

Abstract
Efficient metal catalyzed C–H arylation of 8-alkyl-thiazolo[5,4-f]-quinazolin-9-ones was explored for SAR studies. Application of this powerful chemical tool at the last stage of the synthesis of kinase inhibitors allowed the synthesis of arrays of molecules inspired by fragment-growing studies generated by molecular modeling calculations. Among the potentially active compounds designed through this strategy, FC162 (4c) exhibits nanomolar IC50 values against some kinases, and is the best candidate for the development as a DYRK kinase inhibitor.
Keywords: thiazolo[5,4-f]quinazolin-9(8H)-ones; microwave-assisted synthesis; C–H arylation; protein kinases; DYRK1A; CDK5; GSK-3; CLK1; CK1
1. Introduction
The increasing presence of sulfur in organic compounds of interest in veterinary applications and in medicine has motivated our investment in research programs dealing with the chemical and pharmacological evaluation of fused thiazoles, mostly inspired by marine alkaloids [1,2,3,4,5]. In this context, the design and synthesis of sulfur-containing heterocycles able to inhibit the catalytic activity of kinases have been explored [6,7,8,9,10,11,12,13,14,15,16]. Phosphorylation of proteins by these enzymes is a universal mechanism used by cells to control major physiological phenomena, and many diseases are associated with abnormal kinase activities [17,18]. In the last decade, 43 kinase inhibitors, mostly tyrosine kinase inhibitors, have been approved by the US Food and Drug Administration (FDA) [19,20,21], mainly for cancer therapy. Nowadays, the field is rapidly expanding towards serine/threonine kinases, including for other therapeutic indications (e.g. neurodegenerative diseases) [22,23]. Our group is now focused on the regulation of dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A), a conserved eukaryotic kinase that belongs to the DYRK family. This family is also comprising DYRK1B, DYRK2, DYRK3, and DYRK4 [24,25,26]. DYRK kinases belong to the CMGC group, which includes cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK), and Ccd2-like kinases (CLKs) [27].
Recently, the kinase inhibitory potency of various N-aryl thiazolo[5,4-f]quinazolin-4-amines has been demonstrated, aiming at the improved treatment of Down syndrome (DS), early Alzheimer’s disease (AD), and cancers [12,13,14]. Specifically, a series of tricyclic aminopyrimidine derivatives was synthesized and evaluated on DYRK1A and DYRK1B. Five derivatives (EHT series) displayed single-digit nanomolar or subnanomolar IC50 values, and were quite specific towards the CMGC group (Scheme 1) [28].
Scheme 1.

6-Aminobenzo[d]thiazole-2,7-dicarbonitrile, a versatile molecular platform for global chemistry strategy and structures of the five best DYRK1A/1B inhibitors. Harmine and Leucettine 41 (L41) are positive control inhibitors.
At the same time, novel thiazolo[5,4-f]quinazolin-9(8H)-ones inspired by the EHT series [15,16] were synthesized by further modifications involving the development of a new synthetic methodology. The novel compounds were evaluated for their ability to modulate activity of kinases of the CGMC group, using in vitro enzyme functional assays. Switching the C9-aminosubstituted quinazolines (EHT series, Scheme 1) for the thiazolo[5,4-f]quinazolin-9(8H)-one analogues slightly reduced the activity, but the key effects of the carbimidate functional group in C2 were highlighted.
Based on the results obtained with the crystal structure of DYRK2 in complex with EHT products [29] (Scheme 1, Figure 1), docking experiments and calculations were performed, and resulting models revealed that 9-oxo-inhibitors displayed binding modes identical to that of their 9-amino-congeners (Figure 1, left). As outlined in Figure 1, the methyl carbimidate function points to the hinge region with its nitrogen group.
Figure 1.

Results of molecular modeling studies (docking experiments) (left) and schematic representation (right) of the predicted binding modes of thiazolo[5,4-f]quinazolin-9(8H)-ones (Scheme 2, in DYRK1A).
On the basis of these new targeted thiazolo[5,4-f]quinazolin-9(8H)-ones, a fragment-growing approach was performed using a novel in silico tool that drills down through, to evaluate hundreds of thousands fragments extracted from co-crystallized kinase/inhibitor complexes. This innovative and appealing tool [30] generated more than a thousand novel compounds, sharing the same non-classical binding mode of the initial scaffold. Interestingly, addition of aromatic fragments on C2 seemed to increase the interaction with the hinge region (Scheme 3). Since the slight modification of the structure triggered by the introduction of an aryl residue upon the skeleton could have a major impact on its biological profile, a library of novel C2-arylated N8-alkyl thiazolo[5,4-f]quinazolin-9(8H)-ones was envisioned by addition of (hetero)-aromatic fragments (Scheme 3).
Scheme 3.

Fragment-growing anchor points envisioned on the thiazolo[5,4-f]quinazolin-9(8H)-one scaffold (left). Schematic representation of the predicted binding modes and retrosynthetic route to C2-arylated derivatives suggested via C–H arylation of the former C2-H thiazole precursor (right).
This article focuses on the design and synthesis of an array of 2-aryl-N8-alkylthiazolo[5,4-f]quinazolin-9(8H)-ones (series 4) which were evaluated as potential kinase inhibitors. According to our recent experience in carbon–carbon bond formation [31,32], a regioselective C–H bond activation was planned to provide the corresponding C2-arylated valuable compounds (Scheme 4). Most of the syntheses described in this paper were achieved under microwave irradiation as a powerful alternative to traditional heating with economic and environmental benefits [33].
Scheme 4.

Retrosynthetic pathway envisioned.
2. Results and Discussion
2.1. Chemistry
The target thiazolo[5,4-f]quinazolin-9(8H)-ones are substituted at N8 position by an aliphatic chain and modified in C2 (Scheme 1) by an aromatic fragment, as predicted by the modeling studies. The envisioned retrosynthetic pathway is depicted in Scheme 4. Given that selective C–H arylation is an ideal strategy for late-stage functionalization, N8-benzylated-thiazolo-quinazolin-9(8H)-one (1) was chosen as model substrate.
The synthesis of the key intermediate 1 was previously reported in six steps (overall yield: 38%), starting from commercially available 5-nitroanthranilic acid under microwave irradiation [16]. The crucial steps were (a) reaction of an aromatic amine with 4,5-dichloro-1,2,3-dithiazolium chloride (Appel’s salt), a very versatile and useful sulfur-containing reactant [34]; (b) Cu-mediated cyclisation of the imino-1,2,3-dithiazole resulting from the nucleophilic attack of the amine allowed access to N8-benzyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carbonitrile, which was decyanated (CN group hydrolysis and decarboxylation) by heating in HBr. This route has the advantage to be scalable, allowing an easy synthesis of the key intermediate 1 at a multigram scale (Scheme 5).
Scheme 5.

Six step synthesis of the key N8-benzylthiazolo[5,4-f]quinazoline (1) from 5-nitroanthranilic acid [16].
The most efficient conditions for the selective C2-H arylation of thiazolo[5,4-f]quinazolin-9(8H)-one 1 with aryl halides were previously determined [31,32]. The starting thiazolo[5,4-f]quinazolin-9(8H)-one 1 was heated in a sealed tube at 120 °C for 10 min in the presence of copper iodide (CuI, 1.0 equiv) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, 2.0 equiv) as a base, in dry DMF. After addition of Pd(OAc)2 (10 mol%) and the appropriate aryl halide (2.0 equiv), the reaction mixture was heated again at 120 °C for 5 h (Scheme 6, Table 1).
Scheme 6.

