

Abstract
O-GlcNAc modification (O-GlcNAcylation) is required for survival in mammalian cells. Genetic and biochemical experiments have found that increased modification inhibits apoptosis in tissues and cell culture and that lowering O-GlcNAcylation induces cell death. However, the molecular mechanisms by which O-GlcNAcylation might inhibit apoptosis are still being elucidated. Here, we first synthesize a new metabolic chemical reporter, 6-Alkynyl-6-deoxy-GlcNAc (6AlkGlcNAc), for the identification of O-GlcNAc-modified proteins. Subsequent characterization of 6AlkGlcNAc shows that this probe is selectively incorporated into O-GlcNAcylated proteins over cell-surface glycoproteins. Using this probe, we discover that the apoptotic caspases are O-GlcNAcylated, which we confirmed using other techniques, raising the possibility that the modification affects their biochemistry. We then demonstrate that changes in the global levels of O-GlcNAcylation result in a converse change in the kinetics of caspase-8 activation during apoptosis. Finally, we show that caspase-8 is modified at residues that can block its cleavage/activation. Our results provide the first evidence that the caspases may be directly affected by O-GlcNAcylation as a potential antiapoptotic mechanism.
Graphical Abstract:
INTRODUCTION
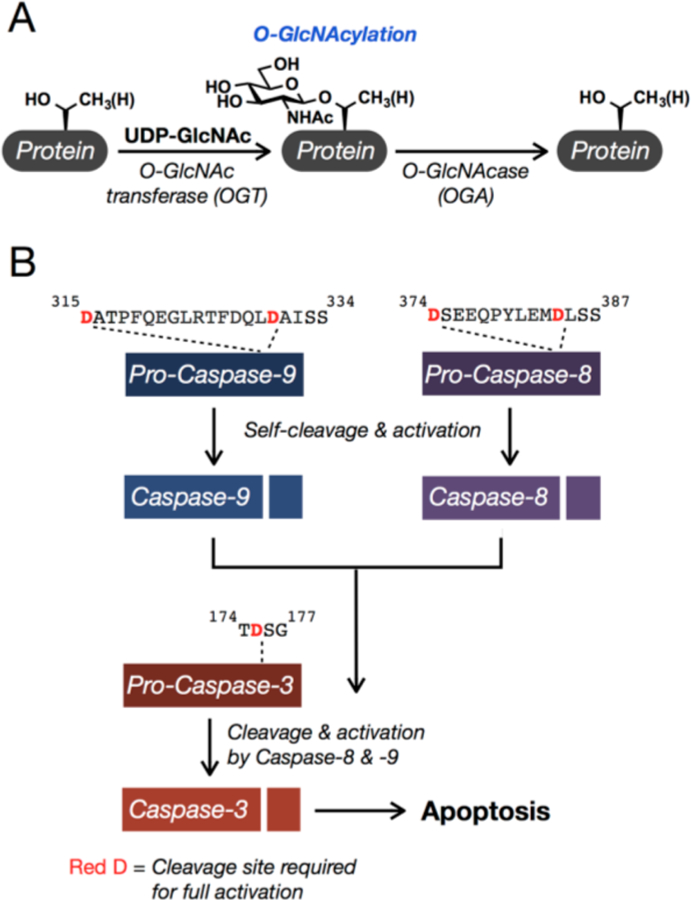
O-GlcNAc modification is found on serine and threonine side-chains of proteins throughout the cytosol, nucleus, and mitochondria of animal and plant cells (Figure 1A).1,2 Unlike many forms of cell-surface glycosylation, it consists only of the addition of the single monosaccharide N-acetylglucosamine that is not elaborated by any additional carbohydrates. O-GlcNAcylation is also dynamic through action of the enzyme O-GlcNAc transferase (OGT), which adds the modification and subsequent removal by the enzyme O-GlcNAcase (OGA).3 Genetic experiments have demonstrated that O-GlcNAcylation is required for development in mice4−6 and Drosophila7 and for survival of mammalian cells in culture. Additionally, changes in the overall cellular levels of this modification directly contribute to a variety of diseases. O-GlcNAcylation levels are consistently elevated in all types of cancer when compared to healthy tissue.8 In contrast, genetic loss of OGT in mouse neurons results in hyperphosphorylated tau,5 and a combination of biochemical and pharmacological experiments has demonstrated that O-GlcNAcylation blocks the aggregation of the neurodegenerative disease associated proteins tau9,10 and α-synuclein.11,12 The general role for O-GlcNAcylation in the modulation of cell survival during stress potentially links these two disease observations. The overall levels of O-GlcNAcyla-tion are significantly elevated by a variety of cellular stressors in both culture and in vivo models, and these increased modification levels promote the survival of both cells and tissue.13–15 Notably, genetic and biochemical experiments have implicated O-GlcNAcylation as an inhibitor of apoptosis. For example, specific genetic deletion of OGT in T-cells caused a large amount of apoptosis in both CD4+ and CD8+ cells,5 and treatment of pancreatic cancer cell lines with a small-molecule inhibitor of OGT resulted in the induction of apoptosis.16 However, the previously identified roles for O-GlcNAcylation in apoptosis that could explain these results are somewhat indirect: O-GlcNAcylation can drive the expression of heat-shock proteins,17 glycosylated phosphofructokinase and glucose-6-phosphate dehydrogenase have altered activities, resulting in the production of NADPH,18,19 and O-GlcNAcy-lated NFκB has increased transcriptional activity.16
Figure 1.
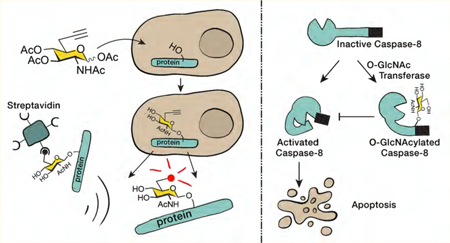
O-GlcNAcylation and the major apoptotic caspases. (A) O-GlcNAcylation is the reversible addition of the monosaccharide N-acetylglucosamine to serine and threonine side-chains of proteins in the cytosol, nucleus, and mitochondria. (B) Apoptosis is carried out by several caspase proteases, including 3, 8, and 9. The caspases are translated as inactive zymogens (pro-caspases) that are activated by cleavage at specific sites, directly after certain aspartic acid, D, residues. Caspases-8 and −9 activate themselves and subsequently activate caspase-3.
While these pathways certainly contribute to cell survival, we were interested in determining if any of the direct components of the apoptotic machinery are affected by O-GlcNAcylation as a more immediate mechanism to inhibit cell death. Apoptosis in mammalian cells is regulated by the activation of a family of cysteine proteases termed caspases.20–23 These enzymes are translated as inactive zymogens (pro-caspases), preventing the uncontrolled activation of cell death. Upon certain stimuli, the caspases are activated by proteolysis in two different pathways (Figure 1B). The intrinsic pathway begins with release of mitochondrial proteins, including cytochrome c, into the cytosol. This results in the formation of a protein complex that contains multiple copies of pro-caspase-9. The proteolytic activity of caspase-9 is increased in this complex, resulting in its self-cleavage and activation. Caspase-9 then cleaves and activates the effector caspases, including caspase-3, which then cleave hundreds of specific substrates, resulting in cell death.24–26 The second pathway, called the extrinsic pathway, is typically initiated from outside the cell through the engagement of death receptors by appropriate ligands. This leads to a similar activation and cleavage of caspase-8 that will then activate the effector caspases. The caspase family members have different substrate preferences, but all require an aspartic acid (D) residue immediately N-terminal to the cleavage site and prefer small amino acids immediately C-terminal.27,28 In addition to the caspases, there are other proteins that play key roles in apoptosis, including numerous receptors, scaffolding proteins, and inhibitors and activators of the pathway.
