

Abstract
In this article we demonstrate how asymmetric total synthesis of (S)-rivastigmine has been achieved using direct asymmetric reductive amination as the key transformation in four steps. The route started with readily available and cheap m-hydroxyacetophenone, through esterification, asymmetric reductive amination, N-diphenylmethyl deprotection and reductive amination, to provide the final (S)-rivastigmine in 82% overall yield and 96% enantioselectivity. In the asymmetric reductive amination, catalysed by the iridium–phosphoramidite ligand complex and helped by some additives, the readily prepared 3-acetylphenyl ethyl(methyl)carbamate directly reductively coupled with diphenylmethanamine to yield the chiral amine product in 96% ee and 93% yield.
Keywords: Alzheimer’s syndrome, rivastigmine, asymmetric reductive amination, asymmetric catalysis, phosphoramidite ligands
1. Introduction
Alzheimer’s disease (AD) is the most common form of dementia. It is characterized by progressive loss of memory and other cognitive functions [1,2,3,4]. It is a severe human health threat with more than 40 million sufferers worldwide, and this number is expected to triple by 2050 [5,6]. Rivastigmine {3-[1-(dimethylamino)-ethyl]phenylethyl (methyl)carbamate)} (Figure 1) is the active pharmaceutical ingredient of Exelon that was developed for the treatment of patients with mild to moderate Alzheimer disease and treatment of dementia caused by Parkinson’s disease and Lewy bodies [7,8,9,10]. Only the (S)-enantiomer exhibits the desired biological activity.
Figure 1.
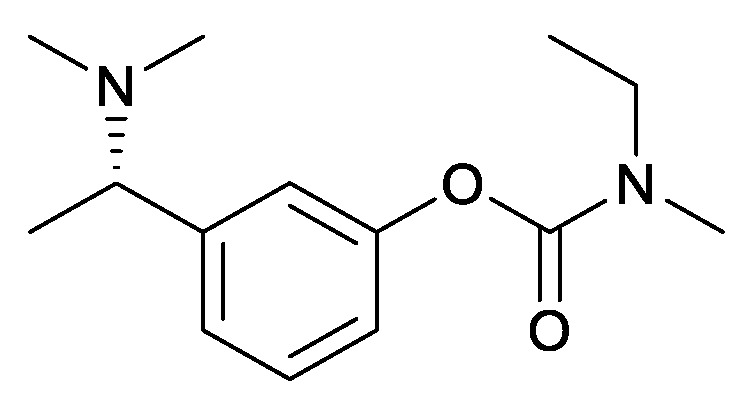
(S)-rivastigmine.
To satisfy the demand of the enantiopure rivastigmine, several preparing routes have been developed. Among these, the method of racemate resolution using tartaric acid derivative is applied in the industrial production of (S)-rivastigmine [11]. Charette et al. reported a novel ligand, based on the bis(phosphine) monoxide framework, along with copper for the addition of Me2Zn to imine to build the key chiral motif [12]. A strategy of copper-catalysed stereoselective hydroamination reactions of alkynes was applied to prepare (S)-rivastigmine by S.L. Buchwald’s group [13]. This synthetic route was concise, but the starting material 3-ethynylphenol was expensive. List group utilized the asymmetric hydrogenation of the N-methyl imine as the core reaction to yield the drug molecule in four steps [14]. M.H. Xie’s group obtained the product in high enantiopurity via six steps and with expensive chiral amine as the starting material [15]. The method of chemoenzymatic asymmetric synthesis was also reported [16,17,18,19]. One scalable route is documented by Che et al. utilizing the asymmetric hydrogenation of corresponding ketone, in which toxic methanesulfonyl chloride was used [20]. Efficient and practical asymmetric synthesis of rivastigmine is still highly desired. Recently we have demonstrated that diphenylmethylamine is a universal amine source for the direct asymmetric reductive amination (DARA) of various ketones, catalyzed by iridium and readily available phosphoramidite ligand based on BINOL [21,22,23]. Here we report the convenient synthesis of (S)-rivastigmine applying the DARA strategy. In DARA, the polar carbamate group on the ketone substrate is well tolerated; the applied chiral ligands were easily prepared from inexpensive BINOL and displayed excellent reactivity and stereoselectivity.
2. Results and Discussion
Starting from readily available and cheap m-hydroxyacetophenone 1, (S)-rivastigmine was synthesized in four steps in high yields and ee (Scheme 1).
