

Abstract
A highly efficient and convenient protocol of imidazolium chloride (30 mol %) catalyzed amidation of amines with moderate to excellent yields was reported. The protocol shows broad substrate scope for aromatic, aliphatic, and heterocyclic primary amines.
Keywords: imidazolium chloride, catalyst, primary amines, transamidation
1. Introduction
Formamide and acetamide are common organic fragments in organic compounds. An amide bond is considered to be one of the most important linkages from a medicinal point of view [1,2,3,4]. In addition, amide is also an important structural core of many FDA approved drugs, such as formoterol [5], itopride [6], trametinib [7], apremilast [8], paracetamol [9], and procainamide [10] (Figure 1). Therefore, finding new methods of amidation would definitely be of major benefit for the drug discovery process. Generally, the classical method of preparing acetamide involves the reaction of an amine with a carboxylic acid derivative [11,12,13]. Alternatively, transamidation has been proved to be an attractive tool and represents one of the most convenient and straightforward methods for the preparation of acetamide [14].
Figure 1.

Amide bonds in some drug molecules.
Stahl [15,16,17,18,19], Williams [20,21], Myers [22], Beller [23], and other groups [24,25,26,27,28] have developed many transamidation synthetic methods employing metallic catalysts such as copper [29], Fe (NO3)3 [30], Pd (OAc)2 [31], ZnCl2 [32], ZnO [33], and Ni(quin)2 [34] as promoters. On the other hand, metal-free methods utilizing catalytic hydroxylamine hydrochloride [35], L-proline [36], and H2SO4.SiO2 [37] have also been reported. Recently, Hudson et al. [38] have developed an alternative and elegant protocol for the N-formylation of amino acid ester hydrochloride with N,N-Dimethylformamide (DMF) employing imidazole as the catalyst. However, further research showed that this synthesis method was not compatible with aromatic amine or aliphatic amine substrates. In view of this, herein, we wish to report a general imidazolium chloride catalyzed transamidation of aromatic or aliphatic amines with satisfactory results (Scheme 1).
Scheme 1.

Methods for transamidation.
2. Results and Discussion
In preliminary reactions, p-toluidine was treated with DMA in the presence of various protic acids or bases such as HAc, PTSA, or Et3N at 150 °C for 6 h, however, with resulting low yields of acetamide (Table 1, entries 4–11). Interestingly, when a reaction was carried out with imidazolium chloride (Table 1, entry 12) at room temperature, traces of product formation was observed. Encouraged by this result, the reaction was carried with 3 equiv. imidazolium chloride under moderate conditions. Upon varying the temperature between 30 °C and 150 °C, the transamidation product increased. No target product was detected at 30 °C (Table 1, entry 17), and 59% yield of product was obtained at 60 °C (Table 1, entry 18), while a satisfactory yield (96%) was observed at 150 °C (Table 1, entry 13), indicating that the reaction was highly sensitive to temperature. Subsequently, the reaction was also found to be efficient with 0.3 equiv. imidazolium chloride, affording a yield of the product with 96% within 2 h (Table 1, entries 15). Further optimization showed that reducing the amount of imidazolium chloride decreased the yield of compound (Table 1, entry 16) and no significant effect on reactivity was observed when the loading was raised from 0.3 to 0.5 equiv. (Table 1, entries 13 and 14). Additionally, the reaction failed to proceed in the absence of imidazolium chloride, suggesting the imidazolium chloride complex may be responsible for the activation of DMA (Table 1, entry 22).
Table 1.
Optimization studies for transamidation a.

| |||||
|---|---|---|---|---|---|
| Entry | Base (equiv.) | Solvent (mL) | Temp (°C) | Time (h) | Yield b (%) |
| 1 | CuCl2 | DMA | 150 | 12 | Trace |
| 2 | Zr(SO4)2 | DMA | 150 | 12 | Trace |
| 3 | CH3COOH(2) | DMA | 150 | 6 | 8 |
| 4 | HCl(2) | DMA | 150 | 6 | 46 |
| 5 | H2SO4(2) | DMA | 150 | 6 | 52 |
| 6 | p-toluenesulfonic acid | DMA | 150 | 6 | 50 |
| 7 | Pyridine(3) | DMA | 150 | 6 | Trace |
| 8 | KOtBu(2) | DMA | 150 | 6 | 12 |
| 9 | NaOH(2) | DMA | 150 | 6 | 18 |
| 10 | 2,6-lutidine(3) | DMA | 150 | 6 | Trace |
| 11 | TEA(3) | DMA | 150 | 6 | Trace |
| 12 | Imidazolium chloride(3) | DMA | rt | 6 | Trace |
| 13 | Imidazolium chloride(3) | DMA | 150 | 2 | 96 |
| 14 | Imidazolium chloride(0.5) | DMA | 150 | 2 | 95 |
| 15 | Imidazolium chloride(0.3) | DMA | 150 | 2 | 96 |
| 16 | Imidazolium chloride(0.1) | DMA | 150 | 6 | 65 |
| 17 | Imidazolium chloride(3) | DMA | 30 | 2 | Trace |
| 18 | Imidazolium chloride(3) | DMA | 60 | 2 | 59 |
| 19 | Imidazolium chloride(3) | DMA | 90 | 2 | 68 |
| 20 | Imidazolium chloride(3) | DMA | 135 | 2 | 85 |
| 21 | Imidazole(3) | DMA | 150 | 12 | Trace |
| 22 | - | DMA | 150 | 12 | NOC |
a Reaction conditions: amine (0.3 g, 2.8 mmol, 1 equiv.), solvent (5.0 mL), temperature 60–150 °C, all reagent and substrate addition was done at room temperature (25 °C), b isolated yield; c no reaction.
Having established the optimal reaction conditions (Table 1, entry 15), we next set out to examine the scope and limitations of this reaction, as shown in Table 2. N,N-Dimethylacetamide (DMA) was chosen as the acetyl donor and a variety of primary amines were studied. To our delight, the aromatic primary amines bearing electron-donating and -withdrawing groups were tolerated well under the reaction conditions, affording the desired products in moderate to good yields (Table 2, entries 1–14). It was noteworthy that the electronic properties of the substituent groups on the phenyl ring played an important role in the reaction. Aromatic primary amine containing electron-donating groups provided the desired N-acetamide product in better yields than those of electron-withdrawing groups (Table 2, entries 2, 3, and 11 vs. entries 12, 13, and 14). The halogen substituted aniline (m/p/o) showed more reactivity in comparison with that of p-nitroaniline and gave corresponding products in relatively high yields (Table 2, entries 4–6 and 7–9 vs. entry 12). Benzlyamines with electron-rich and -deficient substituents were reacted smoothly and produced corresponding transamidation products in good to excellent yields as well (Table 2, entries 15 and 16). In addition, the heterocyclic amines and the aliphatic amines were also compatible and afforded the corresponding products in moderate to good yields (Table 2, entries 17–23).
Table 2.
Synthesis of benzamides via transamidation a.
| ||||
|---|---|---|---|---|
| Entry | Substrate | Time | Product | Yield b (%) |
| 1 |
|
2 h |

