

Abstract
We report here on three constitutionally isomeric peptides, each of which contains two glutamic acid residues and two lysine residues functionalized with S-aroylthiooximes (SATOs), termed peptide–H2S donor conjugates (PHDCs). SATOs decompose in the presence of cysteine to generate hydrogen sulfide (H2S), a biological signaling gas with therapeutic potential. The PHDCs self-assemble in aqueous solution into different morphologies, two into nanoribbons of different dimensions and one into a rigid nanocoil. The rate of H2S release from the PHDCs depends on the morphology, with the nanocoil-forming PHDC exhibiting a complex release profile driven by morphological changes promoted by SATO decomposition. The nanocoil-forming PHDC mitigated the cardiotoxicity of doxorubicin more effectively than its nanoribbon-forming constitutional isomers as well as common H2S donors. This strategy opens up new avenues to develop H2S-releasing biomaterials and highlights the interplay between structure and function from the molecular level to the nanoscale.
Introduction
In proteins, amino acid sequence dictates structure, which in turn regulates biological function. Small changes often have a dramatic effect; for example, hormone-sensitive lipase (HSL) either promotes or prevents lipid hydrolysis depending on the positions of a serine and phosphoserine residue.1 These two lipases, both functional enzymes, are constitutional isomers, i.e., molecules with identical molecular formulas but different connectivity. Constitutional isomers have also been evaluated in synthetic self-assembling peptides,2−7 which are of interest as materials for tissue engineering and regenerative medicine.8−10 For example, peptide amphiphiles with the sequence C16-VVEE (C16 = palmitic acid) form rigid cylindrical nanofibers, while twisted nanoribbons are observed for the constitutionally isomeric sequence C16-EVEV.11 This example highlights sequence dictating nanostructure, but sequence-specific control of nanostructure, with concomitant impact on biological function, has not previously been demonstrated in constitutional isomers. Inspired by Nature’s ability to precisely control biological function in constitutionally isomeric proteins, we aimed here to explore how sequence in constitutionally isomeric self-assembling peptides affects nanostructure and biological activity in the context of hydrogen sulfide (H2S) signaling.
H2S is a vital cellular signaling molecule and one of three established gasotransmitters along with nitric oxide (NO) and carbon monoxide (CO), and it plays critical roles in many physiological and/or pathological processes.12 For example, H2S is involved in heart disease, inflammation, and tumor progression/suppression, among many other diseases and conditions.12−15 Like NO and CO, H2S carries out its signaling functions at low concentrations (generally in the range 10–1000 nM), and physiological production of H2S is tightly controlled by specific enzymes. Modulation of H2S levels, either through inhibition/activation of native enzymes or via administration of exogenous H2S, offers a way for scientists to uncover the role of H2S in biology and holds therapeutic potential.16
Due to the hazards inherent in working with gaseous H2S directly, most of the foundational studies in this area have relied on administration of exogenous H2S, usually as an aqueous solution of Na2S or NaHS.17,18 Recognizing that sulfide salts were not ideal compounds for studying H2S biology, researchers have developed several classes of H2S-releasing compounds (termed H2S donors) over the past several years.16,19−23 Many release H2S in response to specific triggers, including light, biological thiols, pH changes, enzymatic activity, and others. Despite these advances in the development of synthetic H2S donors, most have low water solubility, limited means for modulating release kinetics, and no capacity for targeted delivery, all of which may limit H2S-based treatments due to the reactive nature of this signaling molecule.
To address these limitations, our group and others have begun designing and evaluating bioinspired H2S-releasing materials.24−27 A particularly exciting type of material for use in drug and signal delivery is self-assembling peptides. Designed to aggregate in water into specific nanostructures, self-assembling peptides can be quickly synthesized and purified because they consist only of a single peptide or peptide conjugate.28 Here we report rationally designed peptide–H2S donor conjugates (PHDCs), which spontaneously associate into discrete, stable supramolecular nanostructures with the capacity for self-delivery of H2S (i.e., no additional carriers are needed). To make a series of PHDCs, we appended S-aroylthiooximes (SATOs), a type of thiol-triggered H2S donor, onto short peptide sequences. Specifically, S-benzoylthiohydroxylamine (SBTHA) was added in a condensation reaction to three different peptides, each of which contained two Glu (E) residues and two 4-formylbenzoic acid (FBA)-modified Lys (K) residues. This way, three constitutionally isomeric PHDCs were prepared: K(FBA-SATO)EK(FBA-SATO)E (named KSEKSE for short), K(FBA-SATO)K(FBA-SATO)EE (KSKSEE), and K(FBA-SATO)EEK(FBA-SATO) (KSEEKS) (Figure 1A–C). A control peptide that could not release H2S (KOEEKO) was synthesized in the same way, replacing SBTHA with O-benzylhydroxylamine. Detailed synthetic procedures and characterization can be found in the Supporting Information (Figures S1 and S2).