Scheme 1.

The synthetic route of (S)-rivastigmine.
In the first step, esterification between compound 1 and 2 was carried out under mild conditions to afford 3 in quantitative yield, which is the key intermediate and substrate for next step. On the basis of our group’s early work [21,22,23], the catalytic system of iridium–monodentate phosphoramidite and a few additives were adopted. The monodentate phosphoramidite chiral ligands are based on BINOL back-bone, which is a cheap and readily available bulk chemical. This kind of ligands is easily prepared, air-stable and well documented in a variety of catalytic reactions [24,25,26,27].
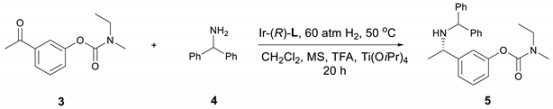
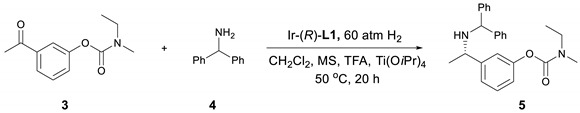
Using DCM as the solvent and PipPhos (Figure 2) as the chiral ligand (Table 1, entry 1), the reaction provided compound 5 in 96% ee and 84% yield. (1H & 13C NMR and HPLC spectra in Supplementary Materials). Common anion additives did not positively affect the reaction (Table 1, entries 2–3) [28,29,30]. With the addition of more TFA, both the stereoselectivity and the yield were improved (Table 1, entry 4). Several other Brønsted acids were tested and the results were not as satisfied as that from TFA (Table 1, entries 5–8). The above results indicated that TFA was crucial in this reaction. Next, a variety of phosphoramidite-type chiral ligands L2–L6 (see Supplementary Materials for the general synthetic procedure) were explored (entries 9–13). Compared with L1, L2 afforded similar ee but lower yield (entry 9). The non-cyclic amine moiety on L3 did not function well (entry 10). More steric hindered ligands L4 and L5 led to poor reactivity (entries 11–12). These results indicate that both the amine moiety and the substituents on the back-bone of the ligands affected their reactivity dramatically. The H8-BINOL-based L6 furnished slightly lower enantiomeric excess than L1 (entry 13). The commercially available MonoPhos was also examined, and it provided moderate to good yield and enantiopurity (entry 14).
Figure 2.

Structures of screened chiral ligands.
Table 1.
The screening of reaction conditions a.
| Entry | Ligand | Acid (Equiv.) | Yield (%) | ee (%) |
|---|---|---|---|---|
| 1 | L1 | TFA (0.5) | 84 | 96 |
| 2 b | L1 | TFA (0.5) | 76 | 96 |
| 3 c | L1 | TFA (0.5) | 45 | 77 |
| 4 | L1 | TFA (1.0) | 93 | 96 |
| 5 | L1 | 4-Cl-C6H4SO3H (1.0) | <15 | - |
| 6 | L1 | TsOH (1.0) | <15 | - |
| 7 | L1 | 4-NO2-C6H4CO2H (1.0) | 65 | 5 |
| 8 | L1 | CCl3CO2H (1.0) | 77 | 92 |
| 9 | L2 | TFA (1.0) | 60 | 90 |
| 10 | L3 | TFA (1.0) | 16 | 58 |
| 11 | L4 | TFA (1.0) | <10 | - |
| 12 | L5 | TFA (1.0) | <10 | - |
| 13 | L6 | TFA (1.0) | 94 | 95 |
| 14 | L7 | TFA (1.0) | 78 | 82 |
a Reaction conditions: [Ir]/L/3/4 = 1:2.2:100:130, 3 0.1 mmol, CH2Cl2 2 mL, 60 atm of H2, 50 °C, 20 h; MS = molecular sieves, 0.1 gram; TFA = trifluoroacetic acid; Ti(OiPr)4 0.2 equiv.; Yields were isolated yields; Enantiomeric excesses were determined by chiral HPLC (see Supplementary Materials for details). b 10 mol% NaBF4 was added. c 10 mol% NH4I was added.