|
92 |
| 2 |

|
2 h |

|
97 |
| 3 |

|
3 h |

|
90 |
| 4 |

|
2 h |

|
75 |
| 5 |

|
2 h |

|
82 |
| 6 |

|
4 h |

|
84 |
| 7 |

|
6 h |

|
83 |
| 8 |

|
6 h |

|
80 |
| 9 |

|
5 h |

|
89 |
| 10 |

|
6 h |
|
90 |
| 11 |

|
4 h |

|
89 |
| 12 |

|
4 h |

|
63 |
| 13 |

|
5 h |

|
57 |
| 14 |

|
4 h |

|
68 |
| 15 |

|
3 h |

|
93 |
| 16 |

|
3 h |

|
94 |
| 17 |

|
5 h |
|
60 |
| 18 |

|
5 h |

|
72 |
| 19 |

|
5 h |

|
75 |
| 20 |

|
3 h |

|
95 |
| 21 |

|
3 h |

|
94 |
| 22 |

|
3 h |

|
95 |
| 23 |

|
3 h |

|
89 |
a Conditions unless otherwise stated: amine (1.0 equiv.), DMA (1.5 mL), Imidazolium chloride (0.3 equiv.) in a sealed tube at 150 °C for 2–8 h. b isolated yield.
In order to explore the scope of transamidation, a variety of aromatic, aliphatic, or benzylic amines were conducted to react with DMF (Table 3) and N,N-Dimethylbenzamide (Table 4). In general, the transamidation of formide with aromatic amines and benzylic amines (electron-deficient, -neutral, -rich) gave corresponding transamidation products in moderate to good yields (2a–2e). Similarly, transamidation of benzamide with N,N-Dimethylbenzamide also gave 3a–3d in excellent yields. It should be noted that the acylation products can be obtained with good quality and purity by a simple filtration procedure in most cases.
Table 3.
N-benzamides of amines 2a–2ea.

| ||||
|---|---|---|---|---|
| Entry | Substrate | Time | Product | Yield b (%) |
| 1 |

|
4 h |

|
89 |
| 2 |
|
4 h |

|
85 |
| 3 |

|
4 h |

|
89 |
| 4 |

|
4 h |

|
71 |
| 5 |

|
3 h |
|
92 |
a Conditions unless otherwise stated: amine (1.0 equiv.), DMF (1.5 mL), Imidazolium chloride (0.3 equiv.) in a sealed tube at 150 °C for 2–8 h. b isolated yield.
Table 4.
N-benzoylation of amines 3a–3da.

| ||||
|---|---|---|---|---|
| Entry | Substrate | Time | Product | Yield b (%) |
| 1 |

|
4 h |

|
89 |
| 2 |

|
3 h |

|
94 |
| 3 |

|
3 h |

|
97 |
| 4 |

|
3 h |
|
93 |
a Conditions unless otherwise stated: amine (1.0 equiv.), N,N-Dimethylbenzamide (2.0 equiv.), Imidazolium chloride (0.3 equiv.) in a sealed tube at 150 °C for 2–4 h. b isolated yield.
To demonstrate the practical utility, the amidation of 4-aminophenol with DMA was carried out on a 50 g scale under the same conditions to give 4-acetamidophenol in 91% satisfactory yield after silica gel chromatography.
Based on our experimental results and previous studies [38], a plausible mechanism is proposed in Scheme 2. Firstly, DMA was activated by H+ to afford intermediate A, which was attacked by imidazole, resulting in the formation of intermediate B; HNMe2 was removed, leading to the formation of the key intermediate C. Next, nucleophilic addition of the primary amine generates a tetrahedral intermediate D which undergoes an elimination of imidazole to afford the target compound.
Scheme 2.

