1. For other complications such as preterm labour, rupture of the membranes and HDP, the investigator will follow the guidelines for obstetric practice in Japan.23 The investigator must provide a report that explains the reason for termination of the pregnancy on the website of this clinical trial (the Clinical Trial Data Management System).
Monitoring safety during the fetal therapy
The investigator must pay close attention to the safety of the fetus and the mother. As shown in the study schedule, the protocol-defined assessments include evaluation of maternal BP and pulse rate, maternal blood and urine tests (blood fibrinogen and antithrombin III levels, liver and renal function tests, serum electrolyte levels, and qualitative urine protein excretion), and maternal serum PlGF and soluble FMS-like tyrosine kinase receptor-1 levels. Other assessments include adverse events assessed by medical consultation and antepartum fetal tests consisting of ultrasonography including Doppler imaging of blood flow (umbilical artery, middle cerebral artery and uterine artery), non-stress test, contraction stress test and biophysical profile scoring depending on GA. The investigator will enter patients’ safety data into the case report form on the website of this clinical trial (the Clinical Trial Data Management System).
Safety Evaluation Committee
The Safety Evaluation Committee is responsible for the overall safety of this clinical study. To ensure the safety of the protocol-defined treatment, the Safety Evaluation Committee will review the adverse events of tadalafil treatment. The safety committee will have blind access to the data. If a seriously adverse event develops, the investigator will provide the Secretariat with the necessary information within 24 hours of its onset, according to the predetermined procedure. Then the Secretariat will forward the obtained information without delay to the Safety Evaluation Committee for review. The Safety Evaluation Committee will notify the investigator of the review results. If the adverse event is definitely or probably related to tadalafil treatment, the ethics committee of Mie University Hospital or each institute will consider possible termination of this clinical study. Special attention must be paid to the reporting requirements stipulated in the Ethical Guidelines for Clinical Studies (Ministry of Health, Labour and Welfare in Japan, 2017). Infants will be followed up and evaluated for physiological and neurological development until 1.5 years of age.
Sample size
The sample will include 140 fetuses and their mothers.
Rationale for the target sample size
Table 3 shows the summary of the distribution of fetal growth velocity from enrolment to birth in our retrospective study.22 We estimate that the distribution of fetal growth velocity of this prospective phase II trial will be similar to that of our retrospective study. When the results of our prospective study are analysed by Wilcoxon rank-sum test and group comparisons, with an α of 0.05, two-sided, we will have 90% power to detect a difference if we randomise 62 women per group. Allowing for a 10% dropout rate, the total sample size required is 140 women.
Table 3.
Distribution of fetal growth velocity from enrolment to birth in the retrospective study conducted at Mie University Hospital
| Fetal growth velocity (g/day) | <5 | ≥5 to <10 | ≥10 to <15 | ≥15 to <20 | ≥20 to <25 | ≥25 |
| Conventional management group (%) | 5.3 | 10.5 | 21.1 | 47.3 | 15.8 | 0 |
| Tadalafil group (%) | 0 | 8.3 | 8.3 | 50.0 | 16.7 | 16.7 |
Statistical analysis
Analysis is done on all randomised fetuses who receive the protocol-defined treatment at least once, as the full analysis set. All randomised participants with outcome data available will be included in the analyses, which will be performed on an intention-to-treat basis, according to the treatment allocation at randomisation. Analysis as per protocol set (ie, removing patients who do not meet the inclusion and exclusion criteria) is done as a secondary analysis population for sensitivity analysis. All outcome measures are presented as summaries of descriptive statistics (mean (SD) or median (minimum, maximum and IQR) for continuous measures, and the numbers and proportions for ordinal and dichotomous measures). Descriptive statistics for the primary endpoint, that is, fetal growth velocity from the first day of the protocol-defined treatment to birth (g/day), are analysed for each treatment arm by the Wilcoxon rank-sum test and group comparisons. All analyses are performed according to a prespecified statistical analysis plan. The Data Coordinating Center in Mie University Hospital supports the data management, statistical analysis and reporting of the study.
Ethics and dissemination
Written informed consent will be obtained from all mothers before they are recruited. This trial has been registered in the UMIN Clinical Trials Registry as UMIN000023778 (https://upload.umin.ac.jp/cgi-open-bin/ctr/ctr_view.cgi?recptno=R000027132). Our findings will be widely disseminated through conference presentations and peer-reviewed publications.