Using Ir–L1 as the reaction catalyst, we further screened other reaction parameters. It is commonly believed that Ti(OiPr)4 can promote the formation of the imine intermediate during the reductive amination process [31]. Our studies also indicated that the amount of tetraisopropoxytitanium displayed a great influence on the reaction. Especially, with the addition of 30%, compound 5 could be obtained in 96% ee. More Ti(OiPr)4 had a negative effect on the enantioselectivity (Table 2, entry 3). As described above, TFA was important for this reaction. It could greatly improve the reactivity (entries 2–7, yield from 86% to 94%) as well as the enantioselectivity (ee from 96% to 97%). When the H2 pressure was decreased to 50 atm, the reaction ee remained the same but the yield dropped to 90% (entry 6). Further lowering the pressure to 30 atm, the yield for the reaction dropped to 75% but the enantioselectivity increased slightly to 98% (entry 7). When the catalyst loading was decreased to 0.5 mol%, the ee and yield of the reaction remained the same; further decreasing the catalyst loading to 0.2 mol% the reaction yield dropped slightly to 88% with ee at 96%; at 60 °C with the catalyst loading at 0.1 mol%, the reaction afforded 70% product. In comparison, L6 outperformed L1 under lower catalyst loading at 0.1 mol%, providing 5 in perfect yield and stereoselectivity (entry 11). Therefore, the optimal reaction conditions (entry 5) for the synthesis of compound 5 were determined based on enantioselectivity, yield and the cost of synthesis.
Table 2.
The screening of the amount of the additives a.
| Entry | Catalyst Loading (mol%) | Ti(OiPr)4 (mol%) | Acid (mol%) | Yield (%) | ee (%) |
|---|---|---|---|---|---|
| 1 | 1 | 20 | 50 | 79 | 93 |
| 2 | 1 | 30 | 50 | 85 | 96 |
| 3 | 1 | 40 | 50 | 87 | 94 |
| 4 | 1 | 30 | 70 | 91 | 96 |
| 5 | 1 | 30 | 80 | 94 | 97 |
| 6 b | 1 | 30 | 80 | 90 | 97 |
| 7 c | 1 | 30 | 80 | 75 | 98 |
| 8 | 0.5 | 30 | 80 | 94 | 96 |
| 9 | 0.2 | 30 | 80 | 88 | 96 |
| 10 d | 0.1 | 30 | 80 | 70 | 94 |
| 11 e | 0.1 | 30 | 80 | 94 | 95 |
a Reaction conditions: Catalyst loading is based on the [Ir] monomer, 1 mol%; 3 0.2 mmol, CH2Cl2 2 mL, 50 °C, 20 h; MS = molecular sieves, 0.2 gram; TFA = trifluoroacetic acid; Yields were isolated yields; Enantiomeric excesses were determined by chiral HPLC. b The H2 pressure was 50 atm. c The H2 pressure was 30 atm. d The reaction temperature was 60 °C. e The applied chiral ligand was L6.
To demonstrate the practical application of this protocol, the asymmetric reductive amination of 3 with 4 was carried out on large scale. The key intermediate 5 was obtained with 93% isolated yield and 96% ee (Scheme 2). Applying (S)-L1 instead of (R)-L1, the corresponding (R)-5 was obtained in the same high yield and ee (Scheme 3), which demonstrates the versatility of asymmetric catalysis. The facile removal of the diphenylmethyl group was carried out with Pd/C as the catalyst and H2 as the reductant leading to primary amine product 6 in 97% yield, without any erosion of the enantioselectivity (Scheme 1).
Scheme 2.

Gram-scale synthesis of (S)-5.
Scheme 3.

Application of (S)-L1 for the synthesis of (R)-5.
Finally, (S)-rivastigmine was obtained through reductive amination of compound 6 with formaldehyde in the presence of sodium triacetoxyborohydride in CH2Cl2 at room temperature. It was purified via column chromatograph to render the pure product in 96% ee and 91% yield. Again, in this step the enantiopurity of the final product was not affected. Through this 4-step procedure, the final product (S)-rivastigmine was synthesized in 82% overall yield and 96% ee. Compared with common (S)-rivastigmine synthetic methods, this procedure is very efficient in terms of operational simplicity and scalability.
The direct asymmetric reductive amination of 3 with dimethylamine 7 was also investigated. Unfortunately, poor yield and moderate ee were achieved (Scheme 4).
Scheme 4.

Application of dimethylamine for the synthesis of (S)-rivastigmine.