Plausible mechanism for N-acetylation using imidazolium chloride catalyst.
3. Materials and Methods
3.1. General Information
All reactions were carried out under normal conditions and no any stringent conditions were used. All reagents were obtained from Aladdin Reagent Co., Ltd. (Shanghai, China), Lagewell Technology Co., Ltd. (Shenzhen, China), Meyer Reagent Co., Ltd. (Shanghai, China), Macklin Reagent Co., Ltd. (Shanghai, China), Chongqing Chuandong Chemical Co., Ltd. (Chongqing, China). etc. without further purification unless otherwise noted. Reactions were monitored by TLC analysis using Merck Silica Gel 60 F-254 thin layer plates. The plates were visualized first with short wavelength UV light followed by iodine stain. 1H and 13C NMR spectra were recorded in CDCl3 and DMSO-d6 on a Bruker Ascend-III 600 MHz spectrometer using TMS as an internal standard. The residual solvent signals were used as references and the chemical shifts converted to the TMS scale (CDCl3: δ H = 7.25–7.30 ppm, δ C = 77.23 ppm; DMSO-d6: δ H = 2.51 ppm, δ C = 39.51 ppm).
3.2. General Procedures for the Synthesis of N-Acetamides 1a–1w
To a mixture of aromatic or aliphatic or heterocyclic amine (3.0 mmol, 1.0 equiv.), Imidazolium chloride (1.0 mmol, 0.3 equiv.), N,N-Dimethyl acetamide (2.0 mL) was added. The mixture was refluxed at 150 °C and the progress of the reaction was monitored by TLC visualized with UV short wavelength followed by iodine stain. After completion, the mixture was diluted with cold water (10 mL) then extracted with EtOAc (10 mL). The EtOAc layer was washed with 1 M hydrochloric acid (3.0 × 15 mL). Adsorption of pigment with activated carbon, filtration of filtrate. The filtrate was dried over anhydrous Na2SO4 and concentrated under vacuum to obtain crude N-acetamide amine, the N-acetamide amine product was isolated by column chromatography eluting with petroleum ether:ethyl acetate (10:1) mixtures.
3.3. General Procedures for the Synthesis of N-Formamides 2a–2e
To a mixture of aromatic or aliphatic or heterocyclic amine (3.0 mmol, 1.0 equiv.), Imidazolium chloride (1.0 mmol, 0.3 equiv.), N,N-Dimethyl formamide (2.0 mL) was added. The mixture was refluxed at 150 °C and the progress of the reaction was monitored by TLC visualized with UV short wavelength followed by iodine stain. After completion, the mixture was diluted with cold water (10 mL) then extracted with EtOAc (10 mL). The EtOAc layer was washed with 1 M hydrochloric acid (3.0 × 15 mL). Adsorption of pigment with activated carbon, filtration of filtrate. The filtrate was dried over anhydrous Na2SO4 and concentrated under vacuum to obtain crude N-formyl amine, the N-formyl amine product was isolated by column chromatography eluting with petroleum ether: ethyl acetate (10:1) mixtures.
3.4. General Procedures for the Synthesis of N-Benzoylation 3a–3d
To a mixture of aromatic or aliphatic or heterocyclic amine (3.0 mmol, 1.0 equiv), Imidazolium chloride (1.0 mmol, 0.3 equiv.), N,N-Dimethylbenzamide (2.0 equiv.) was added. The mixture was refluxed at 150 °C and the progress of the reaction was monitored by TLC visualized with UV short wavelength followed by iodine stain. After completion, the mixture was diluted with cold water (10 mL). The crystallized solid were filtered and washed with water and heptane, dried under vacuum to give the product.
N-Phenylacetamide (1a): The product was obtained as off-white solid in 92% yield (0.37 g); MP: 112–115 °C; 1H NMR (600 MHz, CDCl3) δ 7.82 (s, 1H), 7.54–7.47 (m, 2H), 7.29 (dd, J = 8.5, 7.4 Hz, 2H), 7.12–7.07 (m, 1H), 2.15 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 168.74, 137.90, 128.94, 124.29, 119.97, 24.43.
N-p-Tolylacetamide (1b): The product was obtained as pale yellow solid in 97% yield (0.43 g); MP: 150–153 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.83 (s, 1H), 7.46 (dd, J = 8.7, 2.4 Hz, 2H), 7.08 (d, J = 8.3 Hz, 2H), 2.24 (s, 3H), 2.02 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 168.93, 136.84, 132.67, 129.52, 119.56, 24.16, 20.81.
N-m-Tolyl-acetamide (1c): The product was obtained as dark brown solid in 90% yield (0.40 g); MP: 66–68 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.91 (s, 1H), 7.43 (d, J = 2.0 Hz, 1H), 7.40–7.35 (m, 1H), 7.18 (t, J = 7.8 Hz, 1H), 6.89–6.84 (m, 1H), 2.27 (s, 3H), 2.05 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 168.82, 139.47, 138.30, 128.94, 124.24, 119.96, 116.63, 24.32, 21.60.
