

Abstract
In type 2 diabetes (T2D), the most significant pathological change in pancreatic islets is amyloid deposits, of which a major component is islet amyloid polypeptide (IAPP), also called amylin. IAPP is expressed in β-cells and co-secreted with insulin. Together with the inhibitory effects of synthetic human IAPP (hIAPP) on insulin secretion, our studies, using hIAPP transgenic mice, in which glucose-stimulated insulin secretion was moderately reduced without amyloid deposit, and hIAPP gene-transfected β-cell lines, in which insulin secretion was markedly impaired without amyloid, predicted that soluble hIAPP-related molecules would exert cytotoxicity on β-cells. Human IAPP is one of the most aggregation-prone peptides that interact with cell membranes. While it is widely reported that soluble hIAPP oligomers promote cytotoxicity, this is still a hypothesis since the mechanisms are not yet fully defined. Several hIAPP transgenic mouse models did not develop diabetes; however, in models with backgrounds characterized for diabetic phenotypes, β-cell function and glucose tolerance did worsen, compared to those in non-transgenic models with similar backgrounds. Together with these findings, many studies on metabolic and molecular disorders induced by risk factors of T2D suggest that in T2D subjects, toxic IAPP oligomers accumulate in β-cells, impair their function, and reduce mass through disruption of cell membranes, resulting in β-cell failure. IAPP might be central to β-cell failure in T2D. Anti-amyloid aggregation therapeutics will be developed to create treatments with more durable and beneficial effects on β-cell function.
Keywords: IAPP/amylin, Type 2 diabetes, Islet amyloid, IAPP oligomer, Risk factors of T2D, β-Cell failure
Introduction
Type 2 diabetes mellitus (T2D) is a progressive disease in which poor glycemic control is exacerbated over time and pancreatic β-cell function declines [1, 2]. Most patients require oral antidiabetic drugs in addition to lifestyle intervention, although it remains difficult for them to achieve normoglycemic levels. The majority of patients eventually need the insulin therapy despite treatment by multiple oral antidiabetic drugs [3, 4].
In October 2013, the Global Partnership to Accelerate Diabetes Research, cosponsored by the Endocrine Society and the American Diabetes Association, assembled international experts to examine the foundation of β-cell failure in T2D and to suggest areas for future research on the underlying mechanisms that may lead to improved prevention and treatment [5]. β-Cell failure is a multifactorial process involving many impairments. It remains unclear which pathway is the major process inducing failure. A single molecule, including islet amyloid polypeptide (IAPP), also called amylin, could have risk effects inducing a multifactorial process via a feed-forward loop. In this review, we will discuss the possibility of IAPP, related intermediates, or islet amyloid to induce β-cell failure through a multifactorial process in subjects with risk factors for T2D, presenting the recent research progress made.
Islet amyloid and the human IAPP gene and molecule
In T2D, the most significant pathological change in pancreatic islets is amyloid deposits [6]. Islet amyloid deposits are observed in 70–90% of T2D patients over 40 years of age [7]. A major component of islet amyloids is IAPP, a 37-amino acid peptide [8, 9]. We reported that IAPP-derived amyloid deposits were found in 76% of Japanese T2D patients and increased with the duration of the disease by immune-histochemical techniques using two specific antibodies to hIAPP [10], while a report showed that islet amyloid identified by Congo-red staining was positive in 32 cases (27.1%) in 118 Japanese diabetic patients [11]. The different prevalence of amyloid deposits may be due to the sampling of patients or the method used for survey of the presence of islet amyloids. Amyloid fibrils typically are 5–15 nm in width, are unbranched, are straight over distances approaching 1 micron, and are often many microns long [10, 12]. They contain “cross-β” structures, which are ribbon-like β sheets in which β strand segments run approximately perpendicular to the fibril growth direction. Although these ideas have been studied extensively for amyloid beta (Aβ) and α-synuclein, similar ideas apply to all most proteins whose aggregation is associated with degenerative diseases, including IAPP. Amyloid fibrils are generally polymorphic at the molecular level, with a given peptide or protein being capable of forming a variety of distinct, self-propagating fibril structures [12].
The single gene encoding IAPP in humans is located on the short arm of chromosome 12, and human IAPP (hIAPP) precursors are 89 amino acids in length, and human prepro-IAPP has a typical signal peptide of 22 amino acids followed by a propeptide containing the mature IAPP [13–15]. In humans, cats, and non-human primates, islet amyloid deposits develop in connection with diabetes, whereas no such deposits form in rodents [15]. The amino acid sequence of the central part of IAPP, residues 20–29, varies among species and, therefore, amyloid deposition is considered to be related to this region of the molecule [16, 17] (Fig. 1). The human IAPP molecule itself is responsible for aggregation to amyloid fibrils.
Fig. 1.

Comparison of predicted amino acid sequences for human, cat, rat, mouse, and guinea pig IAPP precursors. The IAPP moiety is boxed. The length of each precursor (number of amino acids) is given in parentheses. The predicted boundaries of the signal peptide and N and C-terminal propeptides are indicated. Dashes indicate amino acid residues that are identical to the human sequence. Colons indicate gaps that were inserted to generate this alignment.
Cited from Nish et al. [16]
IAPP synthesis and the secretory pathway
IAPP is expressed in pancreatic β-cells and is co-secreted with insulin in response to glucose [18, 19]. Prepro-IAPP assembled in the endoplasmic reticulum is processed to its mature 37-amino acid from within the secretory pathway [20]. IAPP is found in the halo region of mature secretory granules, whereas hexameric insulin, together with zinc, constitutes the core of the granules [21]. We also observed the production and secretion of IAPP in β-cell line, MIN6, which expresses the glucose transporter isoform, GLUT2, and retains glucose-inducible insulin secretion [22, 23]. The cells secrete IAPP and insulin in response to rising glucose concentrations but fail, however, to induce increments in IAPP and insulin II mRNA, suggesting that glucose regulates proIAPP synthesis by translational control as well as proinsulin synthesis [23, 24]. Alternatively, the low glucose concentration may maximally stimulate transcription of the IAPP gene, as reported by some studies that glucose has a stimulatory effect on transcription of the insulin gene in insulin-producing cell lines [25, 26].
Increase of IAPP synthesis along with aging and under obese and diabetic conditions
IAPP content and secretion in the pancreatic islet cells increase in addition to insulin in obese and diabetic rodent models, including ventromedial hypothalamic-lesioned obese rats, genetically obese Zucker rats [27], and genetically obese and diabetic mice, both ob/ob and db/db mice [28]. Also, IAPP contents change with increasing age in genetically obese and diabetic mice [29]. Thus, the aberrant biosynthesis and secretion of IAPP may be related to obese and diabetic conditions and change the course of obesity and diabetes. In the case of humans, because of amyloidogenicity, aberrant biosynthesis and secretion may contribute, in part, to the formation of islet amyloid deposits and to the impairment of β-cell function in subjects with T2D.
Inhibitory effect of synthetic hIAPP on insulin secretion
Studies on the effect of hIAPP on insulin secretion have been performed, yielding interesting, however, contradictory results [30]. In 1989, we reported that high doses of synthetic hIAPP (10 μM) inhibited glucose-stimulated insulin secretion from isolated rat pancreatic islets, proposing that the abnormal secretion and deposition of IAPP may be involved in impaired insulin secretion in T2D [31] (Fig. 2a). In the 1990s, several investigators reported inhibition of insulin secretion [32, 33], while others reported no inhibitory effect of IAPP on insulin release [34, 35]. A study showed that at very high plasma concentrations, IAPP affected the insulin response to glucose load in humans, suggesting that IAPP appears unlikely to be a circulating hormone influencing carbohydrate metabolism [36]. At that time, there was no definitive explanation for these varying results and it was later speculated that, at those early dates, the strong tendency for hIAPP to aggregate into amyloid-like fibrils had not been taken into consideration [30]. Thus, the reported effects of early IAPP preparations may be questioned [30]. Alternatively, β-cell dysfunction might be induced by high local concentrations of hIAPP that can be attained near the plasma membrane of β-cells via its co-secretion with insulin, implying that high concentrations of hIAPP might be necessary to induce β-cell toxicity. A recent study showed that the addition of hIAPP oligomers (100 nM–10 μM), concentrations which reflect the local concentration of hIAPP secreted from the granules, decreased in a dose-dependent manner pancreatic RIM-m cell viability and induced apoptosis [37].
Fig. 2.

Aberrant human IAPP (hIAPP) impairs insulin secretion. a Inhibitory effect of synthetic hIAPP on insulin secretion. Islets isolated from rats were incubated with glucose, and 16.7 mM glucose-induced insulin secretion was inhibited by 10 μM synthetic hIAPP [31]. b Impaired insulin secretion in hIAPP transgenic lean mice. Islets isolated from the transgenic mice were incubated with glucose, and 16.7 mM glucose-induced insulin secretion was moderately reduced [41, 42]. Glucose tolerance of intraperitoneal glucose tolerance test (ipGTT) was normal (see Fig. 3b), and islet amyloid deposit was absent in the transgenic mice (see Fig. 3c). c Impaired insulin secretion in β-cell line aberrantly expressing hIAPP. Human IAPP is aberrantly expressed without amyloid fibrils and amyloid deposits in transformed mouse β-cell (MIN6) transfected with hIAPP cDNA, and 16.7 mM glucose-induced insulin secretion was markedly reduced in the clones transfected with hIAPP cDNA [42]
Studies on the diabetogenic role of hIAPP using transgenic hIAPP mouse models and hIAPP gene-transfected β-cell lines
To investigate the diabetogenic role of hIAPP, several transgenic mouse models for hIAPP had been developed [38] but the models did not develop diabetes. Also, insulin concentrations in the peripheral blood did not differ from those in control mice [39–41]. However, we observed a moderate reduction of glucose-stimulated insulin secretion in islets isolated from the transgenic mice, which developed fibril-like materials in secretory granules but did not develop amyloid deposits and diabetes [41, 42] (Fig. 2b). To confirm the impairment of insulin secretion via aberrant expression of hIAPP in pancreatic β-cells, we tried to make an hIAPP-gene-transfected β-cell line which expresses the glucose transporter isoform, GLUT2, and retains glucose-inducible insulin and IAPP secretion [22, 23]. In the β-cell line model, glucose-stimulated insulin secretion was markedly impaired without amyloid deposits, concluding that aberrant expression of hIAPP induced the impairment of insulin secretion without depositing amyloid [42] (Fig. 2c). Our studies using hIAPP transgenic mice and an hIAPP-transfected β-cell line strongly predicted that soluble hIAPP-related molecules aberrantly produced in β-cells had cytotoxic effects on the cell [41, 42]. These reported mouse models expressing hIAPP were heterozygous transgenic mice [39–42]. To demonstrate the effect of an increased burden of hIAPP, homozygous transgenic mice with high rates of expression of hIAPP were developed and the transgenic mice spontaneously developed diabetes, associated with impaired insulin secretion and amorphous IAPP aggregates [43]. Together with the inhibitory effects of high doses of hIAPP on insulin secretion [31–33, 37], these studies suggest that aberrantly expressed hIAPP induces the impairment of insulin secretion, possibly via the cytotoxic effects of hIAPP or the soluble IAPP-related molecules in β-cells.