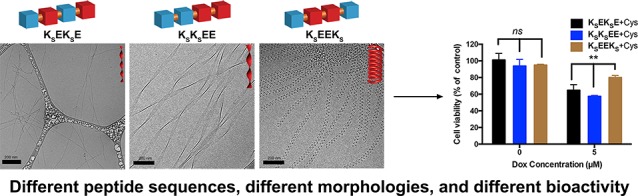
Figure 1.

(A–C) Schematic illustrations of the three isomeric peptide–H2S donor conjugates (PHDCs) in this study. (D–F) Cryo-TEM characterization illustrates the effect of sequence on the self-assembled morphology of constitutionally isomeric PHDCs: (D) cryo-TEM image of twisted ribbons formed by KSEKSE in aqueous solution, (E) cryo-TEM image of twisted ribbons formed by KSKSEE in aqueous solution, and (F) cryo-TEM image of nanocoils formed by KSEEKS in aqueous solution. (G) AFM phase image of nanocoils formed by KSEEKS in aqueous solution. Solution concentration: 1 mM PHDCs in PBS (pH 7.4). (H) SAXS curve of KSEEKS in aqueous solution (1 mM in PBS at pH 7.4) with fitting to a helical model. (I–K) Circular dichroism spectra of (I) KSEKSE, (J) KSKSEE, and (K) KSEEKS in phosphate buffer (pH 7.4) at a concentration of 100 μM.
Results and Discussion
Cryogenic transmission electron microscopy (cryo-TEM) imaging (Figure 1D–F) and conventional TEM (Figure S3) revealed that all PHDCs assembled into one-dimensional nanostructures in aqueous solution. The dominant morphology observed for KSEKSE was twisted ribbons (Figure 1D), as indicated by the varying thickness and grayscale intensity in both the conventional and cryo-TEM images. Widths were 14 ± 3 nm, and lengths were on the scale of a few micrometers. Closer examination of the twisted ribbons revealed that their thickness was 4 ± 1 nm, a value that is equal to the expected thickness of a partially or fully interdigitated bilayer, implying that the observed ribbons are likely bilayer structures caused by association of the aromatic SATO surfaces. This kind of molecular packing behavior has been widely found in peptide systems composed of uniform (AB)n amino acid periodicity (where A and B are polar and nonpolar residues, respectively).11,29−32 Importantly, these images for peptide KSEKSE indicate that non-natural amino acids, such as the derivatized K residues used here, can also promote twisted ribbon formation. The pitch of the twisted ribbons measured from the cryo-TEM micrographs was 103 ± 8 nm.
Peptide KSKSEE also formed twisted ribbons in aqueous solution (Figure 1E), but the dimensions were quite different from those of KSEKSE. The KSKSEE ribbons had widths of 40 ± 6 nm and pitches of 500 ± 10 nm. Both dimensions are three to four times larger than those for the KSEKSE ribbons. With an average thickness of 5 ± 1 nm, these larger twisted ribbons are also bilayer structures. We attribute the increase in width, pitch length, and thickness to the enhanced steric hindrances and electrostatic repulsions among side chains in KSKSEE compared with KSEKSE. The alternating sequence of hydrophobic and hydrophilic residues in KSEKSE allows the peptides to pack tightly during self-assembly because hydrophobic SATO groups and hydrophilic Glu residues display on opposite sides of the peptide backbone. In contrast, the pair of charged, C-terminal Glu residues in KSKSEE likely cause these peptides to repel each other more strongly when two molecules approach during assembly. In order to reach an energy minimum, KSKSEE twists, increasing the distance between assembled molecules, resulting in looser molecular packing within nanostructures than in KSEKSE.