N-(2-Chloro-phenyl)-acetamide (1d): The product was obtained as off-white solid in 75% yield (0.38 g); MP: 87–88 °C; 1H NMR (600 MHz, CDCl3) δ 8.35 (d, J = 8.4 Hz, 1H), 7.64 (s, 1H), 7.36 (d, J = 8.0 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.03 (t, J = 7.7 Hz, 1H), 2.24 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 168.26, 134.61, 128.96, 127.73, 124.62, 122.55, 121.66, 24.88.
N-(3-Chlorophenyl)acetamide (1e): The product was obtained as pale yellowish-brown solid in 82% yield (0.42 g); MP: 74–77 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.13 (s, 1H), 7.94–7.68 (m, 1H), 7.57–7.35 (m, 1H), 7.36–7.27 (m, 1H), 7.14–7.02 (m, 1H), 2.06 (d, J = 0.8 Hz, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.17, 141.01, 133.50, 130.80, 123.17, 118.80, 117.67, 24.39.
N-(4-Chlorophenyl)acetamide (1f): The product was obtained as pale brown solid in 84% yield (0.43 g); MP: 175–177 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.07 (s, 1H), 7.71–7.48 (m, 2H), 7.48–7.23 (m, 2H), 2.04 (d, J = 1.3 Hz, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.00, 138.48, 138.46, 129.01, 127.06, 120.93, 24.31.
N-(3-Bromo-phenyl)-acetamide (1g): The product was obtained as pale brown solid in 83% yield (0.53 g); MP: 85–87 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.11 (s, 1H), 7.95 (d, J = 1.9 Hz, 1H), 7.47–7.44 (m, 1H), 7.26 (t, J = 8.0 Hz, 1H), 7.24–7.19 (m, 1H), 2.05 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.24, 141.11, 131.17, 126.12, 121.98, 121.65, 118.09, 24.38.
N-(2-Bromo-phenyl)-acetamide (1h):The product was obtained as pale brown solid in 80% yield (0.51 g); MP: 97–99 °C; 1H NMR (600 MHz, CDCl3) δ 8.33 (d, J = 8.3 Hz, 1H), 7.61 (s, 1H), 7.53 (dd, J = 8.0, 1.4 Hz, 1H), 7.31 (ddd, J = 8.5, 7.4, 1.5 Hz, 1H), 7.01–6.92 (m, 1H), 2.24 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 168.17, 135.59, 132.19, 128.39, 125.16, 121.87, 113.10, 24.83.
N-(4-Bromophenyl)-acetamide (1i): The product was obtained as pale yellowish-brown solid in 89% yield (0.57 g); MP: 166–169 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.07 (s, 1H), 7.56 (d, J = 8.8 Hz, 2H), 7.47 (d, J = 8.8 Hz, 2H), 2.05 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.12, 138.83, 131.91, 121.36, 115.10, 24.31.
N-(4-Bromo-2-methyl-phenyl)-acetamide (1j): The product was obtained as off-white solid in 90% yield (0.61 g); MP:160–162 °C; 1H NMR (600 MHz, CDCl3) δ 7.68 (d, J = 8.3 Hz, 1H), 7.32 (d, J = 8.8 Hz, 2H), 6.97 (s, 1H), 2.21 (d, J = 17.0 Hz, 6H); 13C NMR (151 MHz, CDCl3) δ 168.26, 134.64, 133.88, 131.14, 129.72, 124.70, 118.08, 24.27, 17.58.
N-(4-Hydroxyphenyl)-acetamide (1k)The product was obtained as white solid in 89% yield (0.40 g); MP: 170–172 °C; 1H NMR (600 MHz, DMSO-d6) δ 9.65 (s, 1H), 9.14 (s, 1H), 7.34 (d, J = 8.8 Hz, 2H), 6.68 (d, J = 8.8 Hz, 2H), 1.98 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 168.09, 153.43, 131.32, 121.27, 115.38, 24.09.
N-(4-Nitro-phenyl)-acetamide (1l): The product was obtained as yellow solid in 63% yield (0.34 g); MP: 209–211 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.21 (dd, J = 9.1, 1.7 Hz, 2H), 7.95–7.65 (m, 2H), 2.12 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.95, 145.66, 142.50, 125.42, 119.01, 24.56.
N-(4-Formyl-phenyl)-acetamide (1m): The product was obtained as white solid in 57% yield (0.28 g); MP: 155–157 °C; 1H NMR (600 MHz, CDCl3) δ 9.92 (s, 1H), 8.07 (s, 1H), 7.95–7.78 (m, 2H), 7.73 (d, J = 8.3 Hz, 2H), 2.24 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 191.27, 168.97, 143.65, 132.12, 131.19, 119.20, 24.72.
N-(4-Acetyl-phenyl)-acetamide (1n): The product was obtained as white solid in 68% yield (0.36 g); MP: 166–168 °C; 1H NMR (600 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.03–7.83 (m, 2H), 7.80–7.60 (m, 2H), 2.52 (s, 3H), 2.09 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 197.15, 169.48, 143.90, 131.97, 129.93, 118.57, 26.84, 24.53.
N-Benzylacetamide (1o): The product was obtained as off-white solid in 93% yield (0.42 g); MP: 60–63 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.48–8.24 (m, 1H), 7.40–7.28 (m, 2H), 7.28–7.17 (m, 3H), 4.26 (dd, J = 9.6, 4.1 Hz, 2H), 1.92–1.83 (m, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.95, 139.88, 128.73, 127.69, 127.21, 42.50, 22.92.