Cytotoxicity and soluble hIAPP oligomers
It has been suggested that insoluble amyloid deposits serve as major pathological agents in degenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease, and T2D. However, recent studies have demonstrated that the number of amyloid deposits does not parallel disease severity and that associated peptide Aβ oligomers are, in fact, more toxic than the mature amyloid fibrils in AD, suggesting that aberrant aggregation of protein into insoluble amyloid fibrils is implicated in these diseases [44–46]. All the soluble oligomers, including Aβ oligomers and hIAPP oligomers, display a common conformation-dependent structure and the in vitro toxicity of soluble oligomers is inhibited by oligomer-specific antibodies, suggesting that the different types of soluble amyloid oligomers have a common structure and they share a common mechanism of cytotoxicity [47]. The corresponding amyloid aggregates display common features, such as the characteristic cross-β structure of the fibrils and cytotoxicity caused by either fibrils or intermediate oligomers, suggesting common amyloid mechanisms [48]. Human IAPP, one of the most aggregation-prone peptides, spontaneously forms amyloid aggregates in vitro within hours at μM concentrations [49].
Toxic hIAPP oligomers and disruption of cell membranes
Human IAPP interacts with negatively charged membranes and this interaction can drastically accelerate misfolding, which is a prerequisite for hIAPP toxicity [50]. The membrane interface of hIAPP has been shown to be mediated by an α-helical structure in the first 20 residues of this peptide [51]. The α-helical structure has important implications for anchoring hIAPP to membranes [52]. The α-helical segment of hIAPP, residues 5–17, is positioned within the hydrophobic lipid environment, whereas the amyloidogenic segment, residue 20–29, is at the interface between the lipid and solvent. Coupling between the two segments might mediate the transition to a toxic structure [53]. The α-helical structure drives the early stages of self-assembly for amyloidogenic hIAPP aggregate formation in membranes [54]. A recent study showed that α-helical hIAPP oligomers induced permeable pores in lipid membranes [55]. It has been speculated that the α-helical structure might be important for hIAPP membrane interactions, leading to high local concentrations and, in turn, promoting intermolecular β-sheet formation and IAPP oligomers, in which toxic oligomers disrupt cell membranes in β-cells. However, there has been considerable debate in the literature as to the primary hIAPP conformation that induces β-cell toxicity [56]. Despite extensive in vitro studies, the process of hIAPP aggregation in vivo is poorly understood, though it is widely reported to promote cytotoxicity. Evidence both for and against toxic hIAPP oligomer hypothesis has been presented; it is apparent that what exactly causes β-cell failure when hIAPP aggregates are still under debate [12, 56–59]. It has been reported that the relationship between IAPP aggregation and toxicity is not simple as both the fresh hIAPP molecules and fully formed fibers showed minimal toxicity [60, 61], and islet amyloid was more common with severe β-cell loss, associated with macrophage infiltration [11].
Human IAPP species and mechanisms of cytotoxicity
A recent review article described that the mechanisms of cytotoxicity are not fully defined and multiple mechanisms for hIAPP induction of β-cell toxicity have been proposed [57], including the ability to perturb membranes, to induce ER stress, to cause defects in autophagy, and to induce inflammatory responses. A recent study showed that fresh and oligomeric hIAPP, as well as mature amyloid, enhanced membrane fluidity and caused losses in cell viability [60]. A study showed that whereas the in-register β strands were perpendicular to the fibril axis, out-of-register β-strands were at an angle of ~ 50° from the perpendicular and that out-of-register amyloid β-sheets and their related cylindrins were part of a toxic amyloid pathway [62]. ER stress has been proposed to be an important contributor to hIAPP-induced β-cell death and exogenously added hIAPP has been reported to induce ER stress [63, 64]. A study showed the protective potential of autophagy for hIAPP-mediated cellular dysfunction, suggesting the involvement of IAPP in autophagy–lysosomal degradation [65]. Human IAPP was reported to activate the NLRP3 inflammasome, inducing a mechanism for enhanced IL-1β in T2D [66]. The mechanism of IAPP oligomerization and amyloid formation is not fully understood due to limitations in the current methodology to characterize hIAPP species and also the mechanisms of cytotoxicity are not fully defined despite extensive investigations.
Studies using hIAPP transgenic rodent models with backgrounds characterized for diabetic phenotypes
Interestingly, hIAPP transgenic mice did not develop diabetes [39–41], while glucose-stimulated insulin secretion was moderately reduced [41, 42], suggesting that aberrantly expressed hIAPP may moderately impair β-cell function but does not induce β-cell failure and, consequently, does not induce diabetes. However, transgenic mice developed diabetes after treatment with diabetogenic drugs associated with impaired insulin secretion and amorphous IAPP aggregates [67]. We produced hIAPP transgenic ob/ob mice by mating a female C57BL/6J mouse transplanted with an ovary of a C57BL/6J ob/ob mouse and a male hIAPP transgenic mouse in which glucose-stimulated insulin secretion was moderately reduced but glucose tolerance was normal [41, 42] (Fig. 3a). We observed that glucose tolerance was worse in hIAPP transgenic ob/ob mice than that in non-transgenic ob/ob mice, while glucose tolerance was normal in both hIAPP transgenic and non-transgenic lean mice (Fig. 3b). Amyloid deposits were absent in the pancreatic islets of both transgenic ob/ob and lean mice (Fig. 3c), again suggesting that soluble hIAPP-related molecules aberrantly produced in β-cells had cytotoxic effects on the cell. The ob/ob mice were grossly overweight and had hyperglycemia, hyperlipidemia, insulin resistance, large islets containing mostly β-cells, marked mitotic figures, and fat accumulation [68]. These phenotypes could enhance the potential of hIAPP cytotoxicity in β-cells. It was also reported that extensive islet amyloid formation, insulin insufficiency, and hyperglycemia were observed in hIAPP transgenic ob/ob mice [69]. A study showed that hIAPP-expressing Avy/a (agouti variable yellow) male mice progressed to severe hyperglycemia relative to their non-transgenic counterparts and induced β-cell apoptosis, suggesting that insulin resistance can induce overt, slow onset diabetes associated with islet amyloid and increased β-cell apoptosis [70, 71]. Human IAPP-knock-in mice with a β-cell-specific autophagy defect had substantial deterioration of glucose tolerance, indicating that reduced autophagy may enhance the toxic potential of hIAPP and enhance β-cell dysfunction and progression of T2D [72]. Mice expressing β-cell hIAPP and lacking β-cell ABCA1, hIAPPTg/o Abca1βKOmice, had increased islet cholesterol, accompanied by fasting hyperglycemia, glucose intolerance, impaired insulin secretion, and increased amyloid area, suggesting that elevations in islet cholesterol may lead to an increase in IAPP aggregation and islet amyloid formation, further worsening β-cell function and glucose homeostasis [73]. Interestingly, in these transgenic mice with backgrounds characterized for diabetic phenotypes, including obesity, hyperglycemia, hyperlipidemia, insulin resistance, islet fat accumulation and defects in unfolded protein removal, glucose tolerance, and β-cell function worsen possibly because of the enhanced potential for hIAPP cytotoxicity in β-cells.
Fig. 3.
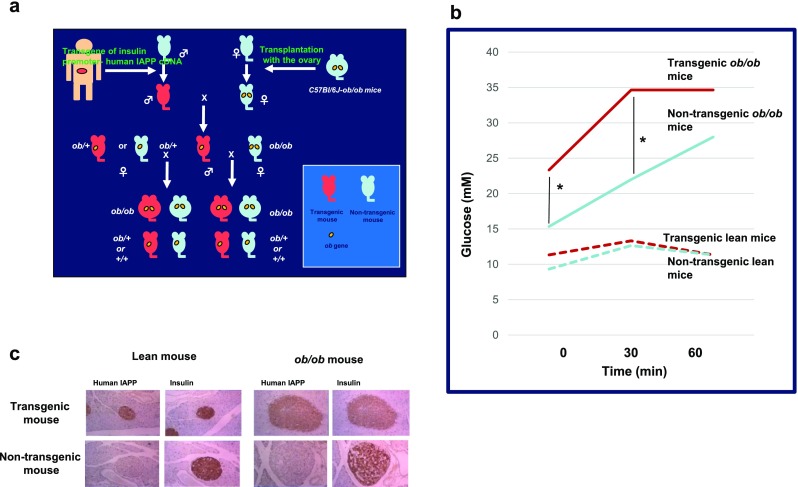
Worse glucose tolerance in human IAPP (hIAPP) transgenic ob/ob mice. a We produced hIAPP transgenic ob/ob mice by mating a female C57BL/6J mouse transplanted with an ovary of a C57BL/6J ob/ob mouse and a male hIAPP transgenic mouse in which glucose-stimulated insulin secretion was moderately reduced but glucose tolerance was normal [41, 42]. b Glucose tolerance of intraperitoneal glucose tolerance test (ipGTT) worsened in hIAPP transgenic ob/ob mice more so than that in non-transgenic ob/ob mice (plasma glucose at 0 and 30 min after the load was higher in hIAPP transgenic ob/ob mice than that in non-transgenic ob/ob mice. *P < 0.01; Student’s t test), while glucose tolerance was normal in both hIAPP transgenic and non-transgenic lean mice. c Immuno-histochemical study using anti-hIAPP antibody showed that hIAPP was expressed in the pancreatic β-cells of both transgenic ob/ob and lean mice, and amyloid deposits were absent in the pancreatic islets of both transgenic mice
Human IAPP processing within the secretory pathway of normal β-cells (Fig. 4)
Fig. 4.