Selective C2-H arylation of 1 with aryl halides.
Table 1.
Chemical structures and yields obtained for the synthesis a of series 2a–j (R1, R2, and R3) and 3a–c b (R, X, and Y).
| Product | R1 | R2 | R3 | X(Ar) | Yield c (%) |
| 2a | H | H | H | Br | 92 |
| 2b | Me | H | H | Br | 86 |
| 2c | MeO | H | H | I | 63 |
| 2d | Cl | H | H | I | 87 |
| 2e | F | H | H | I | 71 |
| 2f | CN | H | H | I | 59 |
| 2h | NMe2 | H | H | Br | 87 |
| 2i | Cl | H | Cl | I | 62 |
| 2j | Cl | Cl | H | I | 64 |
| Product | X | Y | R | X(Ar) | Yield c (%) |
| 3a | CH | N | H | I | 69 |
| 3b | N | N | H | I | 47 |
| 3c | CH | N | OMe | I | 29 |
a Premixing 1, DBU, and CuI, 10 min before adding ArI or ArBr, Pd(OAc)2, and stirring for 5 h; b In the case of 3-iodopyridine, TBD [31] was used as base instead of DBU; c Isolated yields.
For the SAR studies, aryl iodides and bromides screened as coupling partners were mainly inspired by the Topliss tree [35], in order to maximize the chances of synthesizing the most potent compounds of series 2 as early as possible (Scheme 6, Table 1). Extending aryl partners to hetero-aryl halides allowed the synthesis of series 3 compounds in moderate yields. It should be noted that low yields primarily observed in the synthesis of 3a (<20%) was explained by difficult work-up and purification. A better yield of 43% was obtained by using 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) [36] as a base instead of DBU.
The inhibitory potency of series 2 and 3 was evaluated according to standard methods [15,16] on a panel of kinases (for details see kinase profiling paragraph). Among the thiazolo[5,4-f]quinazolin-9(8H)-ones tested, only two molecules of series 3 (3a and 3b) exhibited micromolar IC50 values against kinases CLK1 and GSK3, and nanomolar range inhibition against DYRK1A (see Table 1 and Scheme 6).
Taking these preliminary results into account, series 4 was designed by keeping the 3-pyridinyl moiety in position C2, and modifying the alkyl substituents in position N8 of the thiazolo[5,4-f]quinazolin-9(8H)-ones. The envisioned modifications depicted in Scheme 7 were inspired by previous work on carbimidate derivatives described in Scheme 2 [15,16].
Scheme 7.

The two kinase active derivatives 3a and 3b and new compounds (series 4a–f) suggested by the first active products.
Scheme 2.

Schematic access to thiazolo[5,4-f]quinazolin-9(8H)-ones, a family of multitarget kinase inhibitors.
The target molecules (series 4) were 2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-ones, substituted in position N8 by an aliphatic fragment (Scheme 1). Alkylation of the corresponding debenzylated compound 3 (Scheme 6) was initially envisioned. Usual methods for the cleavage of the benzyl group [31] of compound 3a led to the expected compound in yields up to 42%. Nevertheless, this N8 deprotected derivative was found to be insoluble in most organic solvents, therefore discarding the alkylation pathway. Finally, the target molecules (series 4) were synthesized via the polyfunctionalized methyl 6-amino-2-cyanobenzo[d]thiazole-7-carboxylate (5), a versatile precursor already described in previous studies [16]. Here, again, the key step in the synthesis of 5 involves the sulfur-rich Appel’s salt, and cyclization of the intermediate imino-1,2,3-dithiazole which was transformed into the target benzothiazole (Scheme 8). In this pathway, the quinazolinone part was formed at the last stage of the synthesis.
Scheme 8.

Retrosynthetic route of series 4 products using compound 5 as intermediate.
Treatment of aminoester 5 with 1.5 equivalents of Vilsmeier–Haack reagent in dichloromethane at room temperature gave (E)-methyl 2-cyano-6-([(dimethylamino)methylene]amino)benzo[d]-thiazole-7-carboxylate (6) in excellent yield (90%). This formimidamide was heated at 100 °C under microwave irradiation in the presence of 1.05 equivalents of appropriate amines, in acetic acid. After an irradiation time of 15 min, the corresponding N8-substituted-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carbonitriles (series 7a–f) were obtained in moderate to good yields (43–86%) [15] (Scheme 9, Table 2). Access to C2-H derivatives (series 8a–f) was finally obtained by heating the corresponding precursors in HBr in sealed vials, under microwave irradiation.
Scheme 9.

Synthesis of series 7a–f, 8a–f, and 4a–f.
Table 2.
Chemical structures (R1) and yields obtained for the synthesis of series 7a–f, 8a–f, and 4a–f.
| –R1 | Compound | Yield a (%) | Compound | Yield a (%) | Compound | Yield a (%) |
|---|---|---|---|---|---|---|

|
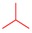
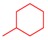
7a | 70 | 8a | 98 | 4a | 14 |
|
7b | 43 | 8b | 99 | 4b | 64 |
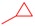
|
7c | 86 | 8c | 86 | 4c | 43 |
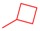
|
7d | 65 | 8d | 97 | 4d | 57 |
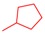
|
7e | 60 | 8e | 98 | 4e | 55 |
|
7f | 50 | 8f | 98 | 4f | 54 |
a Isolated yield.
After the late-stage hetero-arylation, the small library of compounds was evaluated for their kinase inhibitory properties. As described for series 2a–f and 3a–c, the kinase profiling of series 4a–f (for details see kinase profiling paragraph) highlighted the cyclopropyl derivative 4c, which exhibits noteworthy activity against kinases with nanomolar IC50 values for DYRK1A, CLK1 and GSK3 (Scheme 9). Substituting 2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one with alkyl groups, such as methyl, iso-propyl, or cycloakyl containing at least 4 carbons, resulted in complete loss of activity (see Table 2). Following this result, a new series, 9a–j, was envisioned; by analogy with the first series 2a–j, the palladium-catalyzed CH-arylation was applied to 8c, and afforded series 9a–j and compound 10 in moderate to good yields (Scheme 10 and Table 3) [32].
Scheme 10.