Here, we describe a chemical-proteomics approach that enabled the discovery of O-GlcNAcylation on all three of the major apoptotic caspases (3, 8, and 9). To make this discovery, we developed a new, metabolic chemical reporter (MCR), termed Ac36AlkGlcNAc, that shows selectivity for labeling O-GlcNAcylated proteins in mammalian cells. This MCR builds upon our previously published reporter Ac36AzGlcNAc,29 but displays improved detection capabilities because of a superior signal-to-noise ratio due to the reverse orientation of the copper-catalyzed azide−alkyne cycloaddition (CuAAC) chemistry.30 When combined with CuAAC, 6AlkGlcNAc enables the visualization of labeled proteins in as little as 1 h and the proteomic identification of 469 potentially O-GlcNAcylated proteins in embryonic mouse fibroblast cells, including caspases-3 and −8. We subsequently used biochemical methods to confirm O-GlcNAcylation of all three of the major apoptotic caspases in both mouse and human cell lines. We then explored the consequences of modification on caspase-8. Using small-molecule inhibitors to raise and lower global O-GlcNAcylation levels, we find that they are inversely related to the kinetics of caspase-8 activation during apoptosis. Finally, we demonstrate that caspase-8, a key initiator of the extrinsic apoptotic pathway, is modified at its cleavage/activation sites and show that these modifications inhibit cleavage in vitro and potentially in living cells. Collectively, these results provide an advanced MCR for the discovery of potentially O-GlcNAcylated proteins and provide, for the first time, a potential role for O-GlcNAc in the direct inhibition of caspase activation. We predict that this caspase modification may be a function of O-GlcNAcylation in both development and human disease.
RESULTS AND DISCUSSION
Development of a Selective and Robust Metabolic Chemical Reporter for the Identification of Potentially O-GlcNAcylated Proteins.
We and others have demonstrated that the majority of O-GlcNAcylation MCRs are not selective for O-GlcNAc and label other types of glycoproteins. While this does not preclude their use for the characterization of proteins, it does limit the number of potentially O-GlcNAcylated proteins that can be identified in a proteomics experiment, as the enrichment of abundant cell-surface glycoproteins can mask the presence of O-GlcNAcylated proteins.29 We serendipitously overcame this selectivity issue with our previously published MCR (Ac36AzGlcNAc (1), Figure 2A), which was excluded from cell-surface glycoproteins but labeled O-GlcNAcylation substrates.29 We chose to explore a 6-modified MCR because UDP-6AzGlcNAc is a substrate for OGT in vitro31 and because a small-molecule glycoside of 6AzGlcNAc can be hydrolyzed by OGA.32 Notably, we also demonstrated in vitro that Ac36AzGlcNAc (1) can bypass the canonical metabolic salvage pathway by transformation to the 6AzGlcNAc-1-phosphate by the enzyme phosphoacetylglucosamine mutase (AGM1).29 As mentioned above, azido probes in combination with alkyne tags under CuAAC have increased background signal due to nonspecific reactivity of alkyne tags with proteins,30 which reduces the number of proteins that can be identified with confidence. Therefore, we synthesized here the analogous alkyne-bearing MCR Ac36AlkGlcNAc (2, Figure 2A) in eight synthetic steps from commercially available material (Figure S1 (SI)). With this potential MCR in hand, we first compared it to Ac36AzGlcNAc (1), by treating NIH3T3 cells with either reporter or DMSO vehicle for 16 h. Analysis by in-gel fluorescence, following lysis and CuAAC with the appropriate fluorescent tag, demonstrated that Ac36AlkGlcNAc (2) labels a qualitatively similar spectrum of proteins as Ac36AzGlcNAc (1) (Figure 2B) but with notably improved signal-to-noise ratio (Figure 2B, high fluorescence “gain”).
Figure 2.
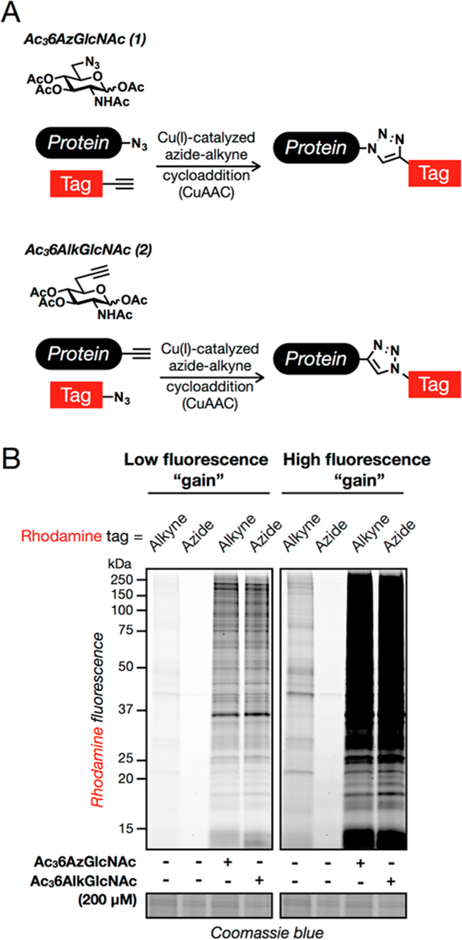
Characterization of the metabolic chemical reporter Ac36AlkGlcNAc (2). (A) Metabolic chemical reporters of O-GlcNAcylation. Treatment of cells with Ac36AzGlcNAc (1) results in the incorporation of an azide functionality onto O-GlcNAcylated proteins, which can be subsequently reacted with alkyne tags using bioorthogonal chemistry. Ac36AlkGlcNAc (2) was hypothesized to function in the same way but with improved signal-to-noise ratio. (B) Ac36AlkGlcNAc (2) has improved signal-to-noise ratio compared to Ac36AzGlcNAc (1). NIH3T3 cells were treated with either DMSO vehicle, Ac36AzGlcNAc (1, 200 μM), or Ac36AlkGlcNAc (2, 200 μM) for 16 h, followed by CuAAC with the appropriate fluorescent tag and analysis by in-gel fluorescence scanning. The data are representative of two biological replicates.