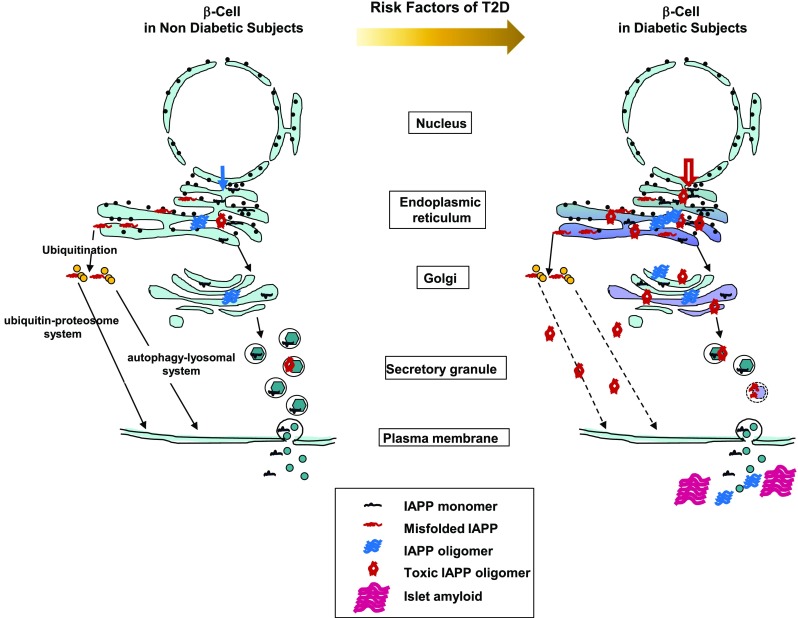
Possible mechanisms for the increase of toxic IAPP oligomers in β-cells in diabetic subjects. In β-cells of non-diabetic subjects with less common genetic variants and clinical risk factors of T2D, production of IAPP molecules and oligomers, including toxic IAPP oligomers, might be low. In β-cells of diabetic subjects with more common genetic variants and clinical risk factors of T2D, production of IAPP molecules and oligomers, including toxic IAPP oligomers, is largely influenced by the risk factors of T2D, including T2D susceptibility genes, aging, obesity, insulin resistance, hyperglycemia, and dyslipidemia. See Table 1
In non-diabetic subjects, Prepro-IAPP is first inserted into the endoplasmic reticulum (ER). Within the ER, secretory proteins are properly folded, modified, and assembled into multiprotein complexes in the ER before they traffic further downstream in the secretory pathway [74]. Notwithstanding the effort of the protein-folding machinery, a substantial fraction of the protein fails to fold properly in the ER. Incompletely folded forms are disposed of through discriminating quality-control systems. Unfolded proteins are removed to the cytosol by subsequent ubiquitination and degradation by the 26S proteasome and also disposed through autophagy [74–76]. Prepro-IAPP molecules are assembled and incompletely folded forms are disposed of through these systems. Then, prepro-IAPP is processed to its mature 37-amino acid form within the secretory pathway [20]; IAPP is found in the halo region of the mature secretory granules [21]. In vitro studies revealed that hIAPP aggregates readily at μM concentrations [77]. However, the peptide is stored in β-cell granules at mM concentrations without apparent formation of amyloid aggregations in healthy individuals [78]. Environmental conditions in β-cell granules, e.g., low pH, high concentrations of zinc ions, and insulin molecules, inhibit the formation of IAPP aggregates [79, 80]. IAPP is co-secreted with insulin in response to glucose and exerts physiological roles as a satiation hormone in the control of food intake and energy expenditure [18, 19, 81].
Risk factors of T2D and β-cell dysfunction (Table 1)
Table 1.
Common genetic variants and clinical risk factors of T2D increase toxic IAPP oligomers in β-cells
| Risk factors | Site | Disorders | References |
|---|---|---|---|
| T2D susceptibility genes | |||
| FTO | N | Incerased insulin and IAPP biosynthesis | [90, 91] |
| ER | Increased ER stress and impaired UPR | [49, 50, 74, 102] | |
| WFS1 | ER | Impaired UPR | [82, 83, 93–98] |
| SG | Altered acidification | [79, 80, 82, 83, 96–98, 104–106] | |
| SLC30A8 | SG | Altered Zn concentration | [41, 42, 63, 80, 105, 107–110] |
| Aging | |||
| Disrupted protein homeostasis | ER | Impaired UPR | [84, 99–101] |
| Ubiquitin–proteasome and autophagy–lysosomal system | Impaired misfolded-IAPP degradation | [84, 99–101] | |
| Obesity and insulin resistance | N | Increased insulin and IAPP biosynthesis | [11, 27–29, 88, 89] |
| ER | Increased ER stress and impaired UPR | [49, 50, 63, 74, 102] | |
| Metabolic disorders | |||
| Hyperglycemia | N | Increased insulin and IAPP biosynthesis | [23, 24, 27–29, 88, 89] |
| ER | Increased ER stress and impaired UPR | [49, 50, 74] | |
| Hyperlipidemia | ER | Impaired UPR | [102, 103] |
T2D type 2 diabetes, IAPP islet amyloid polypeptide, N nucleus, ER endoplasmic reticulum, SG secretory granules, UPR unfolded protein response
T2D is a metabolic and molecular disorder resulting from common genetic variants and clinical risk factors of diabetes. Genome-wide association studies (GWAS) have discovered more than seventy entirely new T2D loci, clearly suggesting that association is not limited to a few candidate genes but many genes are involved in disease pathogenesis [82, 83]. Aging is characterized by a progressive loss of physiological integrity, which is the primary risk factor for major human pathologies, including diabetes and neurodegenerative diseases [84]. Insulin resistance is associated with a cluster of co-morbidities, including obesity, dyslipidemia, and hypertension [85]. Also, β-cells are adversely influenced by dyslipidemia [86]. Chronic exposure to hyperglycemia can lead to β-cell dysfunction that may become irreversible over time, a process that is termed glucotoxicity [87].
Aberrant biosynthesis of hIAPP in subjects with obesity, insulin resistance, and hyperglycemia
As shown in Fig. 4 and Table 1, in subjects with risk factors of T2D, including obesity, insulin resistance, and hyperglycemia, IAPP synthesis might be increased in the early phase of the disease. Increased secretion of IAPP was observed in individuals with obesity or pre-diabetic insulin resistance [88, 89], and islet amyloid volume was correlated with BMI [11]. These findings, together with increased IAPP content in the pancreatic islet cells of obese and diabetic rodent models [27, 28] and glucose-regulated proIAPP and proinsulin synthesis regulated by translational control [23, 24], suggest that aberrant biosynthesis and secretion of IAPP might be related to obese and diabetic conditions. It seems likely that IAPP molecules are largely produced by obesity and insulin resistance as a consequence of both an unhealthy lifestyle and gene susceptibility, for example FTO, and hyperglycemia. Association of FTO with T2D was first discovered by a GWAS for obesity [82] and was later confirmed in replication studies [90, 91]. A common variant of FTO may be related to aberrant biosynthesis and secretion of IAPP with insulin. Human IAPP aberrantly biosynthesized is inserted into the ER and then processed within the secretory pathway. However, excess, misfolded hIAPP progressively aggregates, forming toxic IAPP oligomers. A study using an oligomer immunostaining method showed that toxic IAPP oligomers were present in human β-cells in T2D and, more frequently, with increasing obesity [63].
Accumulation of toxic IAPP oligomers in the ER due to ER stress and insufficient unfolded protein response (UPR) caused by common variants in WFS1
When protein-folding demand in the ER exceeds capacity, aberrantly synthesized hIAPP molecules start to accumulate as incompletely modified and unfolded forms within the ER [74]. Human IAPP is one of the most aggregation-prone peptides and interacts with negatively charged membranes. This interaction can drastically accelerate misfolding [49, 50]. When the UPR in the ER and autophagy are impaired, the accumulation of hIAPP aggregates progresses even further. Wolfram syndrome is an autosomal recessive neurodegenerative disorder associated with juvenile-onset diabetes mellitus, progressive optic atrophy, sensorineural deafness, and diabetes insipidus [92]. WFS1 protein localizes primarily in the ER and is ubiquitously expressed at the highest levels in brain, pancreas, heart, and insulinoma β-cell lines [93, 94]. WFS1 is a component of the UPR and maintains homeostasis of the ER in pancreatic β-cells [95]. WFS1 was included in a list of candidate genes evaluated for association with T2D [82, 83, 96]. A study presented evidence that supported the role of common variants of WFS1 as modest contributors to diabetes risk and these variants act on pancreatic β-cells [97]. The Data from an Epidemiological Study on the Insulin Resistance Syndrome (DESIR) prospective study showed that allelic variations in the WFS1 gene reduced insulin secretion and the most frequent haplotype in the haplotype block containing the WFS1 gene was associated with an increased risk of diabetes [98]. Although the mechanism for the impairment of insulin secretion has not been elucidated, there is a possibility that IAPP aggregation is enhanced through ER stress due to an insufficient UPR caused by common variants of WFS1, increasing IAPP oligomers and, consequently, toxic IAPP oligomers in the ER (Fig. 4 and Table 1).