A dramatically different self-assembled morphology was observed for KSEEKS. PHDC KSEEKS assembled into left-handed nanocoils with an average diameter of 5.6 ± 0.7 nm and a regular twisting pitch of 32 ± 4 nm (Figure 1F). Nanocoil lengths were several micrometers. To the best of our knowledge, this is the first time that this nanocoil morphology, which resembles a telephone cord, has ever been observed in peptide-based materials. Although the term nanocoil has been used in other systems,33−35 reported nanocoils are coiled flat ribbons, while those observed for KSEEKS resembled coiled cylindrical nanofibers. Given the fully extended length of the molecule KSEEKS (∼2.5 nm) and the amphiphilic nature of the design, it is likely that these nanocoils have a core–shell structure. Atomic force microscopy (AFM) was applied to measure the height (Figure S4) of the nanocoils (Figure 1G), indicating that nanocoil heights were 21 ± 3 nm. This value is larger than the thickness of two stacked nanocoils (∼12 nm), implying that the nanocoils were rigid enough to preserve their morphology during sample drying.
To complement the findings from cryo-TEM and AFM, we performed small-angle X-ray scattering (SAXS) on KSEEKS in aqueous solution (Figure 1H). The scattering pattern was fitted to a helical nanostructure model,36 and the diameter and twisting pitch measurements from SAXS matched those observed by imaging (Figure S7). To gain more insight into the molecular requirements for nanocoil formation, we synthesized two control molecules, EKSKSE and KOEEKO (KO represents the oxygen-containing analogue of KS; see Figure S1 for molecular structures). Only ill-defined aggregates were observed for EKSKSE under the same assembly conditions (Figure S5), similar to reported observations for a peptide with a related structure.37 Bundled nanoribbons rather than nanocoils were found for KOEEKO (Figure S6), which indicates that the nanocoil morphology is quite sensitive to molecular architecture.
To understand the assembly differences among these PHDCs, we conducted further experiments to evaluate their critical aggregation concentrations (CACs) and to evaluate packing using multiple spectroscopic techniques. CACs were measured using the Nile Red assay, a common method that relies on the bright fluorescence of the dye when sequestered in a hydrophobic environment. The CAC value for the three PHDCs ranged from 19 to 36 μM (Figure S8 and Table S1). Next, we used circular dichroism (CD) spectroscopy to assess the molecular packing of the self-assembled PHDCs (Figure 1I–K). The secondary structures of these nanoassemblies were different, but all displayed strong signals both in the peptide region (190–240 nm) and in the SATO absorption region (300–360 nm), consistent with SATO absorptions in the corresponding UV–vis spectra (Figure S9). Interestingly, while both KSEKSE and KSKSEE assembled into twisted ribbons, their secondary structures were different on the basis of analysis of the peptide region in their CD spectra. Specifically, KSEKSE assemblies displayed α-helix secondary structures with a slight red shift compared to a typical α-helix spectrum (Figure 1I), while KSKSEE showed a CD spectrum resembling a β-sheet structure (Figure 1J). The larger nanoribbon dimensions for PHDC KSKSEE compared with KSEKSE may be explained by the β-sheet component, which is typically present in extended nanostructures. In sharp contrast, the spectrum for KSEEKS was consistent with a random coil structure with some α-helix contribution (Figure 1K). Taken together, we conclude that the morphological differences between the three PHCDs are rooted in their different constitutionally isomeric sequences, which result in different secondary structures.
We next asked how supramolecular structure in the three constitutionally isomeric PHDCs would affect their ability to release H2S, and ultimately their bioactivity. Given the amphiphilic nature and the assembling ability of these PHDCs, we expected that assembly into nanostructures would shield the SATO components from the external environment, offering a potential mechanism for the controlled release of H2S. The release profiles from the three PHDCs were assessed by an H2S-selective microelectrode (Figure 2A–D and Figure S10). This method allows for real-time monitoring of the concentration of H2S in solution but does not measure cumulative release because H2S oxidizes and volatilizes as it is generated. Thus, H2S release profiles using this method of measurement are typically quantified using peaking time, which is an approximate measure of relative release rates among similar samples.
Figure 2.

(A, C) H2S release profiles and corresponding peaking times of KSEKSE, KSKSEE, and KSEEKS triggered by Cys in PBS (pH = 7.4) at room temperature (rt). Data were collected on an H2S-sensitive electrochemical probe from a solution (110 μL total) of PHDC (1 mM) and Cys (4 mM) sealed in a well with a gas-permeable membrane inside a vial containing PBS (5 mL). Error bars indicate standard deviation of three separate experiments. (B, D) H2S release profiles and corresponding peaking times for PHDCs from a solution of PHDC (40 μM) and Cys (160 μM) in the sealed well. Error bars indicate standard deviation of three separate experiments. (E) Schematic illustration of the proposed release mechanism showing the effect of self-assembly on the relative rates of reaction of the PHDCs with Cys to generate H2S. *P < 0.05 for a comparison of the groups indicated as determined by a one-way analysis of variance (ANOVA) with a Student–Newman–Keuls comparisons posthoc test (n = 3).