(R)-N-methyl-N-phenylacetamide (1p): The product was obtained as yellow solid in 94% yield (0.46 g); MP: 98–101 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.29 (d, J = 8.1 Hz, 1H), 7.33–7.23 (m, 4H), 7.25–7.13 (m, 1H), 4.90 (dd, J = 7.9, 6.8 Hz, 1H), 1.84 (s, 3H), 1.32 (d, J = 7.1 Hz, 3H); 13C NMR (151 MHz, DMSO-d6) δ 168.97, 145.18, 128.69, 127.04, 126.38, 48.26, 23.06, 22.9. = 124 (c = 0.5, CHCl3).
N-(4-Methyl-benzothiazol-2-yl)-acetamide (1q): The product was obtained as white solid in 60% yield (0.37 g); MP: 250–252 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.44 (s, 1H), 7.77 (d, J = 7.8 Hz, 1H), 7.39–6.96 (m, 2H), 2.57 (s, 3H), 2.19 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.84, 157.36, 147.97, 131.44, 130.25, 127.08, 123.95, 119.48, 23.08, 18.36.
N-(6-Methyl-benzothiazol-2-yl)-acetamide (1r): The product was obtained as white solid in 72% yield (0.44 g); MP: 216–218 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.28 (s, 1H), 7.87–7.66 (m, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.24 (dd, J = 8.3, 1.7 Hz, 1H), 2.41 (s, 3H), 2.20 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.78, 157.39, 146.84, 133.46, 131.95, 127.87, 121.67, 120.57, 23.13, 21.38.
N-(6-Chloro-benzothiazol-2-yl)-acetamide (1s): The product was obtained as white solid in 75% yield (0.51 g); MP: 225–227 °C; 1H NMR (600 MHz, DMSO-d6) δ 12.45 (s, 1H), 8.11 (d, J = 2.5 Hz, 1H), 7.73 (dd, J = 8.7, 1.5 Hz, 1H), 7.46 (s, 1H), 2.23 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 170.14, 159.05, 147.75, 133.49, 128.03, 126.89, 122.13, 121.71, 23.09.
N-[2-(4-Methoxy-phenyl)-ethyl]-acetamide (1t): The product was obtained as white solid in 95% yield (0.55 g); MP: 86–87 °C; 1H NMR (600 MHz, DMSO-d6) δ 7.89 (s, 1H), 7.12 (d, J = 8.6 Hz, 2H), 6.96–6.71 (m, 2H), 3.72 (s, 3H), 3.29–3.07 (m, 2H), 2.63 (t, J = 7.5 Hz, 2H), 1.78 (s, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.97, 158.07, 131.75, 130.00, 114.17, 55.39, 40.95, 34.66, 22.97.
N-Cyclohexylacetamide (1u): The product was obtained as off-white solid in 94% yield (0.40 g); MP: 100–103 °C; 1H NMR (600 MHz, CDCl3) 5.57 (s, 1H), 4.00–3.53 (m, 1H), 1.96 (s, 5H), 1.80–1.64 (m, 2H), 1.68–1.53 (m, 1H), 1.48–1.28 (m, 2H),1.23–1.00 (m, 3H); 13C NMR (151 MHz, CDCl3) δ 169.12, 48.15, 33.15, 25.52, 24.88, 23.49.
N-(4-Methyl-cyclohexyl)-acetamide (1v): The product was obtained as white solid in 95% yield (0.44 g); MP: 139–140 °C; 1H NMR (600 MHz, DMSO-d6) δ 7.67 (d, J = 7.9 Hz, 1H), 3.42 (dtd, J = 11.7, 7.7, 3.9 Hz, 1H), 1.81–1.67 (m, 5H), 1.68–1.59 (m, 2H), 1.32–1.19 (m, 1H), 1.19–1.02 (m, 2H), 1.03–0.89 (m, 2H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (151 MHz, DMSO-d6) δ 168.86, 47.99, 34.05, 32.75, 31.94, 23.06, 22.60.
N-(6-Bromo-pyridin-2-yl)-acetamide (1w): The product was obtained as off-white solid in 89% yield (0.57 g); MP: 158–159 °C; 1H NMR (600 MHz, CDCl3) δ 8.16 (d, J = 8.2 Hz, 1H), 8.09 (s, 1H), 7.56 (t, J = 7.9 Hz, 1H), 7.21 (dd, J = 7.7, 0.8 Hz, 1H), 2.21 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 168.65, 151.28, 140.69, 139.12, 123.52, 112.27, 24.67.
N-Phenyl-formamide (2a): The product was obtained as off-white solid in 89% yield (0.34 g); MP: 48–50 °C; 1H NMR (600 MHz, CDCl3) Mixture of rotamers is observed. Ratio: 5/5. Major rotamer: δ 8.47 (s, 1H), 8.35 (d, J = 2.0 Hz, 1H), 7.58 (dd, J = 8.6, 1.0 Hz, 1H), 7.39–7.30 (m, 2H), 7.17–7.09 (m, 2H); Minor rotamer: δ 9.19 (d, J = 9.0 Hz, 1H), 8.72 (d, J = 11.4 Hz, 1H), 7.58 (dd, J = 8.6, 1.0 Hz, 1H), 7.39–7.30 (m, 2H,), 7.22–7.17 (m, 1H), 7.17–7.09 (m, 1H); 13C NMR (151 MHz, CDCl3) δ 162.90, 159.39, 136.88, 125.25, 120.00, 118.69, 77.08.
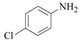
N-(4-Chloro-phenyl)-formamide (2b): The product was obtained as yellow brown solid in 85% yield (0.40 g); MP: 104–106 °C; 1H NMR (600 MHz, DMSO-d6), Mixture of rotamers is observed. Ratio: 5.9/4.1; Major rotamer: δ 8.31 (d, J = 1.2 Hz, 1H), 8.23 (s, 1H), 7.65 (m, 2H), 7.39–7.23 (m, 2H); Minor rotamer: δ 8.82 (d, J = 11.6 Hz, 1H), 7.38 (s, 1H), 7.39–7.23 (m, 2H) 7.21 (m, 2H); 13C NMR (151 MHz, DMSO-d6) δ 162.89, 160.18, 160.11, 160.09, 137.45, 137.43, 129.67, 129.21, 129.20, 127.69, 121.19, 121.11, 119.47, 119.38.
N-(3-Chloro-phenyl)-formamide (2c): The product was obtained as cream white solid in 89% yield (0.42 g); MP: 49–50°C; 1H NMR (600 MHz, DMSO-d6) δ 10.36 (t, J = 4.1 Hz, 1H), 8.62 (t, J = 2.0 Hz, 1H), 8.47–8.20 (m, 2H), 2.21–1.98 (m, 3H); 13C NMR (151 MHz, DMSO-d6) δ 169.85, 144.63, 144.60, 139.25, 137.24, 128.18, 128.09, 120.09, 24.25.