3. Materials and Methods
3.1. Materials
Ethyl acetate (ACS grade), hexanes (ACS grade), methanol (ACS grade) and anhydrous dichloromethane for DARA (ACS grade) were obtained commercially and used without further purification. Toluene and tetrahydrofuran were purified according to standard methods unless otherwise noted. Commercially available reagents were used without further purification. Reactions were monitored by thin layer chromatography (TLC) using silicycle pre-coated silica gel plates (Qingdao Haiyang Chemical Co., Qingdao, China). Flash column chromatography was performed over silica gel (300–400 mesh).
3.2. Characterization
Mass spectra were recorded with Micromass QTOF2Quadrupole/Time-of-Flight Tandem mass spectrometer (Milford, MA, USA) using electron spray ionization. 1H-NMR spectra were recorded on a Bruker AV-400 spectrometer (Bruker, Fällanden, Switzerland) and a Bruker AV-500 (Bruker, Fällanden, Switzerland) spectrometer in chloroform-d. Chemical shifts are reported in ppm with the internal TMS signal at 0.0 ppm as a standard. The data is being reported as (s = singlet, d = doublet, dd = doublet of doublets, t = triplet, dt = doublet of triplets, m = multiplet or unresolved, q = quartet, dq = doublet of quartets, brs = broad singlet, coupling constant(s) in Hz, integration). 13C-NMR spectra were recorded on a Bruker AV-400 spectrometer and a Bruker AV-500 spectrometer in chloroform-d. Chemical shifts are reported in ppm with the internal chloroform signal at 77.0 ppm as a standard.
3.3. Preparation of Compound 3
To a solution of compound 1 (5.11 g, 38 mmol) in acetone (100 mL) was added K2CO3 (10.44 g, 76 mmol) and compound 2 (7.20 g, 58 mmol) subsequently. Then the solution was heated to reflux for 4h. After the reaction solution was cooled to r.t., it was filtered and washed with acetone (30 mL × 3). Then the filtrate was concentrated under reduced pressure and purified via column chromatography (CH2Cl2/MeOH = 40:1 to 20:1 v/v) to afford light yellow oil (Yield: 100%). 1H-NMR (500 MHz, Chloroform-d): δ 7.79 (d, J = 8.0 Hz, 1H, Ar-H), 7.69 (s, 1H, Ar-H), 7.47 (t, J = 7.5 Hz, 1H, Ar–H), 7.35 (d, J = 2.5 Hz, 1H, Ar–H), 3.50 (dq, J = 7.5, 14.5 Hz, 2H, –CH2CH3), 3.08 (d, J = 42.5 Hz, 3H, –NCH3), 2.59 (s, 3H, –COCH3), 1.27 (dt, J = 7.0 Hz, J = 7.0 Hz, 3H, –CH2CH3).
3.4. Preparation of Compound 5
Compound 3 (0.2 mmol), 4 (0.26 mmol, 1.3 equiv.) and TFA (0.8 equiv.) in CH2Cl2 were added to a small vial, followed by Ti(OiPr)4 (0.06 mmol, 0.3 equiv.) and the Ir–L1 (1 mol%) solution in CH2Cl2, which was in situ generated from stirring the solution of [Ir(cod)Cl]2 and L1 in CH2Cl2 for 20 min. The resulting vial was transferred to an autoclave, which was charged with 60 atm of H2, and stirred at 50 °C for 20 h. The reaction was quenched with aqueous sodium bicarbonate solution and extracted with CH2Cl2 (2 mL × 3). The organic phase was dried over anhydrous Na2SO4, concentrated and purified by column chromatography (EtOAc/Hex) to give the chiral amine product 5, which was analyzed by chiral HPLC determine the enantiomeric excess (Yield: 93%, ee: 96%). 1H-NMR (500 MHz, Chloroform-d): δ 7.41–7.36 (m, 7H, ArH), 7.33–7.30 (m, 3H, ArH), 7.25 (t, J = 6.5 Hz, 1H, ArH), 7.14 (d, J = 6.5 Hz, 1H, ArH), 7.09 (s, 2H, ArH), 4.75 (s, 1H, –CH(Ph)2), 3.76 (d, J = 6.0 Hz, 1H, –CHCH3C6H4–), 3.54 (dd, J = 5.5, 2H, –CH2CH3), 3.13 (d, J = 35 Hz, 3H, –NCH3), 1.44 (d, J = 6.5 Hz, 3H, –CHCH3), 1.31–1.27 (m, 3H, –CH2CH3).