Participating institutions
Mie University, Showa University, Osaka University, Nagoya University, Mie Chuo Medical Center, Municipal Yokkaichi Hospital, Ise Red Cross Hospital, St Marianna University, Juntendo University, Jikei University, Toho University, Yokohama City University Medical Center, Kanagawa Children’s Medical Center, Ehime University, Hamamatsu University School of Medicine, Osaka Medical College, Niigata University, Showa University Northern Yokohama Hospital, Showa University Koto Toyosu Hospital, Gifu University, University of the Ryukyu, Shiga University, Shinshu University, Ehime Prefectural Central Hospital, Akita University, Tokyo Metropolitan Bokutoh Hospital, Kyorin University, Tokyo Metropolitan Tama Medical Center, Kuwana East Medical Center, Kanazawa University, Nagasaki Medical Center, University of Toyama, Yamaguchi University, Toyota Memorial Hospital, Kainan Hospital, Dokkyo Medical University, Saga Hospital, Kyoto Prefectural University, Toyama Central Prefectural Hospital, Sapporo City General Hospital, Kagoshima University, Mie Prefectural General Medical Center, Kyoto University, Sakakibara Heart Institute and University of Fukui.
Patient and public involvement
Patients with FGR have helped to prioritise this research question through a James Lind Alliance Priority Setting Partnership, which highlighted this as a priority topic. Patients were not involved in the design of this study and in the recruitment to and conduct of this study. The results of this study will be informed by the home page of Mie University Obstetrics and Gynecology. For randomised controlled trials, there is no burden of the intervention by patients themselves. Patients and/or the public were not involved in this trial.
The original protocol is available in online supplementary file 1.
bmjopen-2017-020948supp001.pdf (2.5MB, pdf)
Discussion
This protocol has already been approved by the Institutional Review Board of Mie University Hospital and 39 institutions in Japan. Fetuses with FGR will be enrolled from these institutions. As the fetal growth velocity from the first day of the treatment to birth has been defined as the primary endpoint and fetuses will be randomly assigned based on an open-label design, timing of delivery should be made on the basis of similar criteria as much as possible. This study is the first nationwide intervention study in the field of obstetrics in Japan. We selected an open-label study design with a strict fetal management algorithm on the basis of the results from a multicentre Japanese survey instead of a placebo-controlled design because of operational challenges including low acceptability by pregnant women in Japan. Each participating medical centre can provide treatment for fetuses with FGR by board-certified members of the Japan Society of Obstetrics and Gynecology, and the investigator will be able to optimally decide timing of delivery according to the guidelines for obstetrical practice in Japan.23 To make more accurate decisions, a fetal indication for delivery is established in this study on the basis of the results from the multicentre survey in Japan, in which 82 level III perinatal centres, including 8 sites participating in this study, were registered (table 1).1 The fetal indication for delivery is divided into three groups depending on infant survival rate in the NICU. As all patients will undergo antepartum fetal tests consisting of evaluation of fetal well-being by ultrasonography, including Doppler imaging of umbilical arterial blood flow, non-stress test, contraction stress test and biophysical profile scoring depending on GA according to the Japanese guidelines, the investigator can easily refer to this indication when deciding timing of delivery. This indication will be used to evaluate fetal baseline condition at enrolment as well. We believe that this approach could take advantage of the strengths and minimise the possible limitations related to open-label trial features.
We retrospectively compared the effect of tadalafil in patients with FGR and demonstrated that both fetal growth velocity from enrolment to birth and birth weight were significantly higher in the tadalafil group than in the conventional management group. The required sample size of this prospective study was estimated based on the results of the retrospective study that used the same primary outcome measure. Since patients with FGR were enrolled in the retrospective study under similar criteria to those in this study, we think that it is reasonable to use the results of the retrospective study for the estimation of the sample size.
Supplementary Material
Acknowledgments
All authors thank Dr Makoto Maeda (Board Certified Member of the
Japan Society of Obstetrics and Gynecology)
and
Dr Yoshiaki Miyake (Board Certified Member of the
Japan Society of Obstetrics and Gynecology)
for their contribution as members of the Safety Evaluation Committee in this trial. We also thank Dr Hirofumi Sawada (Department of Pediatrics, Anesthesiology, and Critical Care Medicine, Mie University Graduate School of Medicine) for his advice on the protocol of paediatric developmental assessment.