N-(3-Nitro-phenyl)-formamide (2d): The product was obtained as yellow solid in 71% yield (0.35 g); MP: 138–139 °C; 1H NMR (600 MHz, DMSO-d6), Mixture of rotamers is observed. Ratio: 7.1/2.9 Major rotamer: δ 8.63 (s, 1H), 8.38 (t, J = 2.0 Hz, 1H), 7.96–7.89 (m, 2H), 7.89–7.88 (m, 2H); (Minor rotamer: δ 8.96 (d, J = 10.4 Hz,1H), 7.97 (d, J = 1.2 Hz, 1H), 7.89–7.88 (m, 1H), 7.87 (dd, J = 8.0 and 1.6 Hz, 1H), 7.66–7.63 (m, 1H); 13C NMR (151 MHz, DMSO-d6) δ 160.85, 148.39, 139.40, 130.88, 125.58, 118.80, 113.77.
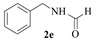
N-Benzyl-formamide (2e): The product was obtained as white solid in 92% yield (0.37 g); MP: 61–62 °C; 1H NMR (600 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.23–8.08 (m, 1H), 7.40–7.19 (m, 5H), 4.34 (s, 2H); 13C NMR (151 MHz, DMSO-d6) δ 161.64, 139.28, 128.82, 127.73, 127.39, 41.12.
N-p-Tolyl-benzamide (3a): The product was obtained as gray solid in 89% yield (0.57 g); MP: 156–158 °C; 1H NMR (600 MHz, CDCl3) δ 7.85 (dt, J = 7.1, 1.4 Hz, 3H), 7.55–7.42 (m, 5H), 7.16 (d, J = 8.2 Hz, 2H), 2.34 (s, 3H); 13C NMR (151 MHz, CDCl3) δ 165.64, 135.27, 135.05, 134.24, 131.74, 129.58, 128.75 , 127.00, 120.35, 120.23, 20.93.
(R)-N-(1-Phenyl-ethyl)-benzamide (3b): The product was obtained as white solid in 94% yield (0.64 g); MP: 137–138 °C; 1H NMR (600 MHz, CDCl3) δ 7.80–7.74 (m, 2H), 7.52–7.47 (m, 1H), 7.45–7.34 (m, 5H), 7.31–7.27 (m, 1H), 5.34 (q, J = 6.9 Hz, 1H), 1.61 (d, J = 6.9 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ 166.54, 143.06, 134.54, 131.51, 128.77, 128.58, 127.50, 126.90, 126.26, 49.13, 21.71. = +19.9 (c = 1.0, CHCl3).
N-[2-(4-Methoxy-phenyl)-ethyl]-benzamide (3c): The product was obtained as white solid in 97% yield (0.74 g); MP: 120–121 °C; 1H NMR (600 MHz, CDCl3) δ 7.75–7.62 (m, 2H), 7.52–7.44 (m, 1H), 7.40 (dd, J = 8.4, 7.0 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.6 Hz, 2H), 3.80 (s, 3H), 3.68 (td, J = 6.9, 5.8 Hz, 2H), 2.87 (t, J = 6.9 Hz, 2H); 13C NMR (151 MHz, CDCl3) δ 167.43, 158.31, 134.62, 131.39, 130.85, 129.75, 128.55, 126.79, 114.12, 55.28, 41.19, 34.75.
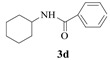
N-Cyclohexyl-benzamide (3d): The product was obtained as white solid in 93% yield (0.57 g); MP: 146–147 °C; 1H NMR (600 MHz, CDCl3) δ 7.87–7.64 (m, 2H), 7.54–7.34 (m, 3H), 5.99 (s, 1H), 3.98 (dt, J = 6.8, 2.8 Hz, 1H), 2.03 (dt, J = 12.5, 3.9 Hz, 2H), 1.91–1.55 (m, 4H), 1.53–1.33 (m, 2H), 1.33–1.09 (m, 3H); 13C NMR (151 MHz, CDCl3) δ 166.60,135.06, 131.25, 128.52, 126.80, 48.56, 33.21, 25.58, 24.91.
Experimental procedures and analytical data of all compounds (1H NMR and 13C NMR), copy of the 1H NMR, 13C NMR and data are available in the Supplementary Materials.
4. Conclusions
In conclusion, a simple method for the transamidation process of primary amines taking imidazolium chloride as the only promoter is reported. This method has wide substrate scope and provides moderate to good yields. Moreover, considering the advantages of imidazolium chloride, including being inexpensive, readily available, and environmentally-friendly, this simple procedure is a valuable addition to the arsenal of amide syntheses.
Acknowledgments
Chongqing University Pharmaceutical Engineering Research Center for support.
Supplementary Materials
The following are available online, general procedure for Transamidation of primary amines reaction and 1H NMR and 13C NMR spectra of all products.
Author Contributions
Q.T. and J.Y. conceived and designed the experiments; Q.T. performed the experiments; W.L. and X.W. analyzed the data; H.W., Z.D. and D.L. contributed reagents/materials/analysis tools; Z.G. wrote the paper.
Funding
Supported by the Fundamental Research Funds for the Chongqing Science and Technology Commission (No. cstc2018jscx-msyb0048) and the Fundamental and Advanced Research Projects of Chongqing City (No. cstc2017jcyjAX0228).
Conflicts of Interest
The authors declare no conflicts of interest.
Footnotes
Sample Availability: Samples of the compounds are not available from the authors.
References
- 1.Cupido T., Tulla-Puche J.J., Albericio F. The synthesis of naturally occurring peptides and their analogs. Curr. Opin. Drug Discov. Dev. 2007;10:768–783. [PubMed] [Google Scholar]
- 2.Lundberg H., Tinnis F., Selander N., Adolfsson H. ChemInform Abstract: Catalytic Amide Formation from Non-Activated Carboxylic Acids and Amines. Chem. Soc. Rev. 2014;43:2714–2742. doi: 10.1039/C3CS60345H. [DOI] [PubMed] [Google Scholar]