Disrupted protein homeostasis along with aging and the accumulation of IAPP oligomers
Aging and some aging-related diseases are linked to impaired protein homeostasis or proteostasis [99]. Proteostasis involves mechanisms for stabilization of correctly folded proteins and mechanisms for the degradation of proteins by the proteasome or the lysosome. There are regulators of age-related proteotoxicity [84] and these systems function in a coordinated fashion to restore the structure of misfolded polypeptides or to remove and degrade them, thus preventing the accumulation of damaged components and assuring the continuous renewal of intracellular proteins. Proteostasis is altered with aging and chronic expression of unfolded, misfolded, or aggregated proteins contributes to the development of some age-related pathologies, such as AD and Parkinson’s disease [99, 100]. It seems likely that along with the mechanisms for these neurodegenerative diseases, chronic expression of aggregated hIAPP contributes to the development of T2D under conditions of impaired proteostasis during aging (Figs. 4, 5 and Table 1). The activities of the autophagy–lysosomal system and the ubiquitin–proteasome system, the two principal proteolytic systems implicated in protein quality control, decline with aging [84, 101]. Decreased activities for stabilizing correctly folded proteins and degrading proteins by the proteasome or the lysosome, along with aging, might promote impaired protein homeostasis or proteostasis. Disrupted protein homeostasis or proteostasis might cause the accumulation of IAPP oligomers and consequently increase toxic IAPP oligomers, enhancing the toxic potential to the cell membranes of β-cells, as shown in Figs. 4, 5 and Table 1.
Fig. 5.
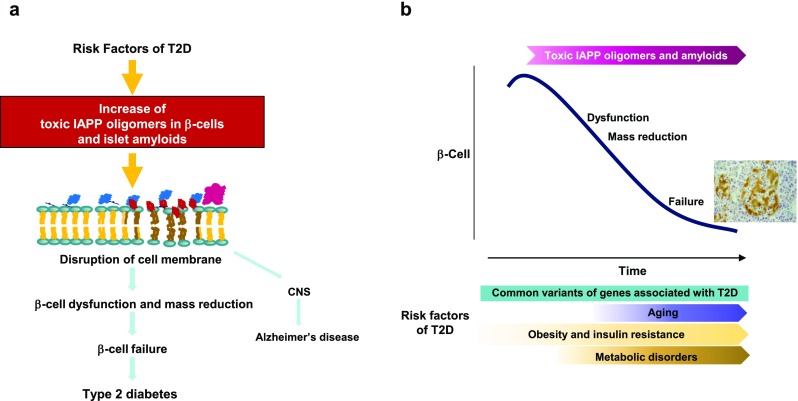
Increased toxic IAPP oligomers induce the disruption of cell membranes, resulting in β-cell failure. a Increased toxic IAPP oligomers induce the disruption of cell membranes, leading to β-cell dysfunction and mass reduction and eventually β-cell failure. IAPP molecules reach CNS, possibly influencing the exacerbation of Alzheimer’s disease. b Toxic IAPP oligomers in β-cells and islet amyloids progressively increase during aging under the influences of risk factors of T2D, inducing β-cell dysfunction and mass reduction, and result in β-cell failure
Disruption of ER homeostasis by hyperglycemia and hyperlipidemia, and accumulation of IAPP oligomers
Physiological fluctuations in the protein-folding load of the ER are a unique translational response of β-cells to variations in blood glucose and blood glucose level changes, leading to a maximum 25-fold increase in proinsulin synthesis. Thus, proinsulin synthesis imposes a heavy biosynthetic burden upon the ER [102]. There are three transmembrane ER-proximal sensors of unfolded proteins that initiate UPR signaling: inositol-requiring protein 1 (IRE1), activating transcription factor 6 (ATF6), and PKP-like ER kinase (PERK). In β-cells, the extracellular glucose level modulates the activity of the UPR sensors; chronically high-glucose concentrations stimulate proinsulin production, leading to the consequence that proinsulin synthesis overcomes the ER folding machinery. There remains some controversy whether chronically high-glucose exposure itself actually causes severe ER stress [103]. Saturated long-chain free fatty acids (FFAs) induce ER stress and thereby cause β-cell failure and cell death, whereas unsaturated long-chain FFAs induce this to a lesser extent [102]. Chronic hyperglycemia and hyperlipidemia, known as important causative factors of T2D, disrupt ER homeostasis to induce unresolvable UPR activation in the ER [103]. This condition enhances the probability of protein misfolding. High glucose and saturated FFAs interfere with ER function, which subsequently disrupts proinsulin synthesis, folding, and processing in the secretory pathway. Thus, it seems likely that during hyperglycemia and hyperlipidemia, misfolded IAPP and IAPP oligomers increasingly accumulate in the ER lumen in β-cells, resulting in an increase of toxic IAPP oligomers and enhancement of the toxic potential of hIAPP in the ER (Fig. 4 and Table 1). The hypothesis that risk factors for T2D, including the common variants of genes associated with T2D, obesity, insulin resistance, hyperglycemia, and hyperlipidemia, promote toxic IAPP oligomer formation in the ER is supported by histological evidence for immunoreactivity to toxic oligomers present in ER membranes in the perinuclear region and in the peripheral ER cisternae of hIAPP transgenic mice and human T2D β-cells [63].
Production of toxic IAPP oligomers in impaired secretory granules
Environmental conditions in β-cell granules, e.g., low pH, high concentrations of zinc ions, and insulin molecules, inhibit the formation of IAPP aggregates [79, 80]. β-Cell secretory granules are acidified through a proton gradient and are established and maintained by coordinated action between the H+-pumping vacuolar-type ATPase and ClC-3, a chloride ion channel [104, 105]. Also, WFS1 localizes not only to the ER but also to dense-core secretory granules in β-cells [106]. Intra-granular acidification is reduced in WFS1-deficient β-cells, indicating that WFS1 has the role of acidifying the secretory granules. WFS1 was included in a list of candidate genes evaluated for association with T2D [82, 83, 96]. It has been reported that allelic variations of the WFS1 gene act on pancreatic β-cells and reduce insulin secretion [97, 98]. Thus, there is a possibility that IAPP aggregation is enhanced due to insufficient acidification in the secretory granules, resulting in an increase of IAPP oligomers and toxic potentiation of hIAPP (Fig. 4 and Table 1). An earlier GWAS identified the SLC30A8 associated with increased T2D risks [107]. The gene encodes the zinc transporter, ZnT8, specific to β-cells [108] and a study using male ZnT8−/− mice showed that the transporter was required for the normal accumulation of zinc by β-cell granules, for normal insulin crystallization and insulin release, suggesting that the defects in carriers of Trp325Arg mutation of ZnT8 may increase T2D risks [109]. Interestingly, a recent GWAS reported that a loss-of-function mutation of SLC30A8 protected against diabetes [110]. Loss-of-function of ZnT8 and subsequent Zn2+ deficiency in β-cell granules shift the insulin-oligomer equilibrium toward insulin monomers and dimers, which bind IAPP monomers efficiently and prevent IAPP from self-association and aggregation, thus reducing T2D risk [80]. Because the function of ZnT8 and the concentration of Zn2+ depends on the variant of the SLC30A8 gene, the IAPP aggregation state could be influenced by the granule function in T2D subjects with the variant SLC30A8 gene (Fig. 4 and Table 1). We observed fibril-like materials in the secretory granules of hIAPP transgenic mice, in which glucose-stimulated insulin secretion from isolated islets was moderately reduced, but they did not develop amyloid deposits and diabetes [41, 42]. It was recently reported that toxic IAPP oligomers were present in secretory granules of hIAPP transgenic mice and human β-cells [63]. These findings suggest that production of toxic IAPP oligomers in the secretory granules is, in part, via altered granule function caused by allelic variations in the WFS1 gene and the SLC30A8 gene.
In subjects with risk factors for T2D, accumulation of toxic IAPP oligomers and disruption of cell membranes during aging
Again, the mechanism of IAPP oligomerization and amyloid formation is not fully understood due to limitations in the current methodology to characterize hIAPP species and also the mechanisms of cytotoxicity are not fully defined, despite extensive investigations. However, findings in studies using hIAPP transgenic rodents with backgrounds characterized for T2D phenotypes and studies on the metabolic and molecular disorders that are induced by common genetic variants and clinical risk factors for T2D suggest that, in subjects with risk factors of T2D, IAPP oligomers, toxic IAPP oligomers, and/or IAPP-derived islet amyloids progressively accumulate intracellularly in β-cells during aging and within islets, they impair β-cell function and reduce β-cell mass though multifactorial processes, possibly disrupting cell membranes and consequently altering the function of organelles, resulting in β-cell failure, as shown in Fig. 5a, b. It seems likely that IAPP is one of the peptides central to β-cell failure in T2D.