Synthetic route to series 9a–j and 10 (R, X, and Y).
Table 3.
Chemical structures and yields obtained for the synthesis a of series 9a–j (R1, R2, and R3).
| Product | R1 | R2 | R3 | X(Ar) | Yield b (%) |
|---|---|---|---|---|---|
| 9a | H | H | H | Br | 58 |
| 9b | Me | H | H | Br | 64 |
| 9c | MeO | H | H | I | 76 |
| 9d | Cl | H | H | I | 67 |
| 9e | F | H | H | I | 52 |
| 9f | CN | H | H | I | - c |
| 9h | NMe2 | H | H | Br | 31 |
| 9i | Cl | H | Cl | I | 65 |
| 9j | Cl | Cl | H | I | 69 |
a Premixing 1, DBU, and CuI, 10 min before adding ArI or ArBr, Pd(OAc)2 and stirring for 5 h; b Isolated yields; c Not prepared.
Microwave-assisted regioselective C–H bond arylation of the thiazolo[5,4-f]quinazolin-9(8H)-one skeleton thus provides an efficient and simplified route towards these valuable sulfur-containing bioactive heterocycles.
2.2. Kinase Profiling
Products of series 2a–j, 3a–c, 4a–f, 9a–i, and 10 were evaluated in five different in vitro kinase assays: CDK5/p25, CK1δ/ε (casein kinase 1), GSK-3α/β, DYRK1A (dual-specificity, tyrosine phosphorylation regulated kinase), and CLK1 [37,38,39].
All compounds were first tested at a final concentration of 10 μM. Compounds showing less than 50% inhibition were considered as inactive (IC50 >10 μM). Compounds displaying more than 50% inhibition at 10 μM were next tested over a wide range of concentrations (usually from 0.01 to 10 μM), and IC50 values were determined from the dose–response curves (Sigma-Plot). Harmine (Table 4), a β-carboline alkaloid (Scheme 1) known to inhibit DYRK1A [40], was used as a positive control.
Table 4.
Kinase inhibitory activity a,b,c of the thiazolo[5,4-f]quinazolin- series (2a–j, 3a–c, 4a–i, and 10).
| Compounds | CDK5/p25 | CK1δ/ε | CLK1 | DYRK1A | GSK-3α/β |
|---|---|---|---|---|---|
| 2a–j | >10 | >10 | >10 | >10 | >10 |
| 3a | >10 | >10 | 2.0 | 0.012 | 3.7 |
| 3b | >10 | >10 | 3.33 | 0.133 | 6.0 |
| 3c | >10 | >10 | >10 | >10 | >10 |
| 4a | n.t. d | >10 | >10 | >10 | >10 |
| 4b | n.t. | >10 | >10 | >10 | >10 |
| 4c (FC162) | n.t. | 6.0 | 0.018 | 0.011 | 0.068 |
| 4d | n.t. | >10 | >10 | >10 | >10 |
| 4e | n.t. | >10 | >10 | >10 | >10 |
| 4f | n.t. | >10 | >10 | >10 | >10 |
| 9a–i | >10 | >10 | >10 | >10 | ≥10 |
| 10 | >10 | >10 | >10 | >10 | ≥10 |
| Harmine | >10 | 1.5 | 0.026 | 0.029 | >10 |
a IC50 values are reported in μM.; b Kinase activities were assayed in triplicate; c Typically, the standard deviation of single data points was below 10%; d n.t., not tested.
Results provided in Table 4 demonstrate that none of the thiazolo[5,4-f]quinazolin-9(8H)-ones synthesized in series 2a–j and 9a–i showed significant inhibitory activity against the set of five kinases.
In series 4a–f, only 8-cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4c) (also called FC162) exhibited nanomolar IC50 values (11, 18, and 68 nM against DYRK1A, CLK1, and GSK3, respectively).
In series 3a–c, two molecules exhibited nanomolar to submicromolar IC50 values against DYRK1A (IC50: 11 nM and 133 nM for 3a and 3b, respectively). It is remarkable that these two derivatives were poor inhibitors of CLK1 and GSK3 compared with 4c, suggesting a high selectivity for DYRK1A. Except for kinase GSK3, the inhibition profile of 4c for CLK1 and DYRK1A was close to that of Harmine.
Docking calculations were next performed in order to predict the molecular interactions of FC162 with DYRK1A. Two main binding modes (Figure 2) were obtained. The first one maintained the initial position of the thiazolo[5,4-f]quinazolin-9(8H)-one skeleton, in which an interaction with the catalytic lysine is retrieved. The pyridine moiety interacts with the hinge region by forming a hydrogen bond with the backbone NH of Leu231. The second possible binding mode showed a complete flip of the molecule in the cavity. Indeed, the pyridine moiety interacted with the catalytic lysine, while the thiazolo[5,4-f]quinazolin-9(8H)-one skeleton interacted with the hinge region through hydrogen bond. The docking score of the two poses was quite similar, thus, both binding modes are equally possible for this compound.
Figure 2.

Predicted binding mode of FC162 by docking calculation. Left, first predicted binding mode (green), with the same orientation of the skeleton, but slightly shifted. Right, second predicted binding mode, in which the skeleton is flipped compared to its initial placement (in brown).
The SAR study revealed that 3a and 4c with a 3-pyridinyl group in position 2 had a higher activity than the series of phenylated derivatives 2 and 9. These results are notably in agreement with the fragment-growing experiments, which suggested replacement of the imidate group by a more stable heteroaromatic substituent (Figure 3). The fact that 3c and 10 were inactive demonstrates the importance of a free 3-pyridinyl group in C2, and may suggest that the planarity between the pyridinyl group and the tricyclic scaffold, broken with an ortho substituent on the pyridinyl group, is compulsory for DYRK inhibitory activity.
Figure 3.