We next determined whether Ac36AlkGlcNAc (2) is able to label proteins in different mammalian cell lines. Accordingly, a small panel of cells was treated with either Ac36AlkGlcNAc (2, 200 μM) or DMSO vehicle for 16 h. The cells were then lysed and subjected to CuAAC conditions with azido-rhodamine. In-gel fluorescence scanning showed a diversity of modified proteins in all the cell lines tested (Figure S2 (SI)), demonstrating the general applicability of Ac36AlkGlcNAc (2) to different mammalian cell lines. We then characterized the dose dependence and kinetics of Ac36AlkGlcNAc (2) labeling and compared it directly to our two previously published MCRs, Ac36AzGlcNAc (1)29 and Ac4GlcNAlk33 (Figures S3 and S4 (SI)). These data show that Ac36AlkGlcNAc (2) labels proteins with faster kinetics than Ac36AzGlcNAc (1) (Figure S3A (SI)), which combined with the low background signal enables proteins to be robustly labeled in as little as 1 h (Figure S3B (SI)). Additionally, Ac36AlkGlcNAc (2) is incorporated more effciently than Ac36AzGlcNAc (1) (Figure S3C (SI)). Ac36AlkGlcNAc (2) labeled cells with slightly slower kinetics compared to Ac4GlcNAlk (Figure S4A (SI)) and at lower overall levels (Figure S4B (SI)). Finally, we looked at the turnover of Ac36AlkGlcNAc-dependent labeling in a pulse-chase experi-ment (Figure S4C (SI)). The associated fluorescent signal was steadily lost over the course of 48 h at a comparable rate to Ac4GlcNAlk labeling. While some of this signal loss is undoubtedly due to protein degradation, the similarity in the kinetics suggests that 6AlkGlcNAc, like GlcNAlk and 6AzGlcNAc, is also a substrate for OGA. We then examined the selectivity of Ac36AlkGlcNAc (2) labeling of different classes of glycoproteins by first using two reporter proteins: GlyCAM-IgG, which is a secreted fusion protein that contains both N-linked and mucin O-linked cell-surface glycans, and FoxO1, which is an O-GlcNAcylated transcription factor. As expected, treatment with Ac4GlcNAlk as a control resulted in labeling of both proteins (Figure 3A), as this MCR is not excluded from cell-surface glycoproteins. However, when these cells were treated with Ac36AlkGlcNAc (2), we observed no labeling of GlyCAM-IgG but robust labeling of FoxO1 (Figure 3A), indicating that Ac36AlkGlcNAc (2) may be selectively labeling potential O-GlcNAcylation substrates. To confirm these results, we also performed an analysis of the entire repertoire of cell-surface glycans using flow cytometry by taking advantage of a copper-chelating azido biotin tag that enables CuAAC on the surface of living cells.34 More specifically, NIH3T3 cells were treated with either Ac36AlkGlcNAc (2) or the nonselective MCR Ac4GlcNAlk or Ac4ManNAlk. Analysis of cell-surface labeling by flow cytometry demonstrated that Ac36AlkGlcNAc (2) treatment resulted in dramatically less global cell-surface labeling (Figure 3B), consistent with its selectivity toward FoxO1 labeling.
Figure 3.
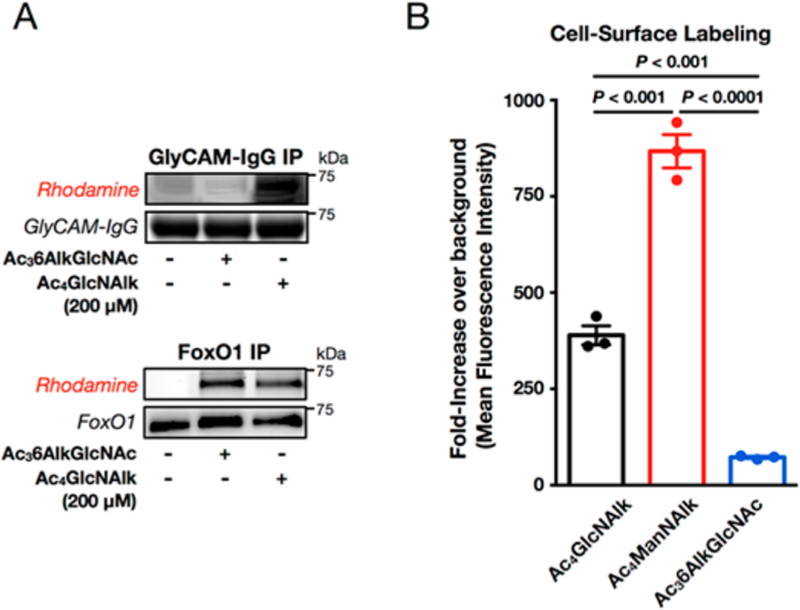
Ac36AlkGlcNAc (2) is largely excluded from cell-surface glycoproteins. (A) Ac36AlkGlcNAc (2) is selective for an O-GlcNAcylated reporter protein. NIH3T3 cells expressing either secreted GlyCAM-IgG or nucleocytoplasmic FoxO1 were treated with DMSO vehicle, Ac36AlkGlcNAc (2, 200 μM), or Ac4GlcNAlk (200 μM) for 24 h. At this time, the proteins were enriched from the media or soluble cell lysate, respectively, subjected to CuAAC with azido-rhodamine, and analyzed by in-gel fluorescence scanning. The data are representative of two biological replicates. (B) Ac36AlkGlcNAc (2) is largely excluded from cell-surface glycosylation. NIH3T3 cells were treated with Ac4GlcNAlk, Ac4ManNAlk, or Ac36AlkGlcNAc (2) (all at 200 μM) for 16 h. The cells were then harvested and subjected to CuAAC with picolyl azido-biotin. After incubation with FITC-streptavidin, cell-surface labeling was measured by flow cytometry. Error bars represent ±SEM from the mean of biological replicates (n = 3), and statistical significance was calculated using a two-tailed Student’s t test.
Finally, we set out to determine if treatment with MCRs reduces cellular proliferation or induces apoptosis. To test these possibilities, NIH3T3 cells were treated in triplicate with either Ac36AlkGlcNAc (2), Ac36AzGlcNAc (1), or Ac4GlcNAlk before analysis using two different, commercially available assays: an MTS assay to measure general cellular proliferation and an apoptosis assay that measures the catalytic activity of caspase-3 and −7 (Figure S5A (SI)). Although there appeared to be a small decrease in the proliferation of the cells, none of the differences were statistically significant and no induction of apoptosis was observed. We next repeated this experiment in HeLa cells under Ac36AlkGlcNAc (2) treatment and observed similar results, with no significant change in cellular proliferation and no induction of apoptosis (Figure S5B (SI)). Together these data demonstrate that Ac36AlkGlcNAc (2) has the potential to be a selective probe for the identification of O-GlcNAcylated proteins that is relatively benign to cells.
Identification of Potentially O-GlcNAcylated Proteins Using Ac36AlkGlcNAc.
Next, we applied Ac36AlkGlcNAc (2) to identify potentially modified proteins and simultaneously compared it to Ac4GlcNAlk in order to uncover any obvious benefits of the increased selectivity. Immortalized mouse embryonic fibroblasts (MEFs) were treated in triplicate with either Ac36AlkGlcNAc (2), Ac4GlcNAlk, or Ac4GlcNAc as a control. The cells were lysed and subjected to CuAAC conditions with an azide-bearing biotin tag. The proteomes were then reduced, alkylated, and subjected to biotin enrichment using streptavidin-conjugated beads. After extensive washing to remove unlabeled proteins, on-bead trypsinolysis afforded peptides that were analyzed using LC-MS/MS. Labeled proteins were identified as those that met threshold criteria for enrichment (see Tables S1 and S2 (SI) for details) based on spectral counting, resulting in 469 proteins identified as potential O-GlcNAcylated proteins that were labeled by Ac36AlkGlcNAc (2) (Figure 4A and Table S1 (SI)) and 433 proteins labeled by Ac4GlcNAlk (Figure 4A and Table S2 (SI)), with many of the proteins showing overlap in the two data sets. Additionally, a large fraction of the identified proteins have been characterized as being potentially O-GlcNAcylated in past proteomic experiments. Although spectral counting is inherently a semiquantitative measurement of protein enrichment, as larger proteins will produce more peptides, we used it because it allowed us to directly compare the identified proteins in this study to our previously published data on azide-bearing MCRs.29
Figure 4.