3.5. Preparation of Compound 6
Compound 5 (0.2 mmol, 78 mg), Pd/C (8 mg, 10%, 50% wetted with water) and MeOH (2 mL) were added to a vial. The resulting vial was transferred to an autoclave, which was charged with 20 atm of H2, and stirred at 40 °C for 17 h. The hydrogen gas was released slowly and the solution was filtered to removed Pd/C. The filtrate was concentrated and purified by flash column chromatography (EtOAc/Hex) to yield the desired product 6 (43 mg, yield: 97%). 1H-NMR (500 MHz, Chloroform-d): δ 7.34–7.31 (m, 1H, ArH), 7.20–7.14 (m, 2H, ArH), 7.01 (s, 1H, ArH), 4.14 (d, J = 6.0 Hz, 1H, –CHCH3), 3.50 (dd, J = 6.0 Hz, J = 6.0 Hz, 2H, –CH2CH3), 3.09 (d, J = 38 Hz, 3H, –NCH3), 1.64 (s, 2H, –NH2), 1.41 (d, J = 7.0 Hz, 3H, –CHCH3), 1.27 (d, J = 26 Hz, 3H, –CH2CH3).
3.6. Preparation of (S)-Rivastigmine
To a solution of compound 6 (64 mg, 0.29 mmol) in CH2Cl2 (6 mL), Na2SO4 (42 mg, 0.30 mmol), NaBH(OAc)3 (504 mg, 2.4 mmol) and formaldehyde (37% in water, 88 μL, 1.0 mmol) were added subsequently. Then the reaction mixture was stirred at −10 °C for 8 h. The reaction was quenched with aqueous K2CO3 solution. The phases were separated, the aqueous phase was extracted with EtOAc (20 mL × 2) and the combined organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure to give crude product, which was purified by flash column chromatography (DCM/MeOH = 20:1) to yield the desired (S)-Rivastigmine as oil (Yield: 91%, ee: 96%). [α] = −29.8 (c = 1.0, EtOH) ([α] = −32.8 (c = 1.3, EtOH))[16]. 1H-NMR (400 MHz, Chloroform-d): δ 7.31–7.27 (m, 1H, ArH), 7.12 (d, J = 9.5 Hz, 1H, ArH), 7.06 (s, 1H, ArH), 7.02 (d, J = 10.0 Hz, 1H, ArH), 3.49–3.38 (m, 2H, –CH2CH3), 3.27 (q, J = 8.5 Hz, J = 16.5 Hz, 1H, –CHCH3), 3.06 (d, J = 37 Hz, 3H, –NCH3), 2.22 (s, 6H, –N(CH3)2), 1.37 (d, J = 8.5 Hz, 3H, –CHCH3), 1.25–1.17 (m, 3H, –CH2CH3); 13C-NMR (100 MHz, Chloroform-d): 153.59, 150.54, 144.72, 127.85, 123.20, 119.74, 64.62, 43.01, 42.18, 33.18, 19.04, 12.21. HRMS for C14H23N2O2: [M + H]+ m/z 251.17540, found m/z 251.17538.
4. Conclusions
In conclusion, (S)-rivastigmine (96% ee) was synthesized via a 4-step route in 82% overall yield, which (to the best of our knowledge) depicts the highest yield route to enantiopure rivastigmine reported to date. In this route, we utilized the highly efficient direct asymmetric reductive amination (DARA) to provide the key intermediate compound 5 in excellent enantioselectivity and yield from the corresponding ketone. DARA is featured with high atom-efficiency and operational simplicity. The chiral ligands we used in the DARA step are easily modulated and readily prepared from inexpensive starting materials. Our strategy opens the door for the practical catalytic asymmetric production of (S)-rivastigmine.
Acknowledgments
We gratefully thank Northwest A&F University and Shandong Normal University for financial support.
Supplementary Materials
Supplementary Materials are available online, spectra for all products and HPLC for intermediate 5 and (S)-rivastigmine.
Author Contributions
Investigation, G.G., S.D., and X.L.; Methodology, Y.Y.; Writing-Original Draft Preparation, G.G.; Writing-Review & Editing, M.C.; Supervision, H.H. and M.C.
Funding
This research was funded by the National Natural Science Foundation of China (No. 21772155, 21675103), the Yangling Bureau of Science & Technology (No. 2016NY-25).
Conflicts of Interest
The authors declare no conflict of interest. funding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Footnotes
Sample Availability: Samples of the compounds (3–6 and rivastigmine) are available from the authors.