H2S release was triggered by cysteine (Cys), which is a small biological thiol that has been used previously as a trigger to release H2S from SATO-based materials.38,39 As Cys also generates a response from the electrochemical probe, we measured PHDC release profiles using a specially made vial that contained an inner well with a volume of 120 μL, into which PHDC and Cys (110 μL total) were added before quickly sealing with a gas-permeable membrane. The experimental setup is shown in Figure S10. PBS solution (5 mL) was then added to the vial above the level of the sealed inner well, and the electrochemical probe was inserted into the outer PBS solution. PHDC solutions in the inner well were held constant at 1 mM with 4 mM total Cys. All three PHDCs exhibited steady H2S release over the course of several hours (Figure 2A,B), while minimal H2S was detected from experimental runs without Cys (Figure S11) and no H2S was detected from the control peptide KOEEKO (Figure S12).
While the H2S release profiles were similar for nanoribbon-forming PHDCs KSEKSE and KSKSEE, nanocoil-forming PHDC KSEEKS exhibited a different profile. Peaking times for KSEKSE and KSKSEE were 143 ± 1 and 127 ± 7 min, respectively, while that for KSEEKS was significantly longer at 182 ± 4 min (Figure 2C). Closer examination of the release profiles revealed that, unlike the profiles for the nanoribbons (KSEKSE and KSKSEE), there was an initial period of 60 min for nanocoils (KSEEKS) where H2S was slowly liberated; after this point the release rate rose sharply. A UV absorption study also showed a similar initial period of slow consumption of SATO groups during H2S release (Figure S13). We attribute this initial period of slower release from KSEEKS to slower diffusion of Cys into the nanocoils compared with the nanoribbons. In order to confirm the impact of supramolecular structure on release rate, we carried out similar H2S release experiments with PHDC concentrations of 40 μM in the inner well, where we expect the peptides to exist more in their monomeric state than in the first experiments. Despite the 25-fold dilution, peaking times were shorter at 40 μM than at 1 mM (Figure 2D). These results are consistent with those from other drug-releasing self-assembling peptides, where dilution enhances the release rate.40 In these dilute release experiments, the release profiles for all three PHDCs were nearly identical (Figure 2B), and there was no significant difference in peaking time (Figure 2D, Table S2). These data collectively highlight the effect of self-assembly on H2S release rate from PHDCs (Figure 2E).
To gain more insight into how morphology affects release of H2S from KSEEKS nanocoils, TEM was used to monitor the morphological transition during the course of release. After addition of Cys, aliquots of KSEEKS solution were removed at different time intervals and drop-cast onto TEM grids (Figure 3). At t = 0, just after addition of Cys, the nanocoil morphology was unaffected (Stage 0). After incubation for 45 min, the nanocoils began to fall apart (Stage 1), as indicated by an increase in pitch from 26 ± 4 to 40 ± 8 nm. In earlier work we found that addition of Cys to SATOs leads to H2S release along with the production of N-benzoyl-Cys and an aldehyde;41 in PHDCs this has the effect of breaking the SATO group in half, leaving only a benzaldehyde fragment on the Lys side chain. We speculate that, after Cys penetrates into the nanostructures and reacts with SATO groups, the strength of the hydrophobic and π–π interactions decreases, leading to less compact molecular packing and loosened nanocoils. Incubation for 90 min revealed a mixture of unwound nanocoils (highlighted by red arrows in Figure 3C) and ill-defined aggregates (Stage 2). At this stage the supramolecular structures had loosened considerably, enabling faster penetration of Cys into the assemblies, which accelerated degradation of nanocoils and dramatically increased the rate of the H2S release. Finally, after 120 min incubation, only ill-defined aggregates were observed (Stage 3). At this point in the process the rate of release begins to peak, indicating that the poorly defined aggregates are the most potent H2S-releasing morphology. On the basis of these results, we conclude that slow H2S release from KSEEKS results from its unique nanostructure, which limits Cys access to the reactive SATO groups.
Figure 3.

(A–D) TEM characterization illustrates the morphological transition of KSEEKS during H2S release in the presence of Cys. The morphology study was carried out with 1 mM KSEEKS in PBS with 4 mM Cys, and aliquots were removed at each time point, diluted, and drop-cast onto TEM grids before staining with 2 wt % uranyl acetate. (E) Schematic illustration of the observed morphological transition during H2S release from KSEEKS nanocoils.