Studies on anti-amyloid aggregation therapeutics
Reducing IAPP molecule-related intermediates and the islet amyloid burden to improve β-cell survival has been the focus of a number of studies using different approaches. One approach is to discover natural products and small molecule inhibitors. Inhibition of IAPP aggregation was caused by the stabilization of small molecular weight IAPP oligomers by the polyphenols, curcumin and resveratrol, offering a general anti-aggregation mechanism for polyphenols and providing a computational framework for the future design of anti-amyloid aggregation therapeutics [111]. Epigallocatechin gallate (EGCG), a green tea extract, inhibits hIAPP aggregation by binding to hIAPP dimers, strongly blocking both the inter-peptide hydrophobic and aromatic-stacking interactions responsible for inter-peptide β-sheet formation and intra-peptide interactions crucial for β-hairpin, an amyloidogenic precursor, formation [112]. A water-miscible tetraphenylethene, BSPOTPE, was able to inhibit hIAPP-induced membrane damage by binding directly to the helical regions of hIAPP and forming oligomers with separated hydrophobic cores and hydrophilic shells, preventing membrane poration [55]. A novel tetraquinolin amide foldamer, ADM-166, has been developed to counter conformational transitions in IAPP [113]. ADM-166 is capable to cross the plasma membrane without assistance from cellular processes and antagonize toxicity long after uptake of IAPP, resulting in specific docking with intracellular IAPP, rescuing β-cells. The IAPP inhibitory peptide, D-ANFLVH, was shown to prevent islet amyloid accumulation in cultured human islets and this peptide, when administered by intraperitoneal injection into hIAPP transgenic mice, was a potent inhibitor of islet amyloid deposition, resulting in reduced islet apoptosis and preservation of β-cells, leading to improved glucose tolerance [114, 115]. A second approach is to exploit enzymes that are capable of degrading hIAPP. Up-regulation of neprilysin in hIAPP transgenic mouse islets reduced amyloid deposition and β-cell apoptosis [116]. Inhibition of insulin-degrading enzyme (IDE) resulted in increased amyloidogenesis in β-cell lines and reduced cell viability when cells were incubated with synthetic hIAPP, suggesting a role for IDE in IAPP clearance and the prevention of IAPP aggregation [117]. Matrix metalloproteinases (MMPs) are proteolytic enzymes and MMP-9 is active in islets and cleaves hIAPP [118]. Adenoviral overexpression of MMP-9 in amyloid-prone islets reduced amyloid deposition and β-cell apoptosis, suggesting that increasing islet MMP-9 activity might be a strategy to limit β-cell loss in T2D [119]. In a third approach, antibodies hold significant potential for inhibiting toxic protein aggregation associated with conformational disorders, such as AD and Huntington’s diseases [120, 121]. Grafting small amyloidogenic peptides into the complementarity-determining regions of a single-domain antibody yields potent domain antibody inhibitors of amyloid formation, and the antibody inhibitors presenting hydrophobic peptides of hIAPP inhibit fibril assembly of the peptide at low sub-stoichiometric concentrations [122]. Antibodies which specifically recognized hIAPP assemblies, but not monomers or amyloid fibrils, were exclusively identified in diabetic patients and were shown to neutralize the apoptotic effect induced by these oligomers, suggesting that hIAPP peptides can form highly toxic oligomers [37]. The newly identified structural epitopes may also provide new mechanistic insights and a molecular target for future therapy [37]. These antibody design approaches also can be extended to generate potent aggregation inhibitors of amyloidogenic polypeptides linked to human disease. These interesting studies provide a framework for the future design of anti-amyloid aggregation therapeutics and hold promise that drugs that protect islets from toxic IAPP oligomers and amyloids will be developed in the near future.
IAPP in neurodegenerative disorders
The most common form of aging-associated dementia is late-onset AD, a neurodegenerative disorder affecting multiple cognitive and behavioral functions [123]. Epidemiological studies have shown that patients with T2D are two to five times more likely to develop AD compared to non-diabetic individuals [124, 125]. A recent cohort study suggests that diabetes is a significant risk factor for all causes of dementia, AD, and probably vascular dementia. Two-hour plasma glucose (PG) levels, but not fasting PG levels, are closely associated with increased risk of all causes of dementia [126]. A review article describes that IAPP may constitute a “second amyloid” in neurodegenerative disorders, such as AD, and that hyperamylinemic conditions may be more relevant for the early processes of amyloid formation in the central nervous system (CNS), whereas hypoamylinemic conditions may be more strongly associated with late stages of central amyloid pathologies [127]. Hypersecretion of IAPP is observed in obese and diabetic rodent models and individuals with obesity and pre-diabetic insulin resistance [27–29, 88, 89]. Peripherally produced IAPP readily crosses the blood–brain barrier [128]. Increased peripheral secretion and central uptake of IAPP may facilitate IAPP oligomerization and eventually amyloid formation in the brain, disrupting the integrity of CNS structures and functions. The IAPP receptor is widely distributed within the CNS and IAPP and Aβ directly activate this G-protein-coupled receptor and trigger multiple common intracellular signal transduction pathways, inducing apoptotic cell death [129]. Hypoamylinemic conditions at late stages of central amyloid pathologies may increase direct activation of Aβ on this G-protein-coupled receptor. A recent study reported a genetic variant within the IAPP/SLCO1A2 genes that modified the effect of cortical Aβ deposition on AD-related cognitive impairment and temporal lobe atrophy, supporting the growing literature on the role of IAPP in AD pathophysiology [130]. Epidemiological studies have established a clear association between T2D and AD. Thus, IAPP might be an important player at the interface between T2D and AD (Fig. 5a).
Conclusions
Progressive loss of β-cell function is central to the development and progression of T2D. The common genetic variants, suggested by GWAS as associated with T2D and the clinical risk factors of diabetes, including aging, obesity, insulin resistance, and metabolic disorders, are associated with reduced β-cell function and mass and implicated in progression to β-cell failure. A single molecule, including an IAPP molecule, could have risk effects inducing multifactorial processes via a feed-forward loop [5]. The metabolic and molecular disorders which are induced by risk factors of T2D might promote the accumulation of soluble IAPP-related molecules and toxic IAPP oligomers in β-cells, impairing function and reducing the mass though disruption of cell membranes, consequently resulting in β-cell failure. IAPP might be one of the peptides central to β-cell failure in T2D. Despite considerable progress, there are inconclusive matters, including 1) the actual mechanism of hIAPP toxicity to β-cells, especially the nature of the toxic species and the initiation site(s) of β-cell toxicity, 2) a substantiated mechanism for the increase of toxic IAPP species in T2D individuals, and 3) the development of effective clinically relevant inhibitors.
A variety of interventions, including weight loss, some oral hypoglycemic drugs, insulin preparations, and GLP-1 receptor agonists, can improve β-cell function temporarily with improved glucose control. However, existing therapy does not arrest progression of β-cell dysfunction in T2D [5]. Anti-IAPP aggregation therapeutics, which protect islets from toxic IAPP oligomers and islet amyloids, will be developed as treatments with more durable beneficial effects on β-cell function.
Acknowledgements
The authors thank the doctors and technicians in the Diabetes Research Group, the Second Department of Internal Medicine, Chiba University School of Medicine, Chiba, Japan.
Compliance with ethical standards
Conflict of interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
References
- 1.UK Prospective Diabetes Study (UKPDS) Group UK prospective diabetes study 16: overview of 6 years’ therapy of type II diabetes: a progressive disease. Diabetes. 1995;44:1249–1258. doi: 10.2337/diab.44.11.1249. [DOI] [PubMed] [Google Scholar]
- 2.Levy J, Atkinson AB, Bell PM, et al. Beta-cell deterioration determines the onset and rate of progression of secondary dietary failure in type 2 diabetes mellitus: the 10-year follow-up of the Belfast Diet Study. Diabet Med. 1998;15:290–296. doi: 10.1002/(SICI)1096-9136(199804)15:4<290::AID-DIA570>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 3.Turner RC, Cull CA, Frighi V, et al. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49) JAMA. 1999;281:2005–2012. doi: 10.1001/jama.281.21.2005. [DOI] [PubMed] [Google Scholar]
- 4.Kahn SE, Haffner SM, Heise ME, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–2443. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- 5.Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, Powers AC, Rhodes CJ, Sussel L, Weir GC. β-Cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. J Clin Endocrinol Metab. 2014;99(6):1983–1992. doi: 10.1210/jc.2014-1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Opie E. The relation of diabetes mellitus to lesion of the pancreas. Hyaline degeneration of the islands of Langerhans. J Exp Med. 1901;5:527–540. doi: 10.1084/jem.5.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark A, Saad MP, Nezzer T, Uren C, Knowler WC, Bennelt PH, et al. Islet amyloid polypeptide in diabetic and non-diabetic Pima Indians. Diabetologia. 1990;33:285–289. doi: 10.1007/BF00403322. [DOI] [PubMed] [Google Scholar]