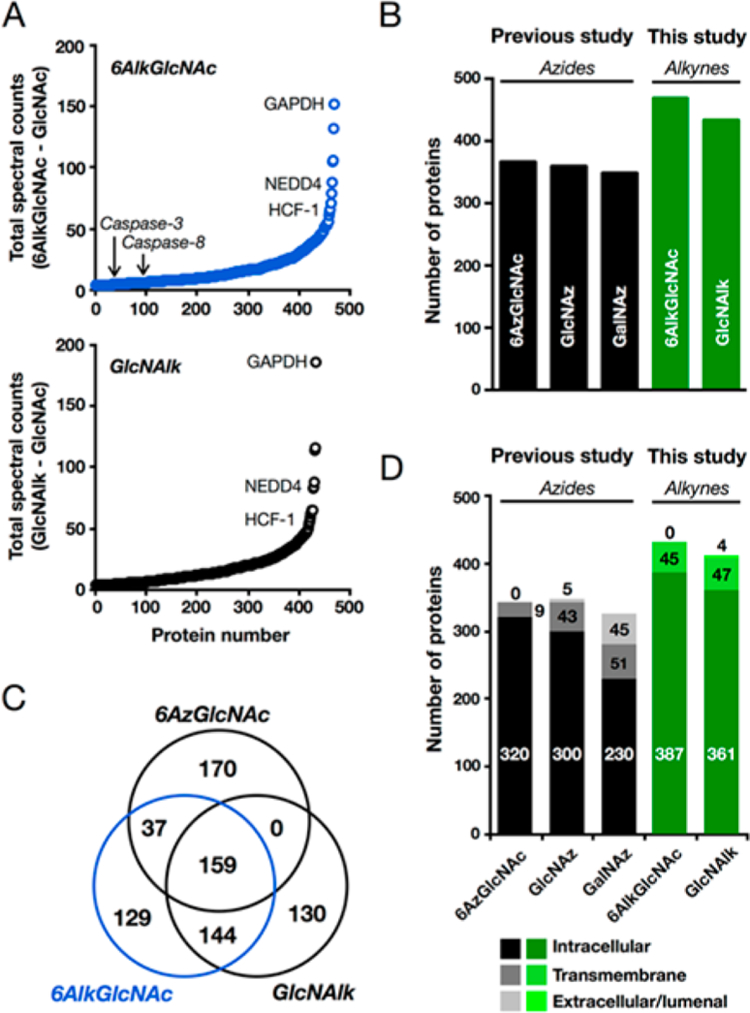
Identification of O-GlcNAcylated proteins, including caspases-3 and −8, using Ac36AlkGlcNAc (2). (A) Mouse embryonic fibroblasts were treated with Ac36AlkGlcNAc (2), Ac4GlcNAlk, or Ac4GlcNAc (all at 200 μM) for 16 h. At this time, the corresponding cell lysates were subjected to CuAAC with azido-biotin, incubation with streptavidin-coated beads, and trypsinolysis. Proteins identified by LC-MS/MS are graphically presented as total number of positive minus total number of control spectral counts. Three known O-GlcNAcylated proteins and identified caspases are annotated. (B) Number of proteins identified as being enriched to a statistically significant amount by MCRs in this study (in green) and a previously published study (in black). (C) Ven diagram showing the overlap in the proteins identified using Ac36AzGlcNAc (1), Ac36AlkGlcNAc (2), and Ac4GlcNAlk. (D) Graphical representation of the enriched proteins from this study (in green) and a previously published experiment (in black) based on their annotated localization. In all experiments, identical cell lines, MCR concentrations, and treatment times were used. The number of proteins in D is less than the number in B due to proteins with unannotated localization.
We first compared the total number of proteins that could be identified at a statistically significant level between the different MCRs (Figure 4B). Not surprisingly the alkyne-MCRs outperformed the azide counterparts because of their improved signal-to-noise ratios, and Ac36AlkGlcNAc (2) enabled the identification of more potentially O-GlcNAcylated proteins compared to Ac4GlcNAlk. We then collated the overlap between the proteins identified by Ac36AzGlcNAc (1), Ac36AlkGlcNAc (2), and Ac4GlcNAlk and found significant overlap between the data sets (Figure 4C). We also annotated the data based on the subcellular localization of the identified proteins as either intracellular or transmembrane, and therefore potential O-GlcNAc substrates, or extracellular/lumenal. Proteins without defined localizations were omitted. In our experiments, Ac36AlkGlcNAc (2) enabled the identification of the most intracellular proteins (Figure 4D), which have the opportunity to be O-GlcNAcylated. Importantly, the previous proteomic identification of proteins labeled with azide-containing MCRs was performed in the same cell line, at identical concentrations and treatment times. Notably, the apoptotic caspases-3 and −8 were identified in our proteomics experiment as potential O-GlcNAc substrates from the Ac36AlkGlcNAc-treated but not Ac4GlcNAlk-treated cells (Figure 4A). To visualize the enrichment of these caspases and three known O-GlcNAcylated proteins, the same cells were treated with Ac36AlkGlcNAc (2) as above, and the labeled proteins were enriched and selectively eluted using a cleavable azido-azo-biotin tag. Western blotting confirmed the enrichment of both caspases and the known O-GlcNAcylated substrates NEDD4–1, pyruvate kinase, and nucleoporin 62 (nup62) (Figure 5A). Importantly, the abundant, non-O-GlcNAcylated protein β-actin was not enriched, demonstrating the potential O-GlcNAc-selective nature of our enrichment. To further compare the utility of Ac36AlkGlcNAc (2) to Ac36AzGlcNAc (1), we repeated this experiment with both MCRs, compared the efficiency of nup62 enrichment, and found that the alkyne probe is indeed superior (Figure 5B). To confirm that caspases-3 and −8 are legitimately O-GlcNAcy-lated, we first used chemoenzymatic labeling (Figure S6 (SI)).35 This method detects endogenous O-GlcNAcylated proteins by using an engineered galactosyltransferase, GalT(Y289L), to modify O-GlcNAc modifications with the azide-bearing monosaccharide N-azidoacetyl-galactosamine. The azide of the resulting disaccharide can then be detected using CuAAC and alkyne visualization- or affinity-tags. Lysates from MEFs were subjected to chemoenzymatic labeling followed by CuAAC with an alkyne-azo-biotin tag (Click Chemistry Tools #1042) to enable enrichment of O-GlcNAcylated proteins. Western blotting after elution from streptavidin beads confirmed that caspases-3 and −8 are indeed endogenously O-GlcNAcylated (Figure 5B).
Figure 5.