References
- 1.Huang Y., Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scarpini E., Scheltens P., Feldman H. Treatment of Alzheimer’s disease: Current status and new perspectives. Lancet Neurol. 2003;2:539–547. doi: 10.1016/S1474-4422(03)00502-7. [DOI] [PubMed] [Google Scholar]
- 3.Frisoni G.B., Fox N.C., Jack C.R., Jr., Scheltens P., Thompson P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010;6:67–77. doi: 10.1038/nrneurol.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berchtold N.C., Cotman C.W. Evolution in the conceptualization of dementia and Alzheimer’s disease: Greco-roman period to the 1960s. Neurobiol. Aging. 1998;19:173–189. doi: 10.1016/S0197-4580(98)00052-9. [DOI] [PubMed] [Google Scholar]
- 5.Scheltens P., Blennow K., Breteler M.M., Strooper B., Frisoni G.B., Salloway S., Van der Flier M. Alzheimer’s disease. Lancet. 2016;388:505–517. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- 6.Panek D., Więckowska A., Wichur T., Bajda M., Godyń J., Jończyk J., Mika K., Janockova J., Soukup O., Knez D., et al. Design, synthesis and biological evaluation of new phthalimide and saccharin derivatives with alicyclic amines targeting cholinesterases, beta-secretase and amyloid beta aggregation. Eur. J. Med. Chem. 2017;125:676–695. doi: 10.1016/j.ejmech.2016.09.078. [DOI] [PubMed] [Google Scholar]
- 7.Farlow M.R., Cummings J.L. Effective Pharmacologic Management of Alzheimer’s Disease. Am. J. Med. 2007;120:388–397. doi: 10.1016/j.amjmed.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 8.Polinsky R.J. Clinical pharmacology of rivastigmine: A new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin. Ther. 1998;20:634–647. doi: 10.1016/S0149-2918(98)80127-6. [DOI] [PubMed] [Google Scholar]
- 9.Rosler M., Anand R., Cicin-Sain A., Gauthier S., Agid Y., Dal-Bianco P., Stahelin H.B., Hartman R., Gharabawi M. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: International randomised controlled trial commentary: Another piece of the Alzheimer’s jigsaw. Br. Med. J. 1999;318:633–638. doi: 10.1136/bmj.318.7184.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spencer C.M., Noble S. Rivastigmine: A review of its use in Alzheimer’s disease. Drugs Aging. 1998;13:391–411. doi: 10.2165/00002512-199813050-00005. [DOI] [PubMed] [Google Scholar]
- 11.Gaitonde A., Mangle M., Pawar S. Novel Processes for the Preparation of Aminoalkyl Phenylcarbamates. WO 2005/061446. International Patent. 2005 Jul 7;
- 12.Boezio A.A., Pytkowicz J., Côté A., Charette A.B. Asymmetric, catalytic synthesis of r-chiral amines using a novel bis(phosphine) monoxide chiral ligand. J. Am. Chem. Soc. 2003;125:14260–14261. doi: 10.1021/ja038291+. [DOI] [PubMed] [Google Scholar]
- 13.Shi S.L., Buchwald S.L. Copper-catalysed selective hydroamination reactions of alkynes. Nat. Chem. 2015;7:38–44. doi: 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wakchaure V.N., Kaib P.S.J., Leutzsch M., List B. Disulfonimide-catalyzed asymmetric reduction of N-alkyl imines. Angew. Chem. Int. Ed. 2015;54:11852. doi: 10.1002/anie.201504052. [DOI] [PubMed] [Google Scholar]
- 15.Hu M., Zhang F.L., Xie M.H. Novel convenient synthesis of rivastigmine. Synth. Commun. 2009;39:1527–1533. doi: 10.1080/00397910802531948. [DOI] [Google Scholar]
- 16.Han K., Kim C., Park J., Kim M.J. Chemoenzymatic synthesis of rivastigmine via dynamic kinetic resolution as a key step. J. Org. Chem. 2010;75:3105–3108. doi: 10.1021/jo9027374. [DOI] [PubMed] [Google Scholar]