Because PHDC KSEEKS possessed the longest H2S release peaking time, we next explored its cardioprotective ability against toxicity induced by the common cancer drug doxorubicin (Dox). As cardiotoxicity is dose-limiting for Dox, reducing its deleterious effects on the heart may enable more effective chemotherapy. H2S (as fast-releasing Na2S) promotes cardiomyocyte viability in the presence of Dox by inhibiting endoplasmic reticulum stress,42,43 but this effect has not been tested on more practical slow-releasing H2S donors. We and others have observed that slow-releasing H2S donors can significantly enhance biological effects compared to Na2S.39,44 Thus, we envisioned that KSEEKS might be effective in rescuing cardiomyocytes in the presence of Cys. First, we established that KSEEKS was nontoxic to H9C2 cardiomyocytes at concentrations up to 200 μM in the presence of Cys (Figure S14). In contrast, Dox induced cytotoxicity at concentrations as low as 2.5 μM (Figure 4A).
Figure 4.

(A) Cell viability of H9C2 cardiomyoctyes pretreated for 30 min with a combination of KSEEKS (200 μM) and Cys (800 μM) followed by exposure to Dox for another 24 h at varying concentrations. *p < 0.01 vs Dox only. (B) Cell viability of H9C2 cardiomyoctyes pretreated with various controls for 30 min before exposure to Dox (5 μM) for 24 h. Control compound concentrations: Cys, 800 μM; KOEEKO and KSEEKS, 200 μM (400 μM in SATO); GYY4137 and Na2S, 400 μM. *p < 0.01 vs Dox group, #p < 0.01 vs Na2S group. (C) Cell viability of H9C2 cardiomyoctyes pretreated with KSEKSE, KSKSEE, or KSEEKS (200 μM) in the presence of Cys (800 μM) for 30 min before exposure to Dox (5 μM) or without Dox. **p < 0.01. Error bars indicate standard deviation of three separate experiments. Group comparisons are indicated as determined by a one-way analysis of variance (ANOVA) with a Student–Newman–Keuls comparisons posthoc test.
In treatment studies, H9C2 cells were pretreated with KSEEKS and Cys for 30 min.42,45 Dox was then added without removing the KSEEKS/Cys solution, and cells were then cultured for another 24 h before analyzing viability. Compared to the Dox only group, cell viability increased significantly when cells were pretreated with KSEEKS and Cys before exposure to Dox (Figure 4A). To further ensure that sustainable H2S release was responsible for imparting protection to the cardiomyocytes in the presence of Dox, several control studies were carried out (Figure 4B). Treatment with Cys alone showed no protective effect, and treatment with a combination of the non-H2S-releasing control peptide (KOEEKO) and Cys did not improve viability compared to Dox alone. We further compared KSEEKS to sodium sulfide (Na2S), a fast-releasing H2S donor, and GYY4137, a slow-releasing H2S donor, under the same experimental conditions. Na2S had a limited ability to rescue cells while GYY4137 had no effect on viability. Interestingly, KSEEKS was more effective at rescuing cells than Na2S, even while Na2S enhanced H9C2 proliferation in the absence of Dox (Figure S14).
Finally, given the significant difference in H2S release peaking time among nanocoils (KSEEKS) and nanoribbons (KSEKSE and KSKSEE) (Figure 2C), we evaluated whether this behavior could influence the cardioprotective capacity of these constitutionally isomeric peptides. H9C2 cells were pretreated with PHDCs KSEEKS, KSEKSE, and KSKSEE in the presence of Cys, and viability was analyzed as before. In the absence of Dox, no difference in cell viability between the three groups was observed (left three columns in Figure 4C). In sharp contrast, KSEEKS was significantly more effective in rescuing cells than KSEKSE and KSKSEE (right three columns in Figure 4C). We attribute this difference in bioactivity to the differences in H2S release profiles, which are influenced by the peptide nanostructures. Interestingly, none of three PHDCs hindered the cytotoxicity of Dox toward MCF-7 breast cancer cells (Figure S15B), indicating that these PHDCs can be used as a powerful adjuvant to reduce the deleterious effect of Dox on the heart. More broadly, these results highlight the importance of controlling the release profile in H2S-based therapies and demonstrate the power of this gas to initiate complex changes in cell behavior.
Conclusions
The self-assembly and bioactivity of the constitutionally isomeric peptides described here reveal how subtle changes in amino acid sequence, such as those employed by proteins, can be harnessed in short peptides to dictate different biological outcomes. Additionally, the use of reactive donor molecules as molecular building units in self-assembly creates new opportunities for the development of biomaterials that release H2S or related species. The ability of such materials to release their chemically trapped payload (e.g., H2S), disassemble, and undergo biodegradation is particularly exciting for applications in regenerative medicine.