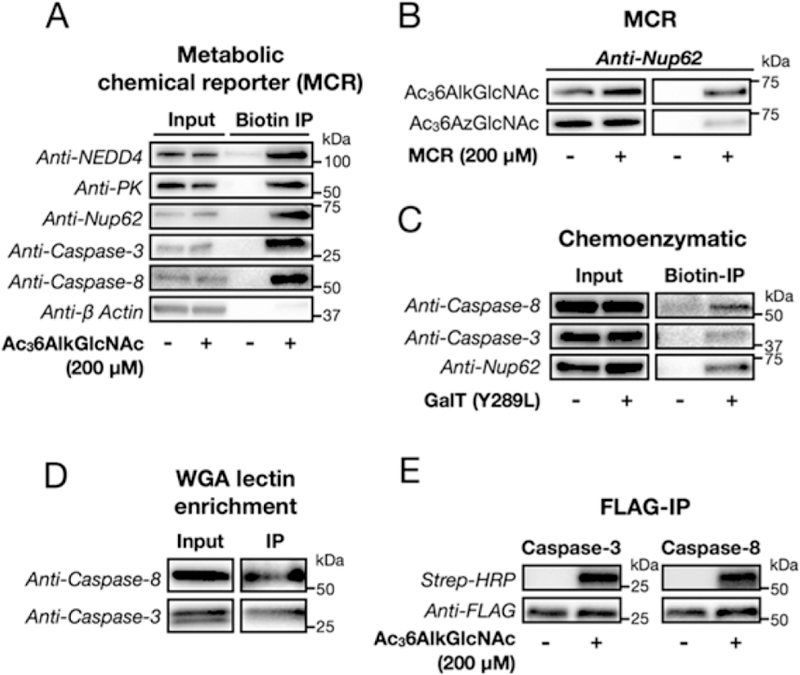
Caspases-3 and −8 are genuine O-GlcNAcylated proteins in mouse cells. (A) Confirmation of Ac36AlkGlcNAc (2) labeling of known O-GlcNAcylated proteins and caspases-3 and −8. Mouse embryonic fibroblasts were treated with either Ac36AlkGlcNAc (2, 200 μM) or DMSO for 16 h, followed by CuAAC with a cleavable azido-biotin tag. After enrichment on streptavidin beads, the labeled proteins were eluted and visualized by Western blotting. The nonglycosylated protein β-actin is a negative control. (B) Nup62 is enriched more efficiently by Ac36AlkGlcNAc (2) labeling when compared to Ac36AzGlcNAc (1). NIH3T3 cells were treated with either Ac36AlkGlcNAc (2, 200 μM), Ac36AzGlcNAc (1, 200 μM), or DMSO for 16 h, followed by CuAAC with a cleavable azido-biotin tag. After enrichment on streptavidin beads, the labeled proteins were eluted and visualized by Western blotting. (C) Chemoenzymatic labeling identifies caspases-3 and −8 as O-GlcNAcylated proteins. MEF cell lysates were subjected to chemoenzymatic modification of endogenous O-GlcNAcylation sites and then CuAAC with alkyne-azo-biotin. Western blotting after enrichment and elution confirmed O-GlcNAcylation of caspases-3 and −8 and the positive control Nup62. The WGA lectin enriches caspases-3 and −8. Cell lysates from NIH3T3 cells that stably express FLAG-tagged caspase-3 or −8 were subjected to two rounds of affinity enrichment, anti-FLAG followed by WGA lectin, before analysis by Western blotting. (E) Reverse confirmation of Ac36AlkGlcNAc labeling of caspases-3 and −8. NIH3T3 cells that stably express FLAG-tagged caspase-3 or −8 were treated with either Ac36AlkGlcNAc (2, 200 μM) or DMSO for 16 h, followed by immunoprecipitation with an anti-FLAG antibody, CuAAC with an azido-biotin tag, and streptavidin and Western blotting. The data in all panels are representative of two biological replicates.
We then stably transformed NIH3T3 cells with FLAG-tagged caspase-3 or caspase-8. Notably, these proteins contained mutations at their active sites to prevent the autoinduction of caspase activity. Immunoprecipitation of these proteins with anti-FLAG beads was performed, followed by a second round of enrichment with the lectin wheat germ agglutinin (WGA) that binds to O-GlcNAc modifications. Western blotting against the caspases showed WGA enrichment of a fraction of the protein, further indicating that these proteins are endogenously O-GlcNAcylated (Figure 5D). The stable cells were then treated with Ac36AlkGlcNAc (2) or DMSO vehicle, followed by immunoprecipitation of the individual caspases using an anti-FLAG antibody. Subsequent CuAAC with azido-biotin, followed by analysis by streptavidin blotting, confirmed the MCR labeling of both caspase −3 and −8 (Figure 5E). Together these data confirm that caspases-3 and −8 are bona fide O-GlcNAcylated proteins in mouse cells.
The Major Apoptotic Caspases Are O-GlcNAcylated in Human Cancer Cell Lines.
To confirm caspase O-GlcNAcylation in human cells, we first treated three cancer cell lines H1299 (lung), HeLa (cervical), and MCF7 (breast) with Ac36AlkGlcNAc (2) and enriched the labeled proteins using the cleavable azido-azo-biotin tag. Western blotting after elution demonstrated MCR labeling of caspase,−3 and −8, as well as caspase-9, which we did not detect in our proteomics experiment (Figure 6A). Notably, the non-O-GlcNAcylated protein β-actin was once again largely unenriched compared to inputs in any of the cell lines, and caspase-3 was absent from MCF7 cells, as this cell line is caspase-3 null. We then used chemoenzymatic labeling followed by biotin enrichment and found that all three caspases are endogenously O-GlcNAcylated in H1299 cells (Figure 6B). We then turned our focus to caspase-8, the initiator caspase of the extrinsic apoptosis pathway, and used chemoenzymatic enrichment to show that caspase-8 is O-GlcNAcylated in all three cancer cell lines (Figure S7 (SI)).
Figure 6.
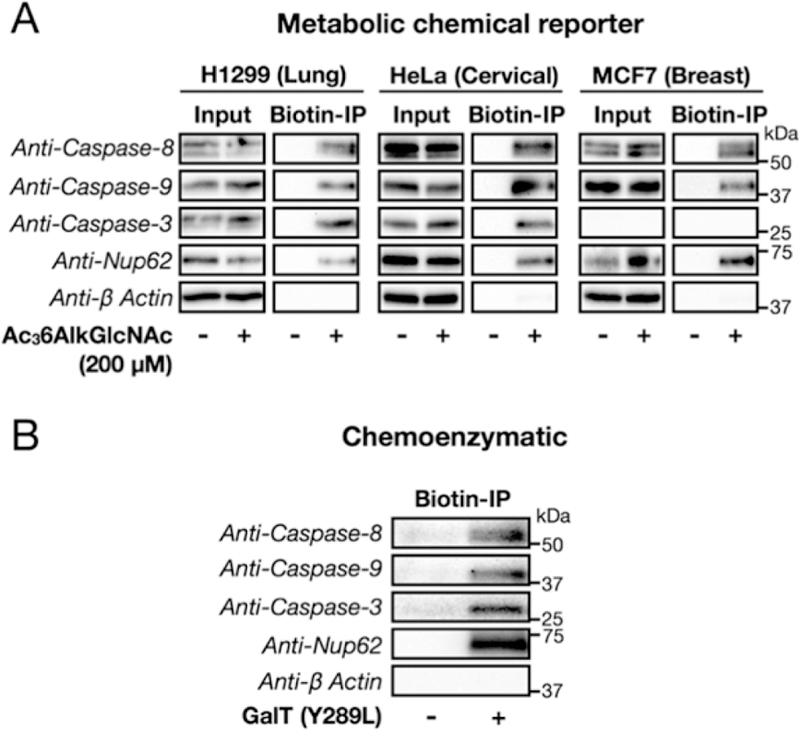
Apoptotic caspases are O-GlcNAcylated in human cancer cell lines. (A) Metabolic chemical reporter enrichment. H1299, HeLa, and MCF7 cells were treated with either DMSO vehicle or Ac36AlkGlcNAc (2, 200 μM) for 16 h. Cell lysates were then subjected to CuAAC with the appropriate cleavable biotin tag, followed by enrichment on streptavidin-coated beads. After extensive washing and elution (Na2S2O4), Western blotting revealed the modification of caspases-3, −8, and −9. (B) Chemoenzymatic modification and enrichment. H1299 cell lysates were subjected to chemoenzymatic modification of endogenous O-GlcNAcylation sites and then CuAAC with alkyne-azo-biotin. Western blotting after enrichment and elution confirmed O-GlcNAcylation of caspases-3, −8, and −9. In A and B, enrichment of the known O-GlcNAcylated protein nucleoporin 62 (Nup62) served as a positive control, and the non-glycosylated protein β-actin was a negative control. In both experiments, the data are representative of two biological replicates.