Footnotes
Contributors: MiK TU wrote the manuscript. ST, YN, MK, CM and MN provided the biostatistical study design. SM, MaK, HT, MN, KT, KO, YK, ME, TaK, ToK, MN, AS and TI helped in the conception of the study.
Funding: This work was supported by the Japan Agency for Medical Research and Development (AMED), JSPS KAKENHI Grant Number 17K16846, and in part by the Takeda Science Foundation.
Competing interests: None declared.
Patient consent: Not required.
Ethics approval: This study was approved by the Institutional Review Board of Mie University Hospital on 25 August 2016 (no 3041) prior to patient enrolment. The study protocol was also approved by each institutional review board of all participating institutions. The study complies with the Helsinki Declaration.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Kusuda S, Fujimura M, Sakuma I, et al. Morbidity and mortality of infants with very low birth weight in Japan: center variation. Pediatrics 2006;118:e1130–e1138. 10.1542/peds.2005-2724 [DOI] [PubMed] [Google Scholar]
- 2. American College of Obstetricians and Gynecologists. ACOG Practice bulletin no. 134: fetal growth restriction. Obstet Gynecol 2013;121:1122–33. 10.1097/01.AOG.0000429658.85846.f9 [DOI] [PubMed] [Google Scholar]
- 3. Hui L, Challis D. Diagnosis and management of fetal growth restriction: the role of fetal therapy. Best Pract Res Clin Obstet Gynaecol 2008;22:139–58. 10.1016/j.bpobgyn.2007.06.004 [DOI] [PubMed] [Google Scholar]
- 4. Coppage KH, Sun X, Baker RS, et al. Expression of phosphodiesterase 5 in maternal and fetal sheep. Am J Obstet Gynecol 2005;193:1005–10. 10.1016/j.ajog.2005.05.054 [DOI] [PubMed] [Google Scholar]
- 5. Rotella DP. Phosphodiesterase 5 inhibitors: current status and potential applications. Nat Rev Drug Discov 2002;1:674–82. 10.1038/nrd893 [DOI] [PubMed] [Google Scholar]
- 6. Wareing M, Myers JE, O’hara M, et al. Effects of a phosphodiesterase-5 (PDE5) inhibitor on endothelium-dependent relaxation of myometrial small arteries. Am J Obstet Gynecol 2004;190:1283–90. 10.1016/j.ajog.2003.12.024 [DOI] [PubMed] [Google Scholar]
- 7. Wareing M, Myers JE, O’Hara M, et al. Sildenafil citrate (Viagra) enhances vasodilatation in fetal growth restriction. J Clin Endocrinol Metab 2005;90:2550–5. 10.1210/jc.2004-1831 [DOI] [PubMed] [Google Scholar]
- 8. Herraiz S, Pellicer B, Serra V, et al. Sildenafil citrate improves perinatal outcome in fetuses from pre-eclamptic rats. BJOG 2012;119:1394–402. 10.1111/j.1471-0528.2012.03430.x [DOI] [PubMed] [Google Scholar]
- 9. Ramesar SV, Mackraj I, Gathiram P, et al. Sildenafil citrate improves fetal outcomes in pregnant, L-NAME treated, Sprague-Dawley rats. Eur J Obstet Gynecol Reprod Biol 2010;149:22–6. 10.1016/j.ejogrb.2009.11.005 [DOI] [PubMed] [Google Scholar]
- 10. Baijnath S, Soobryan N, Mackraj I, et al. The optimization of a chronic nitric oxide synthase (NOS) inhibition model of pre-eclampsia by evaluating physiological changes. Eur J Obstet Gynecol Reprod Biol 2014;182:71–5. 10.1016/j.ejogrb.2014.08.021 [DOI] [PubMed] [Google Scholar]
- 11. Nassar AH, Masrouha KZ, Itani H, et al. Effects of sildenafil in Nω-nitro-L-arginine methyl ester-induced intrauterine growth restriction in a rat model. Am J Perinatol 2012;29:429–34. 10.1055/s-0032-1304823 [DOI] [PubMed] [Google Scholar]
- 12. Cross JC, Hemberger M, Lu Y, et al. Trophoblast functions, angiogenesis and remodeling of the maternal vasculature in the placenta. Mol Cell Endocrinol 2002;187:207–12. 10.1016/S0303-7207(01)00703-1 [DOI] [PubMed] [Google Scholar]