Comparative study of the activity of FC162 (4c) with related molecules.
Other studies are currently in progress, in order to optimize interactions with hinge amino acid residues, and aiming to improve the affinity and selectivity of compounds for the targeted CLK1 and DYRK kinases.
3. Material and Methods
3.1. General Information
All reagents were purchased from commercial suppliers and were used without further purification, except for DMF, which was stored under argon and activated molecular sieves. All reactions were monitored by thin-layer chromatography with silica gel 60 F254 (Merck Ltd., KGaA, Darmstadt, Germany) precoated aluminium plates (0.25 mm). Visualization was performed with a UV light at wavelengths of 254 nm. Purifications were conducted with a flash column chromatography system (Puriflash) equipped with a dual UV/vis spectrophotometer (200–600 nm), a fraction collector (176 tubes), a dual piston pump (1 to 200 mL/min, Pmax = 15 bar), which allowed quaternary gradients, and an additional inlet for air purge (Interchim, Montluçon, France). Melting points of solid compounds were measured with a SMP3 Melting Point instrument (STUART, Bibby Scientific Ltd., Roissy, France) with a precision of 1.5 °C. IR spectra were recorded with a Spectrum 100 Series FTIR spectrometer (PerkinElmer, Villebon S/Yvette, France). Liquids and solids were investigated with a single-reflection attenuated total reflectance (ATR) accessory; the absorption bands are given in cm−1. NMR spectra (1H and 13C) were acquired at 295 K using an AVANCE 300 MHz spectrometer (Bruker, Wissembourg, France) at 300 and 75.4 MHz, using TMS as an internal standard. Coupling constants J are in Hz, and chemical shifts are given in ppm. Signals in 13C spectra were assigned based on the result of 13CDEPT135 experiments (see Supplementary Materials). Mass spectrometry was performed by the Mass Spectrometry Laboratory of the University of Rouen. The mass spectra (ESI, EI, and field desorption (FD)) were recorded with an LCP 1er XR spectrometer (WATERS, Guyancourt, France). Microwave-assisted reactions were carried out in sealed tubes with a Biotage Initiator microwave synthesis instrument, and temperatures were measured by IR-sensor (Biotage, Uppsala, Sweden). Time indicated in the various protocols is the time measured when the mixtures were at the programmed temperature. The purity of all tested compounds was determined by chromatographic analysis performed at 25 °C on Ultimate 3000 (Thermo Scientific, Les Ulis, France) with a quaternary pump equipped with a photodiode array detector (DAD) managed at 254 nm. Column was a Luna C18 (150 mm × 4.6 mm; 3 μm particle size) provided by Phenomenex (Le Pecq, France). The mobile phase was water (A) and acetonitrile (B) (v/v); starting condition is 90% A and 10% B, in which the solvent B changed to 10% to 90% in 4% by min. Flow rate was 0.5 mL/min, and 5 µL were injected. The percentage of purity of all products was more than 98%.
3.2. Chemistry
Compounds 1 and 7a–f were described in ref [15]; compounds 2a–i and 3a,b were described in ref [31]; compounds 2j, 3c, 4c, 9j, and 10 were described in ref [32]; The new products 8a–f and 4a, 4b and 4d–f are described below. The lead molecule FC162 (4a) is described again.
3.2.1. General Procedure for the Synthesis of 8-Alkyl-thiazolo[5,4-f]quinazolin-9(8H)-one (8a–f) from 8-Alkyl-9-oxo-8,9-dihydrothiazolo[5,4-f]quinazoline-2-carbonitrile (7a–f)
In a 2–6 mL sealed tube, a suspension of the corresponding carbonitrile-bearing derivative 7a–f (0.60 mmol, 1 equiv) and hydrobromic acid (48% in water) (2 mL, 0.3 M) was irradiated under microwave for 45–90 min at 100 °C. After cooling, the resulting solution was diluted with dichloromethane, and neutralized with a saturated aqueous solution of NaHCO3 and with solid Na2CO3 (until pH 8–9). The organic layer was then dried over Na2SO4, and concentrated under reduced pressure to provide the corresponding product 8a–f.
8-Methylthiazolo[5,4-f]quinazolin-9(8H)-one (8a): Reaction time: 55 min; Yield: 98%; 112 mg; white solid; Rf: 0.47 (DCM/MeOH, 95:5; v/v); mp 221–224 °C; IR (neat) νmax 3308, 3149, 3077, 1902, 1698, 1664, 1589, 1386 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.22 (1H, s, H2), 8.48 (1H, d, J = 8.8 Hz, H4), 8.20 (1H, s, H7), 7.87 (1H, d, J = 8.8 Hz, H5), 3.73 (3H, s, CH3); 13C{1H}-NMR (CDCl3, 25 °C, 75.4 MHz): δC 160.2 (C), 157.8 (CH), 152.5 (C), 147.1 (C), 146.5 (CH), 130.2 (C), 129.3 (CH), 126.1 (CH), 116.3 (C), 34.3 (CH3). HRMS (ESI+): Calcd for C10H7N3OS [M + H]+: 218.0388; Found: 218.03386.
8-Isopropylthiazolo[5,4-f]quinazolin-9(8H)-one (8b): Reaction time: 45 min; Yield: 99%; 145 mg; beige solid; Rf: 0.69 (DCM/MeOH, 95:5; v/v); mp 237–240 °C; IR (neat) νmax 3053, 2981, 2920, 1897, 1651, 1583, 1445, 1350, 1265, 1172, 827 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.19 (1H, s, H2), 8.44 (1H, d, J = 8.8 Hz, H4), 8.25 (1H, s, H7), 7.84 (1H, d, J = 8.8 Hz, H5), 5.37–5.12 (1H, m, NCH), 1.55 (3H, s, CH3), 1.53 (3H, s, CH3); 13C{1H}-NMR (CDCl3, 25 °C, 75.4 MHz): δC 159.6 (C), 157.7 (CH), 152.5 (C), 146.6 (C), 143.4 (CH), 130.6 (C), 129.4 (CH), 126.0 (CH), 116.4 (C), 47.0 (CH), 22.2(2 × CH3). HRMS (ESI+): Calcd for C12H11N3OS [M + H]+: 246.0701; Found: 246.0708.
8-Cyclopropylthiazolo[5,4-f]quinazolin-9(8H)-one (8c), was described in ref [15], typical data: 1H-NMR (CDCl3, 300 MHz): δ = 9.22 (s, 1H, H2), 8.49 (d, J = 8.7 Hz, 1H, H4), 8.26 (s, 1H, H7), 7.87 (d, J = 8.7 Hz, 1H, H5), 3.50–3.25 (m, 1H, NCH), 1.37–1.17 (m, 2H, CH), 1.12–0.91 (m, 2H, CH). HRMS (ESI+): m/z calcd for C12H10N3OS: 244.0545; found: 244.0542.