Changes in O-GlcNAcylation Levels Directly Affect the Kinetics of Caspase-8 Cleavage/Activation.
We next investigated the consequences of O-GlcNAcylation on caspase-8 biochemistry by first determining whether the levels of its modification change during apoptosis. During the initiation of extrinsic apoptosis, monomers of pro-caspase-8 are brought into close proximity, where they are able to cleave themselves to generate the fully active protease, which then goes on to activate the executioner caspases, such as caspase-3 (Figure 7A). Typically, this happens through first cleavage after aspartic acid residues (D374 and D394) between the large and small subunits of the catalytic domain, removing the “activation loop”, followed by cleavage after at least one other aspartic acid residue (D210, D216, and/or D223), which removes the pro-domain.36 Accordingly, HeLa cells were treated with tumor necrosis factor alpha (TNFα) to induce caspase-8-mediated apoptosis, which also caused a very slight increase in the global levels of O-GlcNAcylation (Figure S8 (SI)). Western blotting with two different caspase-8 antibodies clearly showed the time-dependent cleavage/activation of caspase-8 based on the appearance of a lower molecular weight, activated species (p43/41, Figure 7B). Chemoenzymatic labeling was used to enrich the endogenously O-GlcNAcylated fraction of caspase-8 in this experiment. This modified protein showed less loss (i.e., cleavage) of pro-caspase-8 and very little of the activated p43/41 species (Figure 7B), indicating that O-GlcNAcylation of caspase-8 inhibits the cleavage and activation of caspase-8. Given this result, we next treated HeLa cells with either DSMO, 5SGlcNAc, or Thiamet-G to alter O-GlcNAcylation levels (see Figure S9 (SI) for effects of the inhibitors on O-GlcNAcylation levels). These cells were then treated with TNFα, and the cleavage and activation of caspase-8 and caspase-3 was visualized by Western blotting (Figure 7C and Figure S10 (SI)). In all the biological replicates, treatment with 5SGlcNAc resulted in faster kinetics of caspase activation, while Thiamet-G gave slower rates of activation, when compared to DMSO-treated cells. Notably, quantitation of the percentage of cleaved caspase-8 (p43/41 plus p18) from these replicates confirms a statistically significant difference in the rate of caspase-8 activation that inversely depends on O-GlcNAcylation levels (Figure 7D). Importantly, we did not observe a change in the overall levels of caspase-8 upon inhibitor treatment (Figure S11 (SI)). Additionally, the activation of caspase-8 is through direct engagement of death receptors by TNFα, indicating that the effects of O-GlcNAcylation are in this pathway, and these observed effects must occur upstream of NFκB transcriptional activity, as the cells were also treated with cycloheximide to prevent new protein synthesis. This is in contrast to activation of apoptosis by a stimulus such as cellular stress that could be affected by the established roles for O-GlcNAc in cellular metabolism and heat shock response.
Figure 7.
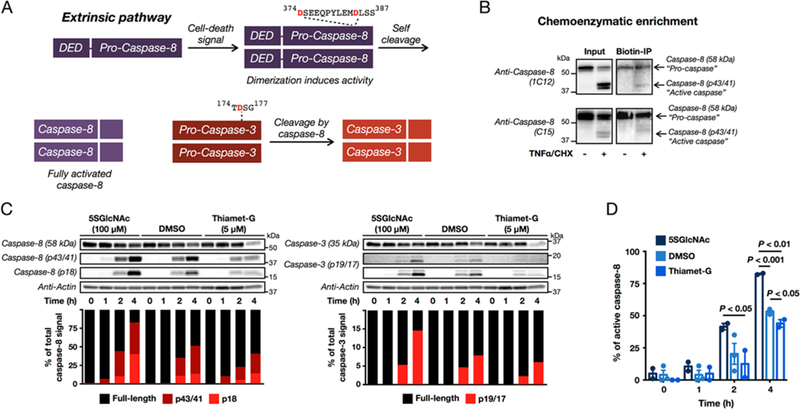
O-GlcNAcylation levels affect caspase-8 cleavage/activation. (A) The extrinsic apoptosis pathway. Upon certain signals, inactive, full-length caspase-8 can be recruited to cell-death receptors, where it is oligomerized. Dimers of inactive caspase-8 have increased activity and self-cleave after aspartic acid residues 374 and 384, removing the activation loop and yielding activated enzyme (p43/p41 and p10 fragments). The p43/41 fragment can then be further cleaved to release the death effector domain (DED), resulting in the formation of a p18 fragment. Active caspase-8 will then go on to cleave the executionerr caspases, such as caspase-3. (B) The endogenously O-GlcNAcylated fraction of pro-caspase-8 has less cleavage during apoptosis. HeLa cells were treated with either vehicle or TNFα (10 ng mL−1) and cycloheximide (1 μg mL−1) to induce apoptosis. After 6 h, the O-GlcNAcylated proteins were enriched using chemoenzymatic modification, and caspase-8 was visualized by Western blotting. (C) Altering O-GlcNAcylation levels changes the kinetics of caspase-8-mediated apoptosis. HeLa cells were treated with TNFα (10 ng mL−1) and cycloheximide (CHX, 1 μg mL−1) to induce apoptosis in the presence or absence of either 5SGlcNAc (OGT inhibitor, 100 μM, 16 h pretreatment) or Thiamet-G (OGA inhibitor, 5 μM, 20 h pretreatment). The kinetics of caspase activation were then visualized after the indicated times by Western blotting. Active caspases were distinguished from their corresponding zymogen form by molecular-weight shift. The different amounts of each caspase species were quantitated and normalized to the total caspase signal in each lane. See Figure S10 for the other biological replicates. (D) Quantitation of the percentage of active caspase-8 (p43/41 plus p18) from the experiments in (C) and Figure S10 (SI). Error bars represent ±SEM from the mean of biological replicates (n = 2 for 5SGlcNAc and Thiamet-G, n = 3 for DMSO), and statistical significance was calculated using a two-tailed Student’s t test.
Caspase-8 Is O-GlcNAcylated near Its Cleavage/Activation Sites.
As mentioned above, the caspases are activated upon self- or cross-cleavage after particular aspartic acid residues. Notably, at or near the cleavage sites required for activation of all three of these caspases are serine (S) or threonine (T) residues that could be O-GlcNAcylated, which might affect the efficiency of cleavage/activation (Figure S12 (SI)). Importantly, these cleavage sites lie in flexible activation loops, and OGT has a strong preference for unstructured regions of its substrate proteins.37–39 Here, we focused on these sites (374DSE376 and 384DLSS387; cleavage after D during activation) in caspase-8 (schematized in Figure 8).36,40,41 Specifically, H1299 cells were cotransfected in duplicate with constructs encoding either HA-tagged wild-type or a FLAG-tagged mutant, either caspase-8(S375A), caspase-8-(S386,387A), or caspase-8(AAA) (374DAE376 and 384DLAA 387) (Figure S13 (SI)). All of these proteins also contained an active-site mutation (C to A) to prevent the autoinduction of apoptosis observed in some caspase-8 overexpression experiments. The entire pool of endogenously O-GlcNAcylated proteins from these cells were enriched using chemoenzymatic labeling (Figure S6), followed by CuAAC with the cleavable biotin tag, enrichment, and elution as above. Western blotting showed that all three mutants were enriched at significantly lower amounts (normalized to inputs) compared to the wild-type protein (Figure 8 and Figure S13 (SI)). Strikingly, this effect was additive, and caspase-8(AAA) was only enriched at ∼10% compared to the wild-type protein, indicating that essentially all of the O-GlcNAcylation occurs at the cleavage sites.