- 8.Westermark P, Wernstedt C, Wilander E, Hayden DW, O’Brien TD, Johnson KH. Amyloid fibrils in human insulinoma and islets Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci USA. 1987;84:3881–3885. doi: 10.1073/pnas.84.11.3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper GJS, Willis AC, Clark A, Turner RC, Sim RB, Reid KBM. Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci USA. 1987;84:8628–8632. doi: 10.1073/pnas.84.23.8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohsawa H, Kanatsuka A, Mizuno Y, Tokuyama Y, Takada K, Mikata A, Makino H, Yoshida S. Islet amyloid polypeptide-derived amyloid deposition increases along with the duration of type 2 diabetes mellitus. Diabetes Res Clin Pract. 1992;15:17–22. doi: 10.1016/0168-8227(92)90062-V. [DOI] [PubMed] [Google Scholar]
- 11.Kamata K, Mizukami H, Inaba W, Tsuboi K, Tateishi Y, Yoshida T, Yagihashi S. Islet amyloid with macrophage migration correlates with augmented β-cell deficits in type 2 diabetic patients. Amyloid. 2014;21:191–201. doi: 10.3109/13506129.2014.937857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tycko R. Amyloid polymorphism: structural basis and neurobiological relevance. Cell Neuron. 2015;86:632–645. doi: 10.1016/j.neuron.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosselman S, Hoppener JWM, Lips CJM, Jansz HS. The complete islet amyloid polypeptide precursor is encoded by two exons. FEBS Lett. 1989;247:154–158. doi: 10.1016/0014-5793(89)81260-8. [DOI] [PubMed] [Google Scholar]
- 14.Sanke T, Bell GI, Sample C, Rubenstein AH, Steiner DF. An islet amyloid peptide is derived from an 89-amino acid precursor by proteolytic processing. J Biol Chem. 1988;263:17243–17246. [PubMed] [Google Scholar]
- 15.Nishi M, Sanke T, Nagamatsu S, Bell GI. Islet amyloid polypeptide A new β cell secretory product related to islet amyloid deposits. J Biol Chem. 1990;265:4173–4176. [PubMed] [Google Scholar]
- 16.Nishi M, Chan SJ, Nagamatsu S, Bell GI, Steiner DF. Conservation of the sequence of islet amyloid polypeptide in five mammals is consistent with its putative role as an islet hormone. Proc Natl Acad Sci USA. 1989;86:5738–5742. doi: 10.1073/pnas.86.15.5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Betsholtz C, Christmanson L, EngstromU Rorsman F, Jordan K, O’Brien TD, et al. Structure of cat islet amyloid polypeptide and identification of amino acid residues of potential significance for islet amyloid formation. Diabetes. 1990;39:118–122. doi: 10.2337/diacare.39.1.118. [DOI] [PubMed] [Google Scholar]
- 18.Kanatsuka A, Makino H, Ohsawa H, Tokuyama Y, Yamaguchi Y, Yoshida H, Adachi M. Secretion of islet amyloid polypeptide in response to glucose. FEBS Lett. 1989;259:199–201. doi: 10.1016/0014-5793(89)81527-3. [DOI] [PubMed] [Google Scholar]
- 19.Ogawa A, Harris V, McCorkle SK, Unger RH, Luskey KL. Amylin secretion from the rat pancreas and its selective loss after streptozotocin treatment. J Clin Invest. 1990;85:973–976. doi: 10.1172/JCI114528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Badman MK, Shenen KI, Jermany JL, Dochewrty K, Clark A. Processing of pro-islet amyloid polypeptide (proIAPP) by the prohormone convertase PC2. FEBS Lett. 1996;378:227–231. doi: 10.1016/0014-5793(95)01460-8. [DOI] [PubMed] [Google Scholar]
- 21.Westermark P, Li ZC, Westermark GT, Leckstrom A, Steiner DF. Effect of beta cell granule components on human islet amyloid polypeptide fibril formation. FEBS Lett. 1996;379:203–206. doi: 10.1016/0014-5793(95)01512-4. [DOI] [PubMed] [Google Scholar]
- 22.Miyazaki J, Araki K, Yamamoto E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic B-cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- 23.Kanatsuka A, Makino H, Yamaguchi T, Ohsawa H, Tokuyama Y, Saitoh T, Yamamura K, Miyazaki J, Yoshida S. Islet amyloid polypeptide/amylin in pancreatic β-cell line derived from transgenic mouse insulinoma. Diabetes. 1992;41:1409–1414. doi: 10.2337/diab.41.11.1409. [DOI] [PubMed] [Google Scholar]
- 24.Itoh N, Okamoto H. Translational control of proinsulin synthesis by glucose. Nature (Lond) 1980;283:100–102. doi: 10.1038/283100a0. [DOI] [PubMed] [Google Scholar]
- 25.Welsh M, Nielsen DA, MacKrell AJ, Steiner DF. Control of insulin gene expression in pancreatic B-cells and in an insulin-producing cell line, RIN-5F cells. J Biol Chem. 1985;260:13590–13594. [PubMed] [Google Scholar]
- 26.Hammonds P, Schofield PN, Stephan JH, Ashcroft SJH. Glucose regulates preproinsulin messenger RNA levels in a clonal cell line of simian virus 40-transformed B cells. FEBS Lett. 1987;213:149–154. doi: 10.1016/0014-5793(87)81481-3. [DOI] [PubMed] [Google Scholar]
- 27.Tokuyama Y, Kanatsuka A, Ohsawa H, Yamaguchi T, Makino H, Yoshida S, Nagase H, Inoue S. Hypersecretion of islet amyloid polypeptide from pancreatic islets ventromedial hypothalamic-lesioned rats and obese Zucker rats. Endocrinology. 1991;128:2739–2744. doi: 10.1210/endo-128-6-2739. [DOI] [PubMed] [Google Scholar]
- 28.Tokuyama Y, Kanatsuka A, Yamaguchi T, Ohsawa H, Makino H, Nishimura M, Yoshida S. Islet amyloid polypeptide/amylin in pancreata increase in genetically obese and diabetic mice. Horm Metab Res. 1993;25:289–291. doi: 10.1055/s-2007-1002100. [DOI] [PubMed] [Google Scholar]
- 29.Takada K, Kanatsuka A, Tokuyama Y, Yagui K, Nishimura, Saito Y, Makino H. Islet amyloid polypeptide/amylin contents in pancreas change with increasing age in genetically obese and diabetic mice. Diabetes Res Clin Pract. 1996;33:153–158. doi: 10.1016/0168-8227(96)01294-6. [DOI] [PubMed] [Google Scholar]
- 30.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 31.Ohsawa H, Kanatsuka A, Yamaguchi T, Makino H, Yoshida S. Islet amyloid polypeptide inhibits glucose-stimulated insulin secretion from isolated rat pancreatic islets. Biochem Biophys Res Commun. 1989;160:961–967. doi: 10.1016/0006-291X(89)92529-1. [DOI] [PubMed] [Google Scholar]
- 32.Degano P, Silvestre RA, Salas M, Peiro E. Amylin inhibits glucose-induced insulin secretion in a dose-dependent manner. Study in the perfused rat pancreas. Regul Pept. 1993;43:91–96. doi: 10.1016/0167-0115(93)90411-Z. [DOI] [PubMed] [Google Scholar]
- 33.Sandler S, Stridsberg M. Chronic exposure of cultured rat pancreatic islets to elevated concentrations of islet amyloid polypeptide (IAPP) causes a decrease in islet DNA content and medium insulin accumulation. Regul Pept. 1994;3:103–109. doi: 10.1016/0167-0115(94)90611-4. [DOI] [PubMed] [Google Scholar]
- 34.Broderick CL, Brooke CS, DiMarchi RD, Gold G. Human and rat amylin have no effect on insulin secretion in isolated rat pancreatic islets. Biochem Biophys Res Commun. 1991;177:932–938. doi: 10.1016/0006-291X(91)90628-K. [DOI] [PubMed] [Google Scholar]
- 35.O’Brien TD, Westermark P, Johnson KH. Islet amyloid polypeptide (IAPP) does not inhibit glucose-stimulated insulin secretion from isolated perfused rat pancreas. Biochem Biophys Res Commun. 1990;170:1223–1228. doi: 10.1016/0006-291X(90)90524-Q. [DOI] [PubMed] [Google Scholar]
- 36.Brethorton-Watt D, Gilbey SG, Ghatei MA, Beacham J, Macrae AD. Very high concentrations of islet amyloid polypeptide are necessary to alter the insulin response to intravenous glucose in man. J Clin Endocrinol Metab. 1992;74:1032–1035. doi: 10.1210/jcem.74.5.1569151. [DOI] [PubMed] [Google Scholar]
- 37.Bram Y, Frydman-Maram A, Yanai I, Gilead S, Shaltiel-Karyo R, Amdulsky N, Gazit E. Apoptosis induced by islet amyloid polypeptide soluble oligomers is neutralized by diabetes-associated specific antibodies. Sci Rep. 2014;4:4267. doi: 10.1038/srep04267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matveyenko AV, Butler PC. Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J. 2006;47:225–233. doi: 10.1093/ilar.47.3.225. [DOI] [PubMed] [Google Scholar]
- 39.Fox N, Schrementi J, Nishi M, Ohgi S, Chan SJ, Heisserman JA, Westermark GT, Leckstrom A, Westermark P, Steiner DF. Human islet amyloid polypeptide transgenic mice as a model of non-insulin-dependent diabetes mellitus (NIDDM) FEBS Lett. 1993;323:40–44. doi: 10.1016/0014-5793(93)81444-5. [DOI] [PubMed] [Google Scholar]
- 40.Koning EJP, Hoppener JWM, Verbeek JS, Oosterwijk C, Hulst KL, Baker CA, Lips CJM, Morris JF, Clark A. Human islet amyloid polypeptide accumulates at similar sites in islets of transgenic mice and humans. Diabetes. 1994;43:640–644. doi: 10.2337/diab.43.5.640. [DOI] [PubMed] [Google Scholar]
- 41.Yagui K, Yamaguchi T, Kanatsuka A, Shimada F, Huang CI, Tokuyama Y, Ohsawa H, Tamamura K, Miyazaki K, Mikata A, Yoshida S, Makino H. Formation of islet amyloid fibrils in beta-secretory granules of transgenic mice expressing human islet amyloid polypeptide/amylin. Eur J Endocrinol. 1995;132:487–496. doi: 10.1530/eje.0.1320487. [DOI] [PubMed] [Google Scholar]
- 42.Tokuyama T, Yagui K, Yamaguchi T, Huang CI, Kuramoto N, Shimada F, Miyazaki J, Horie H, Saito Y, Makino H, Kanatsuka A. Expression of human islet amyloid polypeptide/amylin impairs insulin secretion in mouse pancreatic β cells. Metabolism. 1997;46:1044–1051. doi: 10.1016/S0026-0495(97)90276-6. [DOI] [PubMed] [Google Scholar]
- 43.Janson J, Soeller WC, Roche PC, Nelsoin RT, Torchia AJ, Kreuter DK, Butler PC. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci USA. 1996;93:7283–7288. doi: 10.1073/pnas.93.14.7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snowdon DA. Aging and Alzheimer’s disease: lessons from the Nun study. Gerontology. 1997;37:150–156. doi: 10.1093/geront/37.2.150. [DOI] [PubMed] [Google Scholar]