8-Cyclobutylthiazolo[5,4-f]quinazolin-9(8H)-one (8d): Reaction time: 45 min; Yield: 97%; 148 mg; white solid; Rf: 0.76 (DCM/MeOH, 95:5; v/v); mp 242–245 °C; IR (neat) νmax 3045, 2992, 2957, 2879, 1655, 1601, 1585, 1349, 1277, 1163, 856 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.20 (1H, s, H2), 8.46 (1H, d, J = 8.8 Hz, H4), 8.31 (1H, s, H7), 7.86 (1H, d, J = 8.8 Hz, H5), 5.21–5.06 (1H, m, NCH), 2.71–2.55 (2H, m, CH2), 2.54–2.35 (2H, m, CH2), 2.05–1.90 (2H, m, CH2); 13C-NMR (CDCl3, 25 °C, 75.4 MHz): δC 159.0 (C), 157.8 (CH), 152.6 (C), 146.8 (C), 143.8 (CH), 130.5 (C), 129.5 (CH), 126.1 (CH), 116.3 (C), 50.9 (CH), 29.9 (2 × CH2), 15.5 (CH2). HRMS (ESI+): Calcd for C13H11N3OS [M + H]+: 258.0701; Found: 258.0701.
8-Cyclopentylthiazolo[5,4-f]quinazolin-9(8H)-one (8e): Reaction time: 45 min; Yield: 98%; 160 mg; white solid; Rf: 0.69 (DCM/MeOH, 95:5; v/v); mp 214–217 °C; IR (neat) νmax 2960, 2874, 1661, 1585, 1348, 1259, 1154, 841, 818 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.22 (1H, s, H2), 8.49 (1H, d, J = 8.8 Hz, H4), 8.27 (1H, s, H7), 7.87 (1H, d, J = 8.8 Hz, H5), 5.36–5.20 (1H, m, NCH), 2.38–2.21 (2H, m, CH2), 2.12–1.74 (2H, m, CH2); 13C{1H}-NMR (CDCl3, 25 °C, 75.4 MHz): δC 159.8 (C), 157.7 (CH), 152.3 (C), 146.4 (C), 144.1 (CH), 130.4 (C), 129.2 (CH), 125.8 (CH), 116.1 (C), 56.4 (CH), 32.1 (2 × CH2), 24.5 (2 × CH2). HRMS (ESI+): Calcd for C14H13N3OS [M + H]+: 272.0858; Found: 272.0855.
8-Cyclohexylthiazolo[5,4-f]quinazolin-9(8H)-one (8f): Reaction time: 90 min; Yield: 98%; 168 mg; white solid; Rf: 0.75 (DCM/MeOH, 95:5; v/v); mp 227–230 °C; IR (neat) νmax 3082, 3053, 2932, 2850, 1895, 1665, 1588, 1466, 1448, 1273, 1135, 845, 814 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.19 (1H, s, H2), 8.44 (1H, d, J = 8.8 Hz, H4), 8.25 (1H, s, H7), 7.83 (1H, d, J = 8.8 Hz, H5), 5.03–4.71 (1H, m, NCH), 2.20–1.87 (4H, m, CH2), 1.84–1.62 (3H, m, CH2), 1.62–1.41 (2H, m, CH2), 1.36–1.17 (1H, m, CH2); 13C{1H}-NMR (CDCl3, 25 °C, 75.4 MHz): δC 159.6 (C), 157.7 (CH), 152.5 (C), 146.5 (C), 143.7 (CH), 129.4 (CH), 126.0 (CH), 116.4 (C), 54.3 (CH), 32.7 (2 × CH2), 26.0 (2 × CH2), 25.3 (CH2). HRMS (ESI+): Calcd for C15H15N3OS [M + H]+: 286.114; Found: 286.1021.
3.2.2. General Procedure for the Synthesis of 8-Alkyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4a–f) from 8-Alkyl-thiazolo[5,4-f]quinazolin-9(8H)-one (8a–f)
Thiazolo[5,4-f]quinazolin-9(8H)-one 8a–f (0.341 mmol), copper iodide (0.065 g, 0.341 mmol, 1 equiv), and TBD (95 mg, 0.682 mmol, 2.0 equiv) in dry DMF (850 µL) were added to a 2 mL glass vial, which was sealed under argon atmosphere. The mixture was stirred under microwave irradiation at 120 °C for 10 min. Then, Pd(OAc)2 (7.6 mg, 0.034 mmol, 10 mol%) and 3-iodopyridine (0.140 g, 0.682 mmol, 2.0 equiv) were added to the mixture and purged with argon. The reaction was then stirred under microwave irradiation at 120 °C for 5 h. The resulting solution was diluted with dichloromethane, and washed three times with a 5% aqueous ammonia solution, then with water and brine. The organic layer was dried over Na2SO4 and concentrated under vacuum. The crude product was purified by flash chromatography on silica gel with MeOH/CH2Cl2 as eluent (1/0 to 95:5; v/v), to afford the corresponding product.
8-Methyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4a): Yield: 14%; 14 mg; Yellow solid; Rf: 0.36 (DCM/MeOH, 95:5; v/v); mp 272–275 °C; IR (neat) νmax 3068, 1651, 1588, 1446, 1346, 699 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.39 (1H, br s, HAr), 8.73 (1H, d, J = 4.8 Hz, HAr), 8.58–8.36 (2H, m, HAr + H4), 8.19 (1H, s, H7), 7.86 (1H, d, J = 8.8 Hz, H5), 7.47 (1H, dd, J = 8.0, 4.8 Hz, HAr), 3.73 (3H, s, CH3); 13C{1H}-NMR (CDCl3, 25 °C, 75.4 MHz): δC 168.36 (C), 160.40 (C), 153.29 (C), 151.82 (CH), 148.66 (CH), 147.20 (C), 146.60 (CH), 134.71 (CH), 131.54 (C), 129.77 (C), 129.25 (CH), 126.60 (CH), 124.06 (CH), 116.48 (C), 34.37 (CH3). HRMS (ESI+): Calcd for C15H10N4OS [M + H]+: 295.0654; Found: 295.0668.
8-Isopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4b): Yield: 64%; 70 mg; Yellow solid; Rf: 0.46 (DCM/MeOH, 95:5; v/v); mp 202–205 °C; IR (neat) νmax 3540, (C), 151.82 (CH), 148.66 (CH), 147.20 (C), 146.60 (CH), 134.71 (CH), 131.54 (C), 129.77 (C), 129.25 (CH), 126.60 (CH), 124.06 (CH), 116.48 (C), 34.37 (CH3). HRMS (ESI+): Calcd for C15H10N4OS [M + H]+: 295.0654; Found: 295.0668.
8-Cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4c or FC162) [34]: yield: 69%; 91 mg; beige powder; Rf: 0.45 (DCM/ EtOAc, 1/1, v/v); mp: 263–266°C; IR (neat) νmax 3059, 3012, 2114, 1659, 1588, 1449, 1344, 1295, 1024, 837 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.41 (d, J = 2.3 Hz, 1H, HAr), 8.75 (d, J = 4.9 Hz, 1H, HAr), 8.54–8.38 (m, 2H, HAr + H4), 8.26 (s, 1H, H7), 7.88 (d, J = 8.7 Hz, 1H, H5), 7.48 (dd, J = 8.0, 4.9 Hz, 1H, CHAr), 3.45–3.26 (m, 1H, NCH), 1.36–1.24 (m, 2H, CH), 1.14–0.96 (m, 2H, CH). 13C-NMR (CDCl3, 75.4 MHz): δ = 168.5 (C), 161.3 (C), 153.4 (C), 151.9 (CH), 148.7 (CH), 146.7 (CH), 146.6 (C), 134.7 (CH), 131.7 (C), 129.8 (C), 129.3 (CH), 126.6 (CH), 124.1 (CH), 116.4 (C), 29.8 (CH), 6.7(2 × CH2). HRMS (ESI+): m/z calcd for 12C171H1214N416O32S [M + H]+: 321.0807; Found: 321.0810.
8-Cyclobutyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4d): Yield: 57%; 64 mg; Yellow solid; Rf: 0.45 (DCM/MeOH, 95:5; v/v); mp 251–254 °C; IR (neat) νmax 3072, 2992, 2948, 2865, 1906, 1655, 1584, 1347, 848 cm−1; 1H-NMR (CDCl3 + D2O, 25 °C, 300 MHz): δH 9.39 (1H, br s, HAr), 8.72 (1H, d, J = 4.8, HAr), 8.45–8.29 (2H, m, HAr + H4), 8.30 (1H, s, H7), 7.86 (1H, d, J = 8.8 Hz, H5), 7.46 (1H, dd, J = 8.0, 4.8 Hz, HAr), 5.18–5.09 (1H, m, NCH), 2.70–2.60 (2H, m, CH2), 2.54–2.40 (2H, m, CH2), 2.04–1.94 (2H, m, CH2); 13C{1H}-NMR (CDCl3 + D2O, 25 °C, 75.4 MHz): δC 168.2 (C), 160.0 (C), 153.2 (C), 151.8 (CH), 148.6 (CH), 146.7 (C), 143.8 (CH), 134.7 (CH), 131.6 (C), 129.8 (C), 129.2 (CH), 126.5 (CH), 124.1 (CH), 116.3 (C), 51.1 (CH), 29.8 (2 × CH2), 15.5 (CH2). HRMS (ESI+): Calcd for C18H14N4OS [M + H]+: 335.0967; Found: 335.0974.