- 3.Pattabiraman V.R., Bode J.W. Rethinking amide bond synthesis. Nature. 2011;480:471–479. doi: 10.1038/nature10702. [DOI] [PubMed] [Google Scholar]
- 4.Valeur E., Bradley M. Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 2009;40:606–631. doi: 10.1039/B701677H. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi Y., Mercado N., Millerlarsson A., Barnes P.J., Ito K. Increased corticosteroid sensitivity by a long acting β2 agonist formoterol via β2 adrenoceptor independent protein phosphatase 2A activation. Pulm. Pharmacol. Ther. 2012;25:201–207. doi: 10.1016/j.pupt.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Iwanaga Y., Kimura T., Miyashita N., Morikawa K., Nagata O., Itoh Z. Characterization of acetylcholinesterase-inhibition by itopride. Jpn. J. Pharmacol. 1994;66:317–322. doi: 10.1254/jjp.66.317. [DOI] [PubMed] [Google Scholar]
- 7.Abe H., Kikuchi S., Hayakawa K., Iida T., Nagahashi N., Maeda K. Discovery of a highly potent and selective mek inhibitor: Gsk1120212 (jtp-74057 dmso solvate) ACS Med. Chem. Lett. 2011;2:320–324. doi: 10.1021/ml200004g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perez-Aso M., Montesinos M.C., Mediero A., Wilder T., Schafer P.H., Cronstein B. Apremilast, a novel phosphodiesterase 4 (pde4) inhibitor, regulates inflammation through multiple camp downstream effectors. Arthritis Res. Ther. 2015;17:249. doi: 10.1186/s13075-015-0771-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hinz B., Cheremina O., Brune K. Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB J. 2008;22:383–390. doi: 10.1096/fj.07-8506com. [DOI] [PubMed] [Google Scholar]
- 10.Zamponi G.W., Sui X., Codding P.W., French R.J. Dual actions of procainamide on batrachotoxin-activated sodium channels: Open channel block and prevention of inactivation. Biophys. J. 1993;65:2324–2334. doi: 10.1016/S0006-3495(93)81291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gernigon N., Alzoubi R.M., Hall D.G. Direct amidation of carboxylic acids catalyzed by ortho-iodo arylboronic acids: Catalyst optimization, scope, and preliminary mechanistic study supporting a peculiar halogen acceleration effect. J. Org. Chem. 2012;77:8386–8400. doi: 10.1021/jo3013258. [DOI] [PubMed] [Google Scholar]
- 12.Ueki H., Ellis T.K., Martin C.H., Boettiger T.U., Bolene S.B., Soloshonok V.A. Improved synthesis of proline-derived Ni (II) complexes of glycine: Versatile chiral equivalents of nucleophilic glycine for general asymmetric synthesis of alpha-amino acids. J. Org. Chem. 2003;68:7104–7107. doi: 10.1021/jo0301494. [DOI] [PubMed] [Google Scholar]
- 13.Ueki H., Ellis T.K., Martin C.H., Soloshonok V.A. Efficient large-scale synthesis of picolinic acid-derived Ni (II) complexes of glycine. Eur. J. Org. Chem. 2010;2003:1954–1957. doi: 10.1002/ejoc.200200688. [DOI] [Google Scholar]
- 14.Lanigan R.M., Sheppard T.D. Recent developments in amide synthesis: Direct amidation of carboxylic acids and transamidation reactions. Eur. J. Org. Chem. 2013;33:7453–7465. doi: 10.1002/ejoc.201300573. [DOI] [Google Scholar]
- 15.Kissounko D.A., Hoerter J.M., Guzei I.A., Cui Q., Gellman S.H., Stahl S.S. Ti(iv)-mediated reactions between primary amines and secondary carboxamides: Amidine formation versus transamidation. J. Am. Chem. Soc. 2007;129:1776–1783. doi: 10.1021/ja0650293. [DOI] [PubMed] [Google Scholar]
- 16.Stephenson N.A., Zhu J., Gellman S.H., Stahl S.S. Catalytic transamidation reactions compatible with tertiary amide metathesis under ambient conditions. J. Am. Chem. Soc. 2009;131:10003–10008. doi: 10.1021/ja8094262. [DOI] [PubMed] [Google Scholar]
- 17.Hoerter J.M., Otte K.M., Gellman S.H., Cui Q., Stahl S.S. Discovery and mechanistic study of AlIII-catalyzed transamidation of tertiary amides. J. Am. Chem. Soc. 2008;130:647–654. doi: 10.1021/ja0762994. [DOI] [PubMed] [Google Scholar]
- 18.Hoerter J.M., Otte K.M., Gellman S.H., Stahl S.S. Mechanism of AlIII-catalyzed transamidation of unactivated secondary carboxamides. J. Am. Chem. Soc. 2006;128:5177–5183. doi: 10.1021/ja060331x. [DOI] [PubMed] [Google Scholar]
- 19.Kissounko D.A., Guzei I.A., And S.H.G., Stahl S.S. Titanium(iv)-mediated conversion of carboxamides to amidines and implications for catalytic transamidation. Organometallics. 2005;24:5208–5210. doi: 10.1021/om050768y. [DOI] [Google Scholar]