Figure 8.
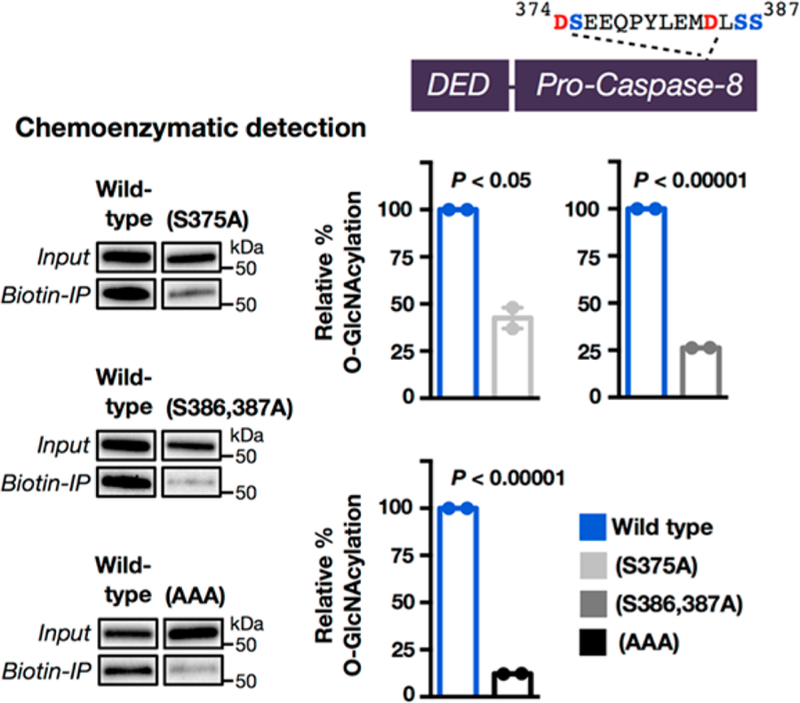
Caspase-8 is O-GlcNAcylated near its cleavage/activation sites. Schematic of wild-type caspase-8 containing two cleavage sites (374DSE376 and 384DLSS387). O-GlcNAcylated proteins from H1299 cells transfected with both HA-tagged wild-type or FLAG-tagged versions of the indicated mutant caspase-8 proteins were enriched using chemoenzymatic transfer and visualized by Western blotting. The HA and FLAG blots were visualized simultaneously to enable a quantitative comparison between the two blots. Results are the average of two separate biological experiments that were imaged simultaneously (see Figure S13 (SI)). Error bars represent ±SEM, and statistical significance was calculated using a two-tailed Student’s t test.
O-GlcNAcylation Blocks Caspase-8 Cleavage/Activation in Vitro and Potentially in Living Cells.
To determine if O-GlcNAcylation can directly affect caspase-8 self-cleavage/activation, we first performed an in vitro cleavage assay. As mentioned above, pro-caspase-8 cleaves itself to generate the fully active protease. Therefore, active caspase-8 is able to cleave peptides that correspond to these cleavage sites in vitro. We synthesized peptides surrounding the cleavage site that were either unmodified (1 and 3) or O-GlcNAcylated (2 and 4, Figure 9A). These peptides were then incubated in triplicate with recombinant, active human caspase-8 before analysis using reverse-phase HPLC (RP-HPLC), which separated any cleavage product from the starting peptide (Figure S14 (SI)), and the absolute identity of the peptides was confirmed by ESI-MS. Caspase-8 was able to readily cleave both of the unmodified peptides. In contrast, O-GlcNAcylation almost completely prevented the formation of the cleavage product, with the unmodified peptides 1 and 3 cleaving approximately 28 and 16 times more efficiently than the O-GlcNAcylated peptides 2 and 4, respectively (Figure 9A). This leads to a 20-times reduction in the relative removal of the activation loop (Figure 9B), indicating that O-GlcNAcylation at these sites would inhibit the self-activation of caspase-8 during cell death. We next wanted to test if O-GlcNAcylation at these sites could affect caspase-8 cleavage in living cells. During apoptosis in cells both dimerization and caspase-8 cleavage have been shown to be required for the generation of the fully active caspase-8 enzyme.39 To determine the consequences of O-GlcNAcylation, we planned to test this cleavage using both a mutant that cannot be O-GlcNAcylated (i.e., serine to alanine) and a mutant that could approximate the steric bulk of O-GlcNAc (serine to tryptophan). We chose tryptophan as a surrogate because there is no way to site-specifically incorporate O-GlcNAc using artificial amino acids. To test if these mutations affected caspase-8 cleavage, we first synthesized the corresponding serine to alanine (StoA) and serine to tryptophan (StoW) peptides (5−8) and repeated the in vitro cleavage assay described above. In this experiment, we found the native peptide 1, containing 374DSE376, to be fully cleaved by caspase-8. This increased cleavage compared to the previous assay could be due to the addition of DMSO, which was required to solubilize the mutant peptides. Consistent with previous experiments,28 we found that substitution of the serine at position P1′ in this peptide by either alanine (5, 374DAE376) or tryptophan (6, 374DWE376) inhibited the cleavage of this peptide, although the inhibition by StoW was more pronounced (Figures S15 and S17 (SI)). To our knowledge the effects of mutations at the P2′ and P3′ positions on caspase-8-mediated cleavage have never been systematically tested. Native peptide 2, containing 384DLSS 387, again showed notable cleavage, while the alanine peptide (7, 384DLAA387) showed increased cleavage and the tryptophan peptide (8, 384DLWW387) displayed less (Figures S15 and S17 (SI)). We then quantitated this data to again determine the efficiency of removal of the activation loop (Figure 9C). We found that the alanine mutations had an overall small inhibitory effect on caspase-8 activation loop cleavage by 1.2 times compared to the corresponding serine residues, indicating that the effect of the mutation alone would be to slow caspase-8 activation slightly. The tryptophan mutations inhibited cleavage of the loop by 3 times, making these mutations a reasonable, but less effective, analogue of O-GlcNAcylation for the inhibition of caspase-8 cleavage.
Figure 9.