Acknowledgments
This work was supported by the National Science Foundation (DMR-1454754), the National Institutes of Health (R01GM123508), and the United States–Israel Binational Science Foundation (2016096). We also thank the Dreyfus Foundation for support of this work through a Camille Dreyfus Teacher-Scholar Award to J.B.M. We thank Chadwick R. Powell for synthesis of GYY4137, Prof. Tijana Grove (Virginia Tech) and her students for experimental assistance, Prof. Matthew J. Webber (University of Notre Dame) for valuable discussions, Dr. Einat Nativ-Rot from the electron microscopy unit at the Ilse Katz Institute for Nanoscale Science and Technology at BGU for cryo-TEM images, and Ms. Tingting Chen from Zhejiang University (China) for assistance with the ribbon drawing. The authors also acknowledge use of facilities within the Nanoscale Characterization and Fabrication Laboratory at Virginia Tech.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b09320.
Detailed experimental section, additional cell studies, and additional characterization (ESI-MS, conventional TEM, AFM, CAC measurements, UV–vis, circular dichroism) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Garton A. J.; Yeaman S. J. Identification and role of the basal phosphorylation site on hormone-sensitive lipase. Eur. J. Biochem. 1990, 191, 245–250. 10.1111/j.1432-1033.1990.tb19116.x. [DOI] [PubMed] [Google Scholar]
- Zhao F. Y.; Guo H.; Zhang Z. D.; Ye J.; Liu L. L.; Zhao C. X.; Shao Z. Z. Conformation and self-assembly changes of isomeric peptide amphiphiles influenced by switching tyrosine in the sequences. J. Mater. Chem. B 2017, 5, 5189–5195. 10.1039/C7TB00736A. [DOI] [PubMed] [Google Scholar]
- Kar S.; Tai Y. Marked difference in self-assembly, morphology, and cell viability of positional isomeric dipeptides generated by reversal of sequence. Soft Matter 2015, 11, 1345–1351. 10.1039/C4SM02537G. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Dobson E. T.; Buettner C. J.; Nicholl M. J.; Goldberger J. E. Programming pH-triggered self-assembly transitions via isomerization of peptide sequence. Langmuir 2014, 30, 15383–15387. 10.1021/la5037663. [DOI] [PubMed] [Google Scholar]
- Fu I. W.; Nguyen H. D. Sequence-dependent structural stability of self-assembled cylindrical nanofibers by peptide amphiphiles. Biomacromolecules 2015, 16, 2209–2219. 10.1021/acs.biomac.5b00595. [DOI] [PubMed] [Google Scholar]
- Frederix P. W. J. M.; Scott G. G.; Abul-Haija Y. M.; Kalafatovic D.; Pappas C. G.; Javid N.; Hunt N. T.; Ulijn R. V.; Tuttle T. Exploring the sequence space for (tri-) peptide self-assembly to design and discover. Nat. Chem. 2015, 7, 30–37. 10.1038/nchem.2122. [DOI] [PubMed] [Google Scholar]
- Clarke D. E.; Parmenter C. D. J.; Scherman O. A. Tunable pentapeptide self-assembled β-sheet hydrogels. Angew. Chem., Int. Ed. 2018, 57, 7709–7713. 10.1002/anie.201801001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks M. P.; Sato K.; Palmer L. C.; Stupp S. I. Supramolecular assembly of peptide amphiphiles. Acc. Chem. Res. 2017, 50, 2440–2448. 10.1021/acs.accounts.7b00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H. G.; Webber M. J.; Stupp S. I. Self-assembly of peptide amphiphiles: from molecules to nanostructures to biomaterials. Biopolymers 2010, 94, 1–18. 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao K.; Levin A.; Adler-Abramovich L.; Gazit E. Fmoc-modified amino acids and short peptides: simple bio-inspired building blocks for the fabrication of functional materials. Chem. Soc. Rev. 2016, 45, 3935–3953. 10.1039/C5CS00889A. [DOI] [PubMed] [Google Scholar]