- 17.Mangas-Sánchez J., Rodríguez-Mata M., Busto E., Gotor-Fernández V., Gotor V. Chemoenzymatic synthesis of rivastigmine based on lipase-catalyzed processes. J. Org. Chem. 2009;74:5304–5310. doi: 10.1021/jo900784g. [DOI] [PubMed] [Google Scholar]
- 18.Fuchs M., Koszelewski D., Tauber K., Sattler J., Banko W., Holzer A.K., Pickl M., Kroutil W., Faber K. Improved chemoenzymatic asymmetric synthesis of (S)-rivastigmine. Tetrahedron. 2012;68:7691–7694. doi: 10.1016/j.tet.2012.06.031. [DOI] [Google Scholar]
- 19.Fuchs M., Koszelewski D., Tauber K., Kroutil W., Faber K. Chemoenzymatic asymmetric total synthesis of (S)-rivastigmine using ω-transaminases. Chem. Commun. 2010;46:5500–5502. doi: 10.1039/c0cc00585a. [DOI] [PubMed] [Google Scholar]
- 20.Yan P.C., Zhu G.L., Xie J.H., Zhang X.D., Zhou Q.L., Li Y.Q., Shen W.H., Che D.Q. Industrial scale-up of enantioselective hydrogenation for the asymmetric synthesis of rivastigmine. Org. Process. Res. Dev. 2013;17:307–312. doi: 10.1021/op3003147. [DOI] [Google Scholar]
- 21.Huang H., Liu X., Zhou L., Chang M., Zhang X. Direct asymmetric reductive amination for the synthesis of chiral β-arylamines. Angew. Chem. Int. Ed. 2016;55:5309–5312. doi: 10.1002/anie.201601025. [DOI] [PubMed] [Google Scholar]
- 22.Huang H., Zhao Y., Yang Y., Zhou L., Chang M. Direct catalytic asymmetric reductive amination of aliphatic ketones utilizing diphenylmethanamine as coupling partner. Org. Lett. 2017;19:1942–1945. doi: 10.1021/acs.orglett.7b00212. [DOI] [PubMed] [Google Scholar]
- 23.Huang H., Wu Z., Gao G., Zhou L., Chang M. Iridium-catalyzed direct asymmetric reductive amination of aromatic ketones. Org. Chem. Front. 2017;4:1976–1980. doi: 10.1039/C7QO00400A. [DOI] [Google Scholar]
- 24.Claver C., Fernandez E., Gillon A., Heslop K., Hyett D.J., Martorell A., Orpen A.G., Pringle P.G. Biarylphosphonites: A class of monodentatephosphorus(III) ligands that outperform their chelatinganalogues in asymmetric hydrogenation catalysis. Chem. Commun. 2000;46:961–962. doi: 10.1039/B001638L. [DOI] [Google Scholar]
- 25.Berg van den M., Minnaard A.J., Schudde E.P., Esch J.V., de Vries A.H.M., de Vries J.G., Feringa B.L. Highly enantioselective rhodium-catalyzed hydrogenation with monodentate ligands. J. Am. Chem. Soc. 2000;122:11539–11540. doi: 10.1021/ja002507f. [DOI] [Google Scholar]
- 26.Minnaard A.J., Feringa B.L., Lefort L., de Vries J.G. Asymmetric hydrogenation using monodentate phosphoramidite ligands. Acc. Chem. Res. 2007;40:1267–1277. doi: 10.1021/ar7001107. [DOI] [PubMed] [Google Scholar]
- 27.Fu W., Tang W. Chiral monophosphorus ligands for asymmetric catalytic reactions. ACS Catal. 2016;6:4814–4858. doi: 10.1021/acscatal.6b01001. [DOI] [Google Scholar]
- 28.Hong L., Sun W., Yang D., Li G., Wang R. Additive effects on asymmetric catalysis. Chem. Rev. 2016;116:4006–4123. doi: 10.1021/acs.chemrev.5b00676. [DOI] [PubMed] [Google Scholar]
- 29.Li C., Villa-Marcos B., Xiao J. Metal-brønsted acid cooperative catalysis for asymmetric reductive amination. J. Am. Chem. Soc. 2009;131:6967–6969. doi: 10.1021/ja9021683. [DOI] [PubMed] [Google Scholar]
- 30.Chang M., Liu S., Zhang X. Direct catalytic asymmetric reductive amination of simple aromatic ketones. Org. Lett. 2013;15:4354–4357. doi: 10.1021/ol401851c. [DOI] [PubMed] [Google Scholar]
- 31.Chi Y., Zhou Y., Zhang X. Highly enantioselective reductive amination of simple aryl ketones catalyzed by Ir-f-binaphane in the presence of titanium(IV) isopropoxide and iodine. J. Org. Chem. 2003;68:4120–4122. doi: 10.1021/jo026856z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.