- 13. Watson ED, Cross JC. Development of structures and transport functions in the mouse placenta. Physiology 2005;20:180–93. 10.1152/physiol.00001.2005 [DOI] [PubMed] [Google Scholar]
- 14. von Dadelszen P, Dwinnell S, Magee LA, et al. Sildenafil citrate therapy for severe early-onset intrauterine growth restriction. BJOG 2011;118:624–8. 10.1111/j.1471-0528.2010.02879.x [DOI] [PubMed] [Google Scholar]
- 15. Sharp A, Comforth C, Jackson R, et al. OC01.05: STRIDER UK: a randomised controlled trial of sildenafil therapy in dismal prognosis early-onset intrauterine growth restriction. Ultrasound in Obstetrics & Gynecology 2017;50:3 10.1002/uog.17574 [DOI] [Google Scholar]
- 16. Sahni S, Palkar AV, Rochelson BL, et al. Pregnancy and pulmonary arterial hypertension: a clinical conundrum. Pregnancy Hypertens 2015;5:157–64. 10.1016/j.preghy.2015.01.004 [DOI] [PubMed] [Google Scholar]
- 17. Ladouceur M, Benoit L, Radojevic J, et al. Pregnancy outcomes in patients with pulmonary arterial hypertension associated with congenital heart disease. Heart 2017;103:287–92. 10.1136/heartjnl-2016-310003 [DOI] [PubMed] [Google Scholar]
- 18. Daimon A, Kamiya CA, Iwanaga N, et al. Management of pulmonary vasodilator therapy in three pregnancies with pulmonary arterial hypertension. J Obstet Gynaecol Res 2017;43:935–8. 10.1111/jog.13279 [DOI] [PubMed] [Google Scholar]
- 19. Wilkins MR, Wharton J, Grimminger F, et al. Phosphodiesterase inhibitors for the treatment of pulmonary hypertension. Eur Respir J 2008;32:198–209. 10.1183/09031936.00124007 [DOI] [PubMed] [Google Scholar]
- 20. Forgue ST, Patterson BE, Bedding AW, et al. Tadalafil pharmacokinetics in healthy subjects. Br J Clin Pharmacol 2006;61:280–8. 10.1111/j.1365-2125.2005.02553.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshikawa K, Umekawa T, Maki S, et al. Tadalafil Improves L-NG-Nitroarginine Methyl Ester-Induced preeclampsia with fetal growth restriction-like symptoms in pregnant mice. Am J Hypertens 2017;31:89–96. 10.1093/ajh/hpx130 [DOI] [PubMed] [Google Scholar]
- 22. Kubo M, Umekawa T, Maekawa Y, et al. Retrospective study of tadalafil for fetal growth restriction: Impact on maternal and perinatal outcomes. J Obstet Gynaecol Res 2017;43:291–7. 10.1111/jog.13218 [DOI] [PubMed] [Google Scholar]
- 23. Minakami H, Maeda T, Fujii T, et al. Guidelines for obstetrical practice in Japan: Japan Society of Obstetrics and Gynecology (JSOG) and Japan Association of Obstetricians and Gynecologists (JAOG) 2014 edition. J Obstet Gynaecol Res 2014;40:1469–99. 10.1111/jog.12419 [DOI] [PubMed] [Google Scholar]
- 24. Kubo M, Tanaka H, Maki S, et al. Safety and dose-finding trial of tadalafil administered for fetal growth restriction: a phase-1 clinical study. J Obstet Gynaecol Res 2017;43:1159–68. 10.1111/jog.13345 [DOI] [PubMed] [Google Scholar]
- 25. Cunningham F, Leveno K, Bloom S, et al. Williams obstetrics, 24e. Mcgraw-hill, 2014. [Google Scholar]
- 26. Parer JT, Ikeda T. A framework for standardized management of intrapartum fetal heart rate patterns. Am J Obstet Gynecol 2007;197:26.e1–26.e6. 10.1016/j.ajog.2007.03.037 [DOI] [PubMed] [Google Scholar]
- 27. Shinozuka N. Fetal biometry and fetal weight estimation: JSUM standardization. The Ultrasound Review of Obstetrics & Gynecology 2002;2:156–61. 10.1080/14722240208500478 [DOI] [Google Scholar]
- 28. Berkley E, Chauhan SP, Abuhamad A, et al. Doppler assessment of the fetus with intrauterine growth restriction. Am J Obstet Gynecol 2012;206:300–8. 10.1016/j.ajog.2012.01.022 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2017-020948supp001.pdf (2.5MB, pdf)