8-Cyclopentyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4e): Yield: 65%; 77 mg; Yellow solid; Rf: 0.43 (DCM/MeOH, 95:5; v/v); mp 212–215 °C; IR (neat) νmax 3064, 3964, 2872; 1902, 1645, 1584, 1451, 828 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.40 (1H, br s, HAr), 8.73 (1H, d, J = 4.8 Hz, HAr), 8.50–8.38 (2H, m, HAr + H4), 8.25 (1H, s, H7), 7.86 (1H, d, J = 8.8 Hz, H5), 7.46 (dd, J = 8.0, 4.8 Hz, HAr), 5.39–5.08 (1H, m, NCH), 2.46–2.16 (2H, m, CH2), 2.14–1.67 (4H, m, CH2); 13C{1H}-NMR (CDCl3, 25 °C, 75.4 MHz): δC 168.0 (C), 159.8 (C), 152.9 (C), 151.5 (CH), 148.4 (CH), 146.3, 144.1 (CH), 134.4 (CH), 131.5 (C), 129.8 (C), 128.9 (CH), 126.2 (CH), 124.1 (CH), 116.2 (C), 56.8 (CH), 32.1 (2 × CH2), 24.6 (2 × CH2). HRMS (ESI+): Calcd for C19H16N4OS [M + H]+: 349.1123; Found: 349.1115.
8-Cyclohexyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one (4f): Yield: 54%; 67 mg; Yellow solid; Rf: 0.43 (DCM/MeOH, 95:5; v/v); mp 201–204 °C; IR (neat) νmax 3309, 3146, 1076, 2920, 1902, 1661, 1587, 1350, 1333, 826 cm−1; 1H-NMR (CDCl3, 25 °C, 300 MHz): δH 9.38 (1H, br s, HAr), 8.72 (1H, br s, HAr), 8.56–8.35 (2H, m, HAr + H4), 8.25 (1H, s, H7), 7.84 (1H, d, J = 8.7 Hz, H4), 7.45 (1H, dd, J = 8.0, 4.8 Hz, HAr), 4.96–4.79 (1H, m, NCH), 2.16–1.89 (4H, m, CH2), 1.86–1.65 (3H, m, CH2), 1.65–1.43 (2H, m, CH2), 1.38–1.16 (1H, m, CH2); 13C{1H}-NMR (CDCl3, 25 °C, 75.4 MHz): δC 168.1 (C), 159.6 (C), 153.0 (C), 151.6 (CH), 148.5 (CH), 146.3 (C), 143.7 (CH), 134.5 (CH), 131.7 (C), 129.8 (C), 129.1 (CH), 126.3 (CH), 124.0 (CH), 116.3 (C), 54.5 (CH), 32.6 (2 × CH2), 25.9 (2 × CH2), 25.2 (CH2). HRMS (ESI+): Calcd for C20H18N4OS [M + H]+: 363.1280; Found: 363.1295.
3.3. In Vitro Kinase Preparation and Assays
3.3.1. Buffers
Homogenization buffer: 25 mM MOPS; 15 mM EGTA; 15 mM MgCl2; 60 mM β-glycerophosphate; 15 mM p-nitrophenylphosphate; 2 mM dithiothreitol (DTT); 1 mM Na3VO4; 1 mM NaF; 1 mM di-sodium phenylphosphate; 1× protease inhibitor cocktail; 0.2% Nonidet P-40 substitute.
Buffer A: 10 mM MgCl2; 1 mM EGTA; 1 mM DTT; 25 mM Tris/HCl, and 50 μg/mL heparin.
Buffer B: 60 mM β-glycerophosphate; 30 mM p-nitrophenylphosphate; 25 mM MOPS pH 7.0; 5 mM EGTA; 15 mM MgCl2; 1 mM DTT; and 0.1 mM sodium vanadate.
All chemicals were purchased from Sigma-Aldrich (St. Quentin Fallavier, France), unless otherwise stated, and the protease inhibitor cocktail was from Roche (Boulogne-Billancourt, France).
3.3.2. Kinase Preparations and Assays
Kinase activities were assayed in triplicates, in buffer A or B, at 30 °C, at a final adenosine triphosphate (ATP) concentration of 15 μmol/L. Blank values were subtracted, and activities were expressed in percent (%) of the maximal activity, i.e., in the absence of inhibitors. Controls were performed with appropriate dilutions of dimethyl sulfoxide (DMSO).
CDK5/p25 (Human, recombinant) was prepared as previously described [37]. Its kinase activity was assayed in buffer A, with 1 mg of histone H1/mL, in the presence of 15 μM [γ-33P] ATP (3000 Ci/mmol; 10 mCi/mL) in a final volume of 30 μL. After 30 min incubation at 30 °C, 25 μL aliquots of supernatant were spotted onto sheets of Whatman P81 phosphocellulose paper, and 20 s later, the filters were washed eight times (for at least 5 min each time) in a solution of 10 mL phosphoric acid/L of water. The wet filters were counted in the presence of 1 mL ACS (Amersham) scintillation fluid.
GSK-3α/β (porcine brain, native) was assayed, in buffer A, with 0.5 mg BSA/mL + 1 mM DTT, using GS-1 (YRRAAVPPSPSLSRHSSPHQSpEDEEE) (pS stands for phosphorylated serine), a GSK-3 specific substrate [38], in the presence of 15 μmol/L [γ-33P] ATP (3000 Ci/mmol; 10 mCi/mL) in a final volume of 30 μL. After 30 min incubation at 30 °C, the reaction was stopped by harvesting onto P81 phosphocellulose supernatant (Whatman, Dutscher SAS, Brumath, France) using a FilterMate harvester (PerkinElmer, Courtaboeuf, France), and were washed in 1% phosphoric acid. Scintillation fluid was added, and the radioactivity measured in a Packard counter.
CLK1 (Human, recombinant, expressed in E. coli as GST fusion protein) was assayed in buffer A (+0.15 mg BSA/mL) with RS peptide (GRSRSRSRSRSR) (1 μg/assay).
CK1δ/ε (porcine brain, native) was assayed as described for CDK1, but in buffer B, and using 25 μM CKS peptide (RRKHAAIGpSAYSITA), a CK1-specific substrate [39].
DYRK1A (Human, recombinant, expressed in E. coli as GST fusion proteins) was purified by affinity chromatography on glutathione-agarose and assayed as described for CDK1/cyclin B, with with 0.5 mg BSA/mL + 1 mM DTT and using Woodtide (KKISGRLSPIMTEQ) (1.5 μg/assay) as a substrate, a residue of transcription factor FKHR.
4. Conclusions
This work demonstrates the efficacy of synthetic methodologies, such as C–H arylation of arenes and hetero-arenes for SAR studies. The application of this powerful tool at the last stage of the synthesis of kinase inhibitors allowed the synthesis of arrays of molecules inspired by fragment-growing studies generated by molecular modeling calculations. Among the potential active compounds generated through this strategy, FC162 (4c) was found to be the best candidate for development as a DYRK inhibitor.
Acknowledgments
J.D. and P.B. would like to thank Colin Bournez and Pascal Krezel for their help in using Frags2Drugs, the in silico FBDD tool.
Abbreviations
| ATP | adenosine triphosphate |
| CMGC group | group of kinases including cyclin-dependent kinases (CDKs), mitogen-activated protein kinases (MAP kinases), glycogen synthase kinases (GSK) and Cdc2-like kinases (CLKs) |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DMF | N,N-dimethylformamide |
| DMFDMA | N,N-dimethylformamide dimethyl acetal |
| NBS | N-bromosuccinimide |
| TBD | 1,5,7-triazabicyclo[4.4.0]dec-5-ene |
| SAR | structure–activity relationship |
Supplementary Materials
The following are available online. 1H-NMR and 13C-NMR spectra of new compounds 8a–f and 4a–f.
Author Contributions
T.B. and C.F. conceived the project and designed the experiments. F.C. and M.H. performed the experimental work, accompanied by C.D.-B.; L.B. and E.P. performed spectroscopic analysis for purity of the compounds tested. J.D. and P.B. were involved in the molecular modeling and fragment-growing experiments. L.M. designed and supervised the biological experiments. T.B. wrote the manuscript with the cooperation of C.F. All authors discussed the results and commented on the manuscript.