- 20.Watson A.J., Maxwell A.C., Williams J.M. Ruthenium-catalyzed oxidation of alcohols into amides. Org. Lett. 2009;11:2667–2670. doi: 10.1021/ol900723v. [DOI] [PubMed] [Google Scholar]
- 21.Atkinson B.N., Chhatwal A.R., Lomax H.V., Walton J.W., Williams J.M.J. Transamidation of primary amides with amines catalyzed by zirconocene dichloride. Chem. Commun. 2012;48:11626–11628. doi: 10.1039/c2cc37427g. [DOI] [PubMed] [Google Scholar]
- 22.Dineen T.A., Zajac M.A., Myers A.G. Efficient transamidation of primary carboxamides by in situ activation with N,N-dialkylformamide dimethyl acetals. J. Am. Chem. Soc. 2007;38:16406–16409. doi: 10.1002/chin.200717039. [DOI] [PubMed] [Google Scholar]
- 23.Zhang M., Imm S., Bähn S., Neubert L., Neumann H., Beller M. Efficient copper (II)-catalyzed transamidation of non-activated primary carboxamides and ureas with amines. Angew. Chem. Int. Ed. 2012;124:3905–3975. doi: 10.1002/anie.201108599. [DOI] [PubMed] [Google Scholar]
- 24.Tamura M., Tonomura T., Shimizu K., Satsuma A. Transamidation of amides with amines under solvent-free conditions using a CeO2 catalyst. Green Chem. 2012;14:717–724. doi: 10.1039/c2gc16316k. [DOI] [Google Scholar]
- 25.Becerra-Figueroa L., Ojeda-Porras A., Gamba-Sanchez D. Transamidation of carboxamides catalyzed by Fe (III) and water. J. Org. Chem. 2015;45:4544–4552. doi: 10.1021/jo500562w. [DOI] [PubMed] [Google Scholar]
- 26.Singh P., Singh D.P., Tiwari K., Mishra M., Singh A.K., Singh V.P. Synthesis, structural investigations and corrosion inhibition studies on Mn (II), Co (II), Ni (II), Cu (II) and Zn (II) complexes with 2-amino-benzoic acid (phenyl-pyridin-2-yl-methylene)-hydrazide. RSC Adv. 2015;5:45217–45230. doi: 10.1039/C4RA11929K. [DOI] [Google Scholar]
- 27.Nageswara R.S., Chandra M.D., Adimurthy S. Chitosan: An Efficient Recyclable Catalyst for Transamidation of Carboxamides with Amines under Neat Conditions. Green Chem. 2014;16:4122–4126. doi: 10.1039/C4GC01402B. [DOI] [Google Scholar]
- 28.Pathare S., Jain A., Akamanchi K. Sulfated tungstate: A highly efficient catalyst for transamidation of carboxamides with amines. RSC Adv. 2013;3:7697–7703. doi: 10.1039/c3ra00127j. [DOI] [Google Scholar]
- 29.Liu H., Liu J., Zhang Y., Shao C., Yu J. copper-catalyzed amide bond formation from formamides and carboxylic acids. Chin. Chem. Lett. 2015;46:11–14. doi: 10.1016/j.cclet.2014.09.007. [DOI] [Google Scholar]
- 30.Becerrafigueroa L., Ojedaporras A., Gambasánchez D. Transamidation of Carboxamides Catalyzed by Fe (III) and Water. J. Org. Chem. 2014;79:4544–4552. doi: 10.1021/jo500562w. [DOI] [PubMed] [Google Scholar]
- 31.Gu D.-W., Guo X.-X. Synthesis of N-arylcarboxamides by the efficient transamidation of dmf and derivatives with anilines. Tetrahedron. 2015;71:9117–9122. doi: 10.1016/j.tet.2015.10.008. [DOI] [Google Scholar]
- 32.Shekhar A.C., Kumar A.R., Sathaiah G., Paul V.L., Sridhar M., Rao P.S. Facile N-formylation of amines using lewis acids as novel catalysts. Tetrahedron Lett. 2009;50:7099–7101. doi: 10.1016/j.tetlet.2009.10.006. [DOI] [Google Scholar]
- 33.Hosseini-Sarvari M., Sharghi H. ZnO as a new catalyst for N-formylation of amines under solvent-free conditions. J. Org. Chem. 2006;37:6652–6654. doi: 10.1021/jo060847z. [DOI] [PubMed] [Google Scholar]
- 34.Sonawane R.B., Rasal N.K., Jagtap S.V. Nickel-(II)-Catalyzed N-Formylation and N-Acylation of Amines. Org. Lett. 2017;19:2078–2081. doi: 10.1021/acs.orglett.7b00660. [DOI] [PubMed] [Google Scholar]
- 35.Allen C.L., Atkinson B.N., Williams J.M. Transamidation of primary amides with amines using hydroxylamine hydrochloride as an inorganic catalyst. Angew. Chem. Int. Ed. 2012;51:1383–1386. doi: 10.1002/anie.201107348. [DOI] [PubMed] [Google Scholar]
- 36.Rao S.N., Mohan D.C., Adimurthy S. l-Proline: An Efficient Catalyst for Transamidation of Carboxamides with Amines. Org. Lett. 2013;15:1496–1499. doi: 10.1021/ol4002625. [DOI] [PubMed] [Google Scholar]
- 37.Rasheed S., Rao D.N., Reddy A.S. Sulphuric acid immobilized on silica gel (H2SO4-SiO2) as an eco-friendly catalyst for transamidation. RSC Adv. 2015;5:10567–10574. doi: 10.1039/C4RA16571C. [DOI] [Google Scholar]
- 38.Suchý M., Elmehriki A.A., Hudson R.H. A remarkably simple protocol for the N-formylation of amino acid esters and primary amines. Org. Lett. 2011;13:3952–3955. doi: 10.1021/ol201475j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.