- Cui H. G.; Cheetham A. G.; Pashuck E. T.; Stupp S. I. Amino acid sequence in constitutionally isomeric tetrapeptide amphiphiles dictates architecture of one-dimensional nanostructures. J. Am. Chem. Soc. 2014, 136, 12461–12468. 10.1021/ja507051w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- Szabo C. Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat. Rev. Drug Discovery 2016, 15, 185–203. 10.1038/nrd.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King A. L.; Polhemus D. J.; Bhushan S.; Otsuka H.; Kondo K.; Nicholson C. K.; Bradley J. M.; Islam K. N.; Calvert J. W.; Tao Y. X.; Dugas T. R.; Kelley E. E.; Elrod J. W.; Huang P. L.; Wang R.; Lefer D. J. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 3182–3187. 10.1073/pnas.1321871111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid. Redox Signaling 2014, 20, 783–793. 10.1089/ars.2013.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartle M. D.; Pluth M. D. A practical guide to working with H2S at the interface of chemistry and biology. Chem. Soc. Rev. 2016, 45, 6108–6117. 10.1039/C6CS00212A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W. M.; Zhang J.; Lu Y. J.; Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous K-ATP channel opener. EMBO J. 2001, 20, 6008–6016. 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha S.; Calvert J. W.; Duranski M. R.; Ramachandran A.; Lefer D. J. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. Am. J. Physiol.-Heart Circul. Physiol. 2008, 295, H801–H806. 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C. R.; Dillon K. M.; Matson J. B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. 10.1016/j.bcp.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Biggs T. D.; Xian M. Hydrogen sulfide (H2S) releasing agents: chemistry and biological applications. Chem. Commun. 2014, 50, 11788–11805. 10.1039/C4CC00968A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Henthorn H. A.; Pluth M. D. Kinetic insights into hydrogen sulfide delivery from caged-carbonyl sulfide isomeric donor platforms. J. Am. Chem. Soc. 2017, 139, 16365–16376. 10.1021/jacs.7b09527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan P.; Bora P.; Ravikumar G.; Jos S.; Chakrapani H. Esterase activated carbonyl sulfide/hydrogen sulfide (H2S) donors. Org. Lett. 2017, 19, 62–65. 10.1021/acs.orglett.6b03336. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Wang H.; Xian M. Cysteine-activated hydrogen sulfide (H2S) donors. J. Am. Chem. Soc. 2011, 133, 15–17. 10.1021/ja1085723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y.; Matson J. B. Gasotransmitter delivery via self-assembling peptides: treating diseases with natural signaling gases. Adv. Drug Delivery Rev. 2017, 110, 137–156. 10.1016/j.addr.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Feng S.; Zhao Y.; Xian M.; Wang Q. Biological thiols-triggered hydrogen sulfide releasing microfibers for tissue engineering applications. Acta Biomater. 2015, 27, 205–213. 10.1016/j.actbio.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquhart M. C.; Ercole F.; Whittaker M. R.; Boyd B. J.; Davis T. P.; Quinn J. F. Recent advances in the delivery of hydrogen sulfide via a macromolecular approach. Polym. Chem. 2018, 9, 4431–4439. 10.1039/C8PY00938D. [DOI] [Google Scholar]
- Xiao Z. Y.; Bonnard T.; Shakouri-Motlagh A.; Wylie R. A. L.; Collins J.; White J.; Heath D. E.; Hagemeyer C. E.; Connal L. A. Triggered and tunable hydrogen sulfide release from photogenerated thiobenzaldehydes. Chem. Eur. J. 2017, 23, 11294–11300. 10.1002/chem.201701206. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Cheetham A. G.; Angacian G.; Su H.; Xie L. S.; Cui H. G. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv. Drug Delivery Rev. 2017, 110, 112–126. 10.1016/j.addr.2016.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthington P.; Langhans S.; Pochan D. Beta-hairpin peptide hydrogels for package delivery. Adv. Drug Delivery Rev. 2017, 110, 127–136. 10.1016/j.addr.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore A. N.; Hartgerink J. D. Self-assembling multidomain peptide nanofibers for delivery of bioactive molecules and tissue regeneration. Acc. Chem. Res. 2017, 50, 714–722. 10.1021/acs.accounts.6b00553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport H.; Kjaer K.; Jensen T. R.; Leiserowitz L.; Tirrell D. A. Two-dimensional order in beta-sheet peptide monolayers. J. Am. Chem. Soc. 2000, 122, 12523–12529. 10.1021/ja002238t. [DOI] [Google Scholar]