Funding
Financial support from the MESR (French Ministère de l’Enseignement Supérieur & de la Recherche) is gratefully acknowledged for the doctoral fellowships to F.C. and M.H.; T.B. and his co-workers (F.C., M.H., C.D.-B., L.B., E.P., P.B. and C.F.) thank the LABEX SynOrg (ANR-11-LABX-0029) for financial support. This research was supported by grants from the “Fonds Unique Interministériel” (FUI) TRIAD project and Conseil Régional de Bretagne (L.M.) and the “Fondation Jérôme Lejeune” (L.M.).
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Footnotes
Sample Availability: Samples of compound FC162 (4c) are available from the authors for academic studies with Material Transfer Agreement (MTA).
References
- 1.Bénéteau V., Besson T. Synthesis of novel pentacyclic pyrrolothiazolobenzoquinolinones, analogs of natural marine alkaloids. Tetrahedron Lett. 2001;42:2673–2676. doi: 10.1016/S0040-4039(01)00258-1. [DOI] [Google Scholar]
- 2.Lamazzi C., Chabane H., Thiéry V., Pierre A., Léonce S., Pfeiffer B., Renard P., Guillaumet G., Besson T. Synthesis and cytotoxic evaluation of novel thiazolocarbazoles. J. Enz. Inhib. Med. Chem. 2002;17:397–401. doi: 10.1080/1475636021000005703. [DOI] [PubMed] [Google Scholar]
- 3.Frère S., Thiéry V., Bailly C., Besson T. Novel 6-substituted benzothiazol-2-yl indolo[1,2-c]quinazolines and benzimidazo[1,2-c]quinazolines. Tetrahedron. 2003;59:773–779. doi: 10.1016/S0040-4020(02)01593-4. [DOI] [Google Scholar]
- 4.Chabane H., Pierre A., Léonce S., Pfeiffer B., Renard P., Thiéry V., Guillaumet G., Besson T. Synthesis and cytotoxic activity of thiazolofluorenone derivatives. J. Enzym. Inhib. Med. Chem. 2004;19:567–575. doi: 10.1080/14756360400004607. [DOI] [PubMed] [Google Scholar]
- 5.Testard A., Picot L., Fruitier-Arnaudin I., Piot J.M., Chabane H., Domon L., Thiéry V., Besson T. Microwave-assisted synthesis of novel thiazolocarbazoles and evaluation as potential anticancer agents. Part III. J. Enz. Inhib. Med. Chem. 2004;19:467–473. doi: 10.1080/14756360412331280491. [DOI] [PubMed] [Google Scholar]
- 6.Logé C., Testard A., Thiéry V., Lozach O., Blairvacq M., Robert J.-M., Meijer L., Besson T. Novel 9-oxo-thiazolo[5,4-f]quinazoline-2-carbonitrile derivatives as dual cyclin-dependent kinase 1 (CDK1)/glycogen synthase kinase-3 (GSK-3) inhibitors: Synthesis, biological evaluation and molecular modeling studies. Eur. J. Med. Chem. 2008;43:1469–1477. doi: 10.1016/j.ejmech.2007.09.020. [DOI] [PubMed] [Google Scholar]
- 7.Testard A., Logé C., Léger B., Robert J.-M., Lozach O., Blairvacq M., Meijer L., Thiéry V., Besson T. Thiazolo[5,4-f]quinazolin-9-ones, inhibitors of glycogen synthase kinase-3. Bioorg. Med. Chem. Lett. 2006;16:3419–3423. doi: 10.1016/j.bmcl.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 8.Loidreau Y., Deau E., Marchand P., Nourrisson M.-R., Logé C., Coadou J.M., Loaëc N., Meijer L., Besson T. Synthesis and molecular modelling studies of 8-arylpyrido[3′,2′:4,5]thieno[3,2-d] pyrimidin-4-amines as multitarget Ser/Thr kinases inhibitors. Eur. J. Med. Chem. 2015;92:124–134. doi: 10.1016/j.ejmech.2014.12.038. [DOI] [PubMed] [Google Scholar]
- 9.Loidreau Y., Marchand P., Dubouilh-Benard C., Nourrisson M.-R., Duflos M., Loaëc N., Meijer L., Besson T. Synthesis and biological evaluation of N-aryl-7-methoxybenzo[b]furo[3,2-d] pyrimidin-4-amines and their N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amine analogues as dual inhibitors of CLK1 and DYRK1A kinases. Eur. J. Med. Chem. 2013;59:283–295. doi: 10.1016/j.ejmech.2012.11.030. [DOI] [PubMed] [Google Scholar]
- 10.Loidreau Y., Marchand P., Dubouilh-Benard C., Nourrisson M.-R., Duflos M., Lozach O., Loaëc N., Meijer L., Besson T. Synthesis and biological evaluation of N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amines and their pyrido and pyrazino analogues as Ser/Thr kinase inhibitors. Eur. J. Med. Chem. 2012;58:171–183. doi: 10.1016/j.ejmech.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Foucourt A., Dubouilh-Benard C., Chosson E., Corbière C., Buquet C., Iannelli M., Leblond B., Marsais F., Besson T. Microwave-accelerated Dimroth rearrangement for the synthesis of 4-anilino-6-nitroquinazolines. Application to an efficient synthesis of a microtubule destabilizing agent. Tetrahedron. 2010;66:4495–4502. doi: 10.1016/j.tet.2010.04.066. [DOI] [Google Scholar]
- 12.Foucourt A., Hédou D., Dubouilh-Benard C., Désiré L., Casagrande A.-S., Leblond B., Loaëc N., Meijer L., Besson T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part I. Molecules. 2014;19:15546–15571. doi: 10.3390/molecules191015546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Foucourt A., Hédou D., Dubouilh-Benard C., Désiré L., Casagrande A.-S., Leblond B., Loaëc N., Meijer L., Besson T. Design and synthesis of thiazolo[5,4-f]quinazolines as DYRK1A inhibitors, Part II. Molecules. 2014;19:15411–15439. doi: 10.3390/molecules191015411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leblond B., Casagrande A.-S., Désiré L., Foucourt A., Besson T. DYRK1 inhibitors and uses thereof WO 2013026806. Chem. Abstr. 2013;158:390018. [Google Scholar]