- 45.Dahlgrent KN, Manelli AM, Stine MW, Baker LK, Krafft GA, LadU MJ. Oligomeric and fibrillary species of amyloid-β peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 46.Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 47.Kayed R, Head E, Thompson JL, Mclntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 48.Kayed R, Bernhagen J, Greenfield N, Sweimeh K, Brunner H, Voelter W, Kaspumiotu A. Conformational transitions of islet amyloid polypeptide (IAPP) in amyloid formation in vitro. J Mol Biol. 1999;287:781–796. doi: 10.1006/jmbi.1999.2646. [DOI] [PubMed] [Google Scholar]
- 49.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J Mol Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 50.Willamson JA, Miranker AD. Direct detection of transient α-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brender J, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the Type-2-diabetes-related peptide. J Am Chem Soc. 2008;21:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Apostolidou M, Jayasinghe SA, Langen R. Structure of & α-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J Biol Chem. 2008;283:17205–17210. doi: 10.1074/jbc.M801383200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patil AM, Xu S, Sheftic SR, Alexanderescu AT. Dynamic & α-helix structure of micelle-bound human amylin. J Biol Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pannuzzo M, Raudino A, Milardi D, Rosa CL, Kattunen M. α-Helical structure drive early stages of self-assembly of amyloidogenic amyloid polypeptide aggregate formation in membranes. Sci Rep. 2013;3:2781. doi: 10.1038/srep02781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X, Wan M, Gao L, Fang W. Mechanism of inhibition of human islet amyloid polypeptide-induced membrane damage by small organic fluorogen. Sci Rep. 2016;6:21614. doi: 10.1038/srep21614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zraika S, Hull RL, Verchere CB, Clark A, Potter KJ, Fraser PE, Raleigh DP, Kahn SE. Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence? Diabetologia. 2010;53:1046–1056. doi: 10.1007/s00125-010-1671-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cao P, Marek P, Noor H, Patsalo V, Tu L-H, Wang H, Abedini A, Raleigh DP. Islet amyloid: from fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013;587:1106–1118. doi: 10.1016/j.febslet.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haataja L, Gurlo T, Huang CJ, Peter C, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocrinol Rev. 2008;29:303–316. doi: 10.1210/er.2007-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pithadia A, Brender JR, Fierke CA, Ramamoorthy A. Inhibition of IAPP aggregation and toxicity by natural products and derivatives. J Diabetes Res. 2016;2016:12. doi: 10.1155/2016/2046327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pilkington EH, Gurzov EN, Kakinen A, Litwak SA, Stanley WJ, Davis TP, Ke PC. Pancreatic β-cell membrane fluidity and toxicity induced by human islet amyloid species. Sci Rep. 2016;6:21274. doi: 10.1038/srep21274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schlamadinger DE, Miranker AD. Fiber-dependent and -independent toxicity of islet amyloid polypeptide. Biophys J. 2014;107:2559–2566. doi: 10.1016/j.bpj.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu C, Zhao M, Jiang L, Cheng P-N, Park J, Sawaya MR, Pensalfini A, Gou W, Berk AJ, Glabe CG, Nowic J, Eiseberg D. Out-of-register β-sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci USA. 2012;109:20913–20918. doi: 10.1073/pnas.1218792109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gurio T, Ryazantsev S, Huang C-J, Yeh MW, Reber HA, Hines OJ, O’Brien TD, Glabe CG, Butler PC. Evidence for proteotoxicity in β-cells in type 2 diabetes. Toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol. 2010;176:861–869. doi: 10.2353/ajpath.2010.090532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Casas S, Gomis R, Gribble FM, Altirriba J, Knuutila S, Novials A. Impairment of the ubiquitin–proteasome pathway is a downstream endoplasmic reticulum stress response induced by extracellular human islet polypeptide and contributes to pancreatic β-cell apoptosis. Diabetes. 2007;56:2284–2294. doi: 10.2337/db07-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morita S, Sakagashira S, Shimajin Y, Eberhardt N, Kondo T, Kondo T, Sanke T. Autophagy protects against human islet amyloid polypeptide-associated apoptosis. J Diabetes Invest. 2011;2:48–55. doi: 10.1111/j.2040-1124.2010.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannanhill GM, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Couce M, Kane LA, O’Brien TD, Charlesworth J, Soeller W, McNeish J, Kreutter D, Roche P, Bulter PC. Treatment with growth hormone and dexamethasone in mice transgenic for human islet amyloid polypeptide causes islet amyloidosis and β-cell dysfunction. Diabetes. 1996;45:1094–1101. doi: 10.2337/diab.45.8.1094. [DOI] [PubMed] [Google Scholar]
- 68.Lindstroem P. The physiology of obese-hyperglycemic mice (ob/ob mice) Sci World J. 2007;7:666–685. doi: 10.1100/tsw.2007.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoppener JWM, Oosterwijk C, Nieuwenhuis MG, Posthuma G, Thijssen JHH, Vroom TM, Ahren B, Lips CGM. Extensive islet amyloid formation is induced by development of type II diabetes mellitus and contributes to its progression: pathogenesis of diabetes in a mouse model. Diabetologia. 1999;42:427–434. doi: 10.1007/s001250051175. [DOI] [PubMed] [Google Scholar]
- 70.Soeller WC, Janson J, Hart SE, Parker JC, Carty MD, Stevenson RW, Kreutter DK, Butler PC. Islet amyloid-associated diabetes in obese Avy/a mice expressing human islet amyloid polypeptide. Diabetes. 1998;47:743–750. doi: 10.2337/diabetes.47.5.743. [DOI] [PubMed] [Google Scholar]
- 71.Butler AE, Janson J, Soeler WC, Butler PC. Increased & β-cell apoptosis prevents adaptive increase in β-cell mass in mouse model of type 2 diabetes. Evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52:2304–2314. doi: 10.2337/diabetes.52.9.2304. [DOI] [PubMed] [Google Scholar]
- 72.Shigihara N, Fukunaka A, Hara A, Komiya K, Honda A, Uchida T, Abe H, Toyofuku Y, Tamaki M, Ogihara T, Miyatsuka T, Hiddinga H, Sakagashira S, Koike M, Uchiyama Y, Yoshimori T, Eberhaldt NL, Fujitani Y, Watada H. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J Clin Invest. 2014;124:3634–3644. doi: 10.1172/JCI69866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wijesekara N, Kaur A, Westwell-Roper C, Nackiewiecz D, Soukhatcheva G, Hayden MR, Verchere CB. ABCA1 deficiency and cellular cholesterol accumulation increases islet amyloidogenesis in mice. Diabetologia. 2016;59:1242–1246. doi: 10.1007/s00125-016-3907-6. [DOI] [PubMed] [Google Scholar]
- 74.Papa FR. Endoplasmic reticulum stress, pancreatic β-cell degeneration, and diabetes. Cold Spring Harb Perspect Med. 2012;2:a007666. doi: 10.1101/cshperspect.a007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCracken AA, Brodsky JL. Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD) BioEssays. 2003;25:868. doi: 10.1002/bies.10320. [DOI] [PubMed] [Google Scholar]
- 76.Yorimitsu T, Klionsky DJ. Eating the endoplasmic reticulum: quality control by autophagy. Trends Cell Biol. 2007;17:279–285. doi: 10.1016/j.tcb.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 77.Padrick SB, MiraNKER AD. Islet amyloid: phase partitioning and secondary nucleation are central mechanism of fibrillogenesis. Biochemistry. 2002;41:4694–4703. doi: 10.1021/bi0160462. [DOI] [PubMed] [Google Scholar]
- 78.Hutton CG. The insulin secretory granule. Diabetologia. 1989;32:271–281. doi: 10.1007/BF00265542. [DOI] [PubMed] [Google Scholar]
- 79.Jha S, et al. pH dependence of amylin fibrillization. Biochemistry. 2014;53:300–310. doi: 10.1021/bi401164k. [DOI] [PubMed] [Google Scholar]
- 80.Nedumpully-Govidan P, Ding F. Inhibition of IAPP aggregation by insulin depends on the insulin oligomeric state regulated by zinc ion concentration. Sci Rep. 2015;5:8240. doi: 10.1038/srep08240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Isaksson B, Wang F, Permert J, Olsson M, Furin B, Herrington MK. Chronically administered islet amyloid polypeptide in rat serves as an adiposity inhibitor and energy homeostasis. Pancreatology. 2005;5:29–36. doi: 10.1159/000084488. [DOI] [PubMed] [Google Scholar]
- 82.Petretto E, Liu ET, Aitman TJ. A gene harvest revealing the archeology and complexity of human diseases. Nat Genet. 2007;39:1299–1301. doi: 10.1038/ng1107-1299. [DOI] [PubMed] [Google Scholar]
- 83.Sanghera DK, Blackett PR. Type 2 diabetes genetics: beyond GWAS. J Diabetes Metab. 2012;3:1–12. doi: 10.4172/2155-6156.1000198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ford ES, Li C, Sattar N. Metabolic syndrome and incident diabetes: current state of the evidence. Diabetes Care. 2008;61:1898–1904. doi: 10.2337/dc08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kruit JK, Brunham LR, Verchere CB, Hayden MR. HDL and LDL cholesterol significantly influence beta-cell function in type 2 diabetes mellitus. Curr Opin Lipidol. 2010;21:178–185. doi: 10.1097/MOL.0b013e328339387b. [DOI] [PubMed] [Google Scholar]
- 87.Robertson RP, Harmon J, Tran PO, Tanaka Y, Takahashi H. Glucose toxicity in β-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes. 2003;52:581–587. doi: 10.2337/diabetes.52.3.581. [DOI] [PubMed] [Google Scholar]