- Ivnitski D.; Amit M.; Silberbush O.; Atsmon-Raz Y.; Nanda J.; Cohen-Luria R.; Miller Y.; Ashkenasy G.; Ashkenasy N. The strong influence of structure polymorphism on the conductivity of peptide fibrils. Angew. Chem., Int. Ed. 2016, 55, 9988–9992. 10.1002/anie.201604833. [DOI] [PubMed] [Google Scholar]
- Nakagawa M.; Kawai T. Chirality-controlled syntheses of double-helical Au nanowires. J. Am. Chem. Soc. 2018, 140, 4991–4994. 10.1021/jacs.8b00910. [DOI] [PubMed] [Google Scholar]
- Zhang Y. B.; Zheng Y. X.; Xiong W.; Peng C.; Zhang Y. F.; Duan R.; Che Y. K.; Zhao J. C. Morphological transformation between nanocoils and nanoribbons via defragmentation structural rearrangement or fragmentation-recombination mechanism. Sci. Rep. 2016, 6, 27335. 10.1038/srep27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T.; Fukushima T.; Yamamoto Y.; Kosaka A.; Jin W.; Ishii N.; Aida T. Stabilization of a kinetically favored nanostructure: surface ROMP of self-assembled conductive nanocoils from a norbornene-appended hexa-peri-hexabenzocoronene. J. Am. Chem. Soc. 2006, 128, 14337–14340. 10.1021/ja064461h. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Form factor of helical ribbons. Macromolecules 2008, 41, 8948–8950. 10.1021/ma8014917. [DOI] [Google Scholar]
- Hu Y.; Lin R.; Zhang P. C.; Fern J.; Cheetham A. G.; Patel K.; Schulman R.; Kan C. Y.; Cui H. G. Electrostatic-driven lamination and untwisting of beta-sheet assemblies. ACS Nano 2016, 10, 880–888. 10.1021/acsnano.5b06011. [DOI] [PubMed] [Google Scholar]
- Carter J. M.; Qian Y.; Foster J. C.; Matson J. B. Peptide-based hydrogen sulphide-releasing gels. Chem. Commun. 2015, 51, 13131–13134. 10.1039/C5CC04883D. [DOI] [PubMed] [Google Scholar]
- Foster J. C.; Radzinski S. C.; Zou X. L.; Finkielstein C. V.; Matson J. B. H2S-releasing polymer micelles for studying selective cell toxicity. Mol. Pharmaceutics 2017, 14, 1300–1306. 10.1021/acs.molpharmaceut.6b01117. [DOI] [PubMed] [Google Scholar]
- Cheetham A. G.; Zhang P. C.; Lin Y. A.; Lock L. L.; Cui H. G. Supramolecular nanostructures formed by anticancer drug assembly. J. Am. Chem. Soc. 2013, 135, 2907–2910. 10.1021/ja3115983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster J. C.; Powell C. R.; Radzinski S. C.; Matson J. B. S-aroylthiooximes: a facile route to hydrogen sulfide releasing compounds with structure-dependent release kinetics. Org. Lett. 2014, 16, 1558–1561. 10.1021/ol500385a. [DOI] [PubMed] [Google Scholar]
- Wang X. Y.; Yang C. T.; Zheng D. D.; Mo L. Q.; Lan A. P.; Yang Z. L.; Hu F.; Chen P. X.; Liao X. X.; Feng J. Q. Hydrogen sulfide protects H9c2 cells against doxorubicin-induced cardiotoxicity through inhibition of endoplasmic reticulum stress. Mol. Cell. Biochem. 2012, 363, 419–426. 10.1007/s11010-011-1194-6. [DOI] [PubMed] [Google Scholar]
- Chegaev K.; Rolando B.; Cortese D.; Gazzano E.; Buondonno I.; Lazzarato L.; Fanelli M.; Hattinger C. M.; Serra M.; Riganti C.; Fruttero R.; Ghigo D.; Gasco A. H2S-donating doxorubicins may overcome cardiotoxicity and multidrug resistance. J. Med. Chem. 2016, 59, 4881–4889. 10.1021/acs.jmedchem.6b00184. [DOI] [PubMed] [Google Scholar]
- Kang J.; Li Z.; Organ C. L.; Park C.-M.; Yang C.-T.; Pacheco A.; Wang D.; Lefer D. J.; Xian M. pH-controlled hydrogen sulfide release for myocardial ischemia-reperfusion injury. J. Am. Chem. Soc. 2016, 138, 6336–6339. 10.1021/jacs.6b01373. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Sun L.; Wang Y.; Zhao X. A dual-fluorescent whole-well imaging approach for screening active compounds against doxorubicin-induced cardiotoxicity from natural products. RSC Adv. 2015, 5, 106431–106438. 10.1039/C5RA20887D. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.