- 15.Hédou D., Godeau J., Loaëc N., Meijer L., Fruit C., Besson T. Synthesis of Thiazolo[5,4-f]quinazolin-9(8H)-ones as Multi-Target Directed Ligands of Ser/Thr Kinases. Molecules. 2016;21:578. doi: 10.3390/molecules21050578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hédou D., Dubouilh-Benard C., Loaëc N., Meijer L., Fruit C., Besson T. Synthesis of Bioactive 2-(Arylamino)thiazolo[5,4-f]-quinazolin-9-ones via the Hügershoff Reaction or Cu-Catalyzed Intramolecular C-S Bond Formation. Molecules. 2016;21:794. doi: 10.3390/molecules21060794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin L., Latypova X., Wilson C.M., Magnaudeix A., Perrin M.-L., Terro F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013;12:289–309. doi: 10.1016/j.arr.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Flajolet M., He G., Heiman M., Lin A., Nairn A.C., Greengard P. Regulation of Alzheimer’s disease amyloid-β formation by casein kinase I. Proc. Nat. Acad. Sci. USA. 2007;104:4159–4164. doi: 10.1073/pnas.0611236104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinmann H., Metternich R. Drug discovery process for kinase Inhibitors. Chembiochem. 2005;6:455–459. doi: 10.1002/cbic.200500034. [DOI] [PubMed] [Google Scholar]
- 20.Wu P., Nielsen T.E., Clausen M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discovery Today. 2016;21:5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 21.Carles F., Bourg S., Meyer C., Bonnet P. PKIDB: A Curated, Annotated and updated database of protein kinase inhibitors in clinical trials. Molecules. 2018;23:908. doi: 10.3390/molecules23040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jarhed D.B., Mashelkar K.K., Kim H.-R., Noh M., Jeong L.S. Dual-dspecificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors as potential therapeutics. J. Med. Chem. 2018;61 doi: 10.1021/acs.jmedchem.8b00185. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen T.L., Fruit C., Herault Y., Meijer L., Besson T. Dual-specificity tyrosine phosphorylation-regulated kinase 1a (dyrk1a) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017;27:1183–1199. doi: 10.1080/13543776.2017.1360285. [DOI] [PubMed] [Google Scholar]
- 24.Abbassi R., Johns T.G., Kassiou M., Munoz L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Ther. 2015;151:87–98. doi: 10.1016/j.pharmthera.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 25.Medda F., Smith B., Gokhale V., Shaw A.Y., Dunckley T., Hulme C. Beyond secretases: Kinase inhibitors for the treatment of Alzheimer’s disease. Annu. Rep. Med. Chem. 2013;48:57–71. doi: 10.1016/B978-0-12-417150-3.00005-3. [DOI] [Google Scholar]
- 26.Smith B., Medda F., Gokhale V., Dunckley T., Hulme C. Recent advances in the design, synthesis, and biological evaluation of selective DYRK1A inhibitors: A new avenue for a disease modifying treatment of Alzheimer’s? ACS Chem. Neurosci. 2012;3:857–872. doi: 10.1021/cn300094k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varjosalo M., Keskitalo S., Van Drogen A., Nurkkala H., Vichalkovski A., Aebersold R., Gstaiger M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3:1306–1320. doi: 10.1016/j.celrep.2013.03.027. [DOI] [PubMed] [Google Scholar]
- 28.Coutadeur S., Benyamine H., Delalonde L., de Oliveira C., Leblond B., Foucourt A., Besson T., Casagrande A.-S., Taverne T., Girard A., et al. A Novel DYRK1A (Dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: Effect on Tau and amyloid pathologies in vitro. J. Neurochem. 2015;133:440–451. doi: 10.1111/jnc.13018. [DOI] [PubMed] [Google Scholar]
- 29.Chaikuad A., Diharce J., Schröder M., Foucourt A., Leblond B., Casagrande A.-S., Désiré L., Bonnet P., Knapp S., Besson T. An unusual binding mode of the methyl 9-anilinothiazolo[5,4-f]quinazoline-2-carbimidates (EHT 1610 and EHT 5372) confers high selectivity for DYRK kinases. J. Med. Chem. 2016;59:10315–10321. doi: 10.1021/acs.jmedchem.6b01083. [DOI] [PubMed] [Google Scholar]
- 30.Bournez C., Gally J.-M., Krezel P., Aci-Sèche S., Bonnet P. Frags2Drugs: An in silico tool to discover new molecules based on 3D fragment network. J. Chem. Inf. Model. 2018 in press. [Google Scholar]
- 31.Harari M., Couly F., Fruit C., Besson T. Late-stage C–H Pd-catalyzed and copper assisted regioselective sequential C2 and C7 arylation of thiazolo[5,4-f]quinazolin-9(8H)-one with aryl halides. Org. Lett. 2016;18:3282–3285. doi: 10.1021/acs.orglett.6b01552. [DOI] [PubMed] [Google Scholar]
- 32.Couly F., Dubouilh-Benard C., Besson T., Fruit C. Arylation of thiazolo[5,4-f]quinazolin-9(8H)-one backbone: Synthesis of an array of potential kinase inhibitors. Synthesis. 2017;49:4615–4622. doi: 10.1055/s-0036-1588434. [DOI] [Google Scholar]
- 33.Besson T., Fruit C. Recent developments in microwave-assisted metal-catalyzed C-H functionalization of heteroarenes for medicinal chemistry and material applications. Synthesis. 2016;48:3879–3889. doi: 10.1039/C6OB00227G. [DOI] [Google Scholar]
- 34.Appel R., Janssen H., Siray M., Knoch F. Synthese und reaktionen des 4,5-dichlor-1,2,3-dithiazolium-chlorids. Eur. J. Inorg. Chem. 1985;118:1632–1643. doi: 10.1002/cber.19851180430. [DOI] [Google Scholar]
- 35.Topliss J.G. Utilization of operational schemes for analog synthesis in drug design. J. Med. Chem. 1972;15:1006–1011. doi: 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- 36.Turočkin A. 1,5,7-Triazabicyclo[4.4.0]dec-5-ene (TBD) as a Lewis Base. Synlett. 2014;25:894–895. doi: 10.1055/s-0033-1340852. [DOI] [Google Scholar]
- 37.Tahtouh T., Elkins J.M., Filippakopoulos P., Soundararajan M., Burgy G., Durieu E., Cochet C., Schmid R.S., Lo D.C., Delhommel F., et al. Selectivity, cocrystal structures and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012;55:9312–9330. doi: 10.1021/jm301034u. [DOI] [PubMed] [Google Scholar]
- 38.Primot A., Baratte B., Gompel M., Borgne A., Liabeuf S., Romette J.L., Jho E.H., Costantini F., Meijer L. Purification of GSK-3 by affinity chromatography on immobilized axin. Protein Expr. Purif. 2000;20:394–404. doi: 10.1006/prep.2000.1321. [DOI] [PubMed] [Google Scholar]
- 39.Reinhardt J., Ferandin Y., Meijer L. Purification of CK1 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2007;54:101–109. doi: 10.1016/j.pep.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 40.Patel K., Gadewar M., Tripathi R., Prasad S.K., Patel D.K. A review on medicinal importance, pharmacological activity and bioanalytical aspects of beta-carboline alkaloid “Harmine”. Asian Pac. J. Trop. Biomed. 2012;2:660–664. doi: 10.1016/S2221-1691(12)60116-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.