- 88.Johnson KH, O’Brien TD, Jordan K, Westermark P. Impaired glucose tolerance is associated with increased islet amyloid polypeptide (IAPP) immunoreactivity in pancreatic beta cells. Am J Pathol. 1989;135:245–250. [PMC free article] [PubMed] [Google Scholar]
- 89.Enoki S, Mitsukawa T, Takemura J, Nakazato M, Aburaya J, Toshomori H. Plasma islet amyloid polypeptide levels in obesity, impaired glucose tolerance and non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract. 1992;15:97–102. doi: 10.1016/0168-8227(92)90074-2. [DOI] [PubMed] [Google Scholar]
- 90.Frailing TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zeggin E, Weedon MN, Lindgren CM, Frailing TM, Elliott KS, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barrett TG, Bundey SE. Wolfram (DIDMOAD) syndrome. J Med Genet. 1997;34:838–841. doi: 10.1136/jmg.34.10.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Takeda K, Inoue H, Tanizawa Y, Matsuzaki Y, Oba J, Watanabew Y, Shinoda K, Oka Y. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neural expression in rat brain. Hum Mol Genet. 2001;10:477–484. doi: 10.1093/hmg/10.5.477. [DOI] [PubMed] [Google Scholar]
- 94.Hofman S, Philbrook C, Gerbitz K-D, Bauer MF. Wolfram syndrome: structural and functional analyses of mutant and wild-type wolframin, the WFS1 gene product. Hum Mol Genet. 2003;12:2003–2012. doi: 10.1093/hmg/ddg214. [DOI] [PubMed] [Google Scholar]
- 95.Fonseka SG, Fukuma M, Lipson KL, Nguyen LX, Allen JR, Oka Y. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the ER in pancreatic β-cells. J Biol Chem. 2005;280:39609–39615. doi: 10.1074/jbc.M507426200. [DOI] [PubMed] [Google Scholar]
- 96.Sandhu MS, Weedon MN, Fawsett KA, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet. 2007;39:951–953. doi: 10.1038/ng2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Florez JC, Jablonski KA, MacAteer J, Sandhu MS, Warenham NJ, Barroso I, Franks PW, Altshuler D, Knowler WC. for the Diabetes Prevention Program Research Group. Testing of diabetes-associated WFS1 polymorphisms in the Diabetes Prevention Program. Diabetologia. 2008;51:451–457. doi: 10.1007/s00125-007-0891-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheurfa N, Brenner GM, Reis AF, Dubois-Laforgue D, Roussel R, Tichet J, Lantieri O, Balkau B, Fumeron F, Timsit J, Marre M, Velho G. Decreased insulin secretion and increased risk of type 2 diabetes associated with allelic variations of the WFS1 gene: the Data from Epidemiological Study on the insulin Resistance Syndrome (DESIR) prospective study. Diabetologia. 2011;54:554–562. doi: 10.1007/s00125-010-1989-0. [DOI] [PubMed] [Google Scholar]
- 99.Powers ET, Morimot RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 100.Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: the importance of exquisite quality control. Ageing Res Rev. 2011;10:205–215. doi: 10.1016/j.arr.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tomaru U, Takahashi S, Ishizu A, Miyatake Y, Gohda A, Suzuki S, Ono A, Ohara J, Baba T, Murata S, et al. Decreased proteasomal activity causes age-related phenotypes and promotes the development of metabolic abnormalities. Am J Pathol. 2012;180:963–972. doi: 10.1016/j.ajpath.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 102.Cnop M, Ladriere L, Igoillo-Esteve M, Moura RF, Cunha DA. Causes and cures for endoplasmic reticulum stress in lipotoxic beta-cell dysfunction. Diabetes Obes Metab. 2010;12(Suppl 2):76–82. doi: 10.1111/j.1463-1326.2010.01279.x. [DOI] [PubMed] [Google Scholar]
- 103.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–793. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schoonderwoert VT, Martens GJ. Proton pumping in the secretory pathway. J Membr Biol. 2001;182:159–169. doi: 10.1007/s00232-001-0040-2. [DOI] [PubMed] [Google Scholar]
- 105.Barg S, Huang P, Eliasson L, Nelson DJ, Obermuller S, Rorsman P, Thevenot F, Renstrom E. Priming insulin granules for exocytosis by granule Cl(−) uptake and acidification. J Cell Sci. 2001;114:2145–2154. doi: 10.1242/jcs.114.11.2145. [DOI] [PubMed] [Google Scholar]
- 106.Hatanaka M, Tanabe K, Yanai A, Ohta Y, Kondo M, Akiyama M, Shinoda K, Oka Y, Tanizawa Y. Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic β-cells. Hum Mol Genet. 2011;20:1274–1284. doi: 10.1093/hmg/ddq568. [DOI] [PubMed] [Google Scholar]
- 107.Sladek S, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 108.Chimienti F, Devergnas S, Favier A, Seve M. Identification and cloning of a β-cell–specific zinc transporter, ZnT-8, localized into insulin secretory granules. Diabetes. 2004;4:2330–2336. doi: 10.2337/diabetes.53.9.2330. [DOI] [PubMed] [Google Scholar]
- 109.Nicolson TJ, Bellomo EA, Wijesekara N, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes. 2009;58:2070–2082. doi: 10.2337/db09-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Flannic K, et al. Loss-of-function mutation in SCL30A8 protect against type 2 diabetes. Nat Genet. 2014;46:357–363. doi: 10.1038/ng.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nudumpully-Govinadan P, Kakinen A, Pilkington EH, Davis TP, Ke PC, Ding F. Stabilizing off-pathway oligomers by polyphenol nanoassemblies for IAPP aggregation inhibition. Sci Rep. 2016;6:19463. doi: 10.1038/srep19463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mo Y, Lei J, Sun Y, Zhang Q, Wei G. Conformational ensemble of hIAPP dimer: insight into a green tea extract inhibits hIAPP aggregation. Sci Rep. 2016;6:33076. doi: 10.1038/srep33076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kumar S, Birol M, Schlamadinger DE, Wojcik SP, Rhoades E, Miranker AD. Foldamer-mediated manipulation of a pre-amyloid toxin. Nat Commun. 2016;7:11412. doi: 10.1038/ncomms11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Potter KJ, Scrocchi LA, Warnock GL, et al. Amyloid inhibitors enhance survival of cultured human islets. Biochem Biophys Acta. 2009;1790:566–574. doi: 10.1016/j.bbagen.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 115.Wijesekara N, Ahren R, Wu L, Ha K, Liu Y, Wheeler MB, Fraser PE. Islet amyloid inhibitors improve glucose homeostasis in a transgenic mouse model of type 2 diabetes. Diabetes Obes Metab. 2015;17:1003–1006. doi: 10.1111/dom.12529. [DOI] [PubMed] [Google Scholar]
- 116.Guan H, Chow KM, Shah R, Rhodes CJ, Hersh LB. Degradation of islet amyloid polypeptide by neprilysin. Diabetologia. 2012;55:2989–2998. doi: 10.1007/s00125-012-2678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bennet RG, Hamel FG, Duckworth WC. An insulin-degrading enzyme inhibitor decreases amylin degradation, increases amyloid-induced cytotoxicity, and increases amyloid formation in insulinoma cell culture. Diabetes. 2003;52:2315–2320. doi: 10.2337/diabetes.52.9.2315. [DOI] [PubMed] [Google Scholar]
- 118.Aston-Mourney K, Zraika S, Udayasakar J, Subramanian SL, Green PS, Kahn SE, Hull RL. Matrix metalloproteinase-9 reduces islet amyloid formation by islet amyloid polypeptide. J Biol Chem. 2013;2013(288):3553–3559. doi: 10.1074/jbc.M112.438457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Meier DT, Tu L-H, Zraika S, Hogan MF, Templin AT, Hull RL, Raleigh DP, Kahn SE. Matrix metalloproteinase-9 protects islet from amyloid-induced toxicity. J Biol Chem. 2015;290:30475–30485. doi: 10.1074/jbc.M115.676692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc Natl Acad Sci USA. 1996;93:452–455. doi: 10.1073/pnas.93.1.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Emadi S, Liu R, Yuan B, Schulz P, McAllister C, Lyubchenko Y, Messer A, Sierks MR. Inhibiting aggregation of alpha-synuclein with human single chain antibody fragments. Biochemistry. 2004;43:2871–2878. doi: 10.1021/bi036281f. [DOI] [PubMed] [Google Scholar]
- 122.Ladiwalaa ARA, Bhattacharyaa M, Perchiaccaa JM, Caob P, Raleighb DP, Abedinic A, Schmidtc AM, Varkeyd J, Langend R, Tessiera PM. Rational design of potent domain antibody inhibitors of amyloid fibril assembly. Proc Natl Acad Sci USA. 2012;109:19965–19970. doi: 10.1073/pnas.1208797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer’s disease. Nat Rev Neurol. 2001;7:137–152. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ott A, Stolk RP, Hofman A, van Harskamp F, Grobee DE, Breteler MM. Association of diabetes mellitus and dementia: the Rotterdam Study. Diabetologia. 1996;39:1392–1397. doi: 10.1007/s001250050588. [DOI] [PubMed] [Google Scholar]
- 125.Li L, Holscher C. Common pathological processes in Alzheimer’s disease and type 2 diabetes: a review. Brain Res Rev. 2007;56:384–402. doi: 10.1016/j.brainresrev.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 126.Ohara T, Doi Y, Ninomiya T, Hirakawa Y, Hata J, Iwaki T, Kanba S, Kiyohara Y. Glucose tolerance status and risk of dementia in the community. The Hisayama Study. Neurology. 2011;77:1126–1134. doi: 10.1212/WNL.0b013e31822f0435. [DOI] [PubMed] [Google Scholar]
- 127.Lutz TA, Meyer U. Amylin at the interface between metabolic and neurodegenerative disorders. Front Neurosci. 2015;9:216. doi: 10.3389/fnins.2015.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Banks WA, Kastin AJ. Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides. 1998;19:883–889. doi: 10.1016/S0196-9781(98)00018-7. [DOI] [PubMed] [Google Scholar]
- 129.Fu W, Patel A, Jhamandas JH. Amylin receptor: a common pathophysiological target in Alzheimer’s disease and diabetes mellitus. Front Aging Neurosci. 2013;5:42. doi: 10.3389/fnagi.2013.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roostaei T, Nazeri A, Felsky D, Jager PLD, Schneider JA, Pollock BG, Bennett DA, Voineskos AN. Genome-wide interaction study of brain beta-amyloid burden and cognitive impairment in Alzheimer’s disease. Mol Psych. 2017;22:287–295. doi: 10.1038/mp.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]