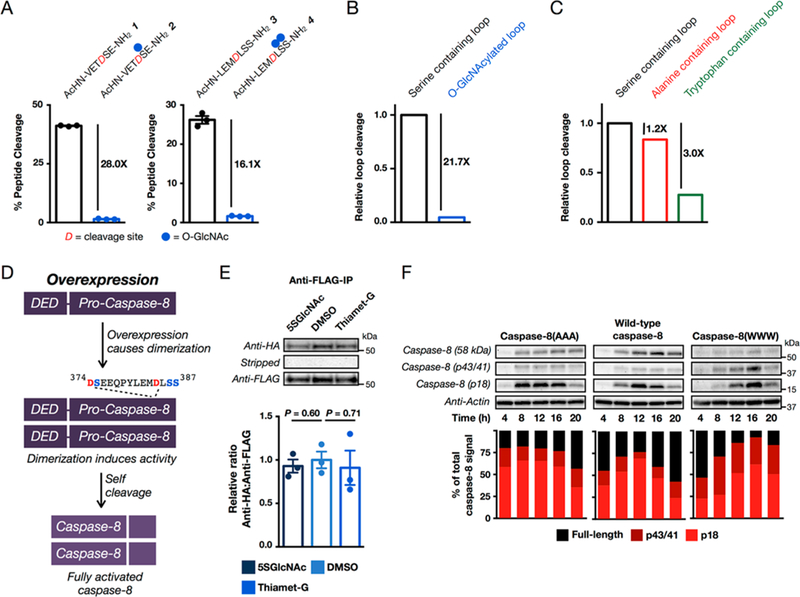
O-GlcNAcylation of the caspase-8 cleavage sites affects its cleavage/activation. (A) O-GlcNAcylation inhibits the self-cleavage of caspase-8 in vitro. The indicated unmodified or O-GlcNAcylated peptides (2 mM) were incubated with recombinant, active caspase-8 for 24 h, and the percent cleavage was determined using reverse-phase high-performance liquid chromatography (RP-HPLC). Results are the mean ± SEM of three separate biological experiments (see Figure S14 (SI)). (B) Comparison of the relative efficiency of caspase-8 activation loop cleavage between unmodified and O-GlcNAcylated peptides. The individual effects of O-GlcNAcylation shown in (A) were averaged to obtain the overall loop cleavage efficiency.Mutation of the O-GlcNAcylation sites in caspase-8 to alanine or tryptophan inhibits overall caspase-8 activation loop cleavage in vitro, with tryptophan having a larger inhibitory effect. The individual effects of the alanine and tryptophan mutations (shown in Figure S17 (SI)) were averaged to obtain overall loop cleavage efficiency. (D) Activation of caspase-8 by transient overexpression. Upon overexpression, caspase-8 will self-dimerize and activate itself without a requirement for cell-death signaling. (E) Changing O-GlcNAcylation levels does not affect the dimerization of caspase-8. Caspase-8 null HeLa cells were cotransfected with plasmids encoding HA- and FLAG-tagged, inactive caspase-8, before being treated with 5SGlcNAc (200 μM), DMSO vehicle, or Thiamet-G (20 μM) to alter O-GlcNAcylation levels. An anti-FLAG immunoprecipitation was then performed before visualization with an anti-HA antibody, stripping of the blot, and visualization with an anti-FLAG antibody. The amount of enriched HA-tagged protein relative to enriched FLAG-tagged protein was then quantified. Results are the average of three separate biological experiments that were imaged simultaneously (Figure S19B (SI)). Error bars represent ±SEM of three biological experiments, and statistical significance was calculated using a two-tailed Student’s t test. (F) Caspase-8 with its O-GlcNAc sites mutated to alanine (caspase-8(AAA)) activates faster than wild-type protein, and mutation to tryptophan (caspase-8(WWW)) slows the rate of cleavage. Caspase-8 null HeLa cells were transiently transfected with plasmids encoding either wild-type caspase-8, caspase-8(AAA), or caspase(WWW) for 6 h. After the indicated lengths of time, cells were collected. Analysis by Western blotting shows autoactivation of all proteins. The different amounts of each caspase species were quantitated and normalized to the total caspase signal in each lane. The data are representative of two biological experiments.
With the peptide mutations characterized in vitro, we set out to test their consequences on caspase-8 activation in living cells. It has been observed that transient transfection of caspase-8 can lead to spontaneous dimerization and cleavage/activation of the protein, presumably due to high cellular concentrations (Figure 9D). This feature can be used to directly test any differences in self-activation caspase-8, without complications from upstream receptor-mediated activation steps. Unfortunately, the endogenous caspase-8 in mammalian cells can participate in this process, so we first used the CRISPR genetic engineering system to generate several clones of a caspase-8 null HeLa cell line (Figure S18 (SI)). With these cells in hand, we next confirmed that overexpression of caspase-8 would cause its autodimerization. To accomplish this goal, caspase-8 null cells were cotransfected with wild-type HA- and FLAG-tagged caspase-8 constructs bearing active-site mutations to prevent autoactivation. Importantly, we first confirmed that the antibodies to these tags do not cross-react (Figure S19A (SI)). Immunoprecipitation with an anti-FLAG antibody resulted in coenrichment of the HA-tagged caspase-8 and vice versa, demonstrating that we were indeed inducing dimerization of caspase-8 upon overexpression (Figure S19A (SI)). To determine if global changes in O-GlcNAcylation could affect this process, we performed the same immunoprecipitation in triplicate in cells that were treated with either 5SGlcNAc, DMSO, or Thiamet-G to change the overall O-GlcNAcylation levels (Figure 9E and Figure S19B (SI)). Notably, the changes in O-GlcNAcylation had no significant effect on the co-immunoprecipitation, indicating it does not alter the ability of caspase-8 to dimerize during its activation. With these control experiments completed, we next tested the effects of mutating the activation loop from serine to alanine (caspase-8(AAA)) to prevent O-GlcNAcylation or from serine to tryptophan (caspase-8(WWW)) to mimic O-GlcNAc. Accordingly, caspase-8 null cells were transiently transfected with wild-type caspase-8, caspase-8(AAA), or caspase-8(WWW). In contrast to previous experiments, these caspase-8 enzymes do not contain active-site mutations. Cells were collected after different lengths of time post-transfection, and analysis by Western blotting showed that at relatively short times (<36 h) this results in a “pulse” of cleaved/activated caspase-8 that is then removed, potentially by death of the transfected cell population (Figure 9F). Using these conditions, side-by-side comparison of wild-type caspase-8, caspase-8(AAA), and caspase-8(WWW) by Western blotting showed that the caspase-8(AAA) mutant self-cleaved/activated with faster kinetics than wild-type protein (Figure 9F). Specifically, cleavage of caspase-8(AAA) could be detected in as little as 4 h post-transfection, essentially as soon as significant amounts of protein were detectable. In contrast, wild-type caspase-8 also self-activated but did not reach peak activation until 12 h post-transfection. This cellular result is notable because our in vitro peptide data suggested that caspase(AAA) should self-cleave/activate with similar, if not slightly slower, kinetics compared to wild-type protein. Finally, caspase-8(WWW) self-activated with the slowest kinetics with a peak at 16 h post-transfection, consistent with our peptide data and tryptophan acting as an O-GlcNAc surrogate. These data indicate that O-GlcNAcylation of caspase-8 can change the kinetics of its activation of living cells by serving as a steric hindrance to caspase-8 self-cleavage. Importantly, on the basis of our peptide data, bona fide O-GlcNAcylation should have an even more pronounced inhibitory effect on cleavage than mutation to tryptophan.
CONCLUSION
Despite the observations that increased O-GlcNAcylation inhibits apoptosis and promotes survival in cell culture and tissues undergoing stress, there have been relatively few mechanistic explanations for this phenotypic association. Here, we demonstrate that the proteins that directly carry out apoptosis, the caspases, are themselves O-GlcNAc modified. In order to make this discovery, we created a new metabolic chemical reporter, Ac36AlkGlcNAc (2), which we show has significantly improved signal-to-noise ratio compared to our previous probe Ac36AzGlcNAc (1)29 while maintaining its selectivity for intracellular proteins over cell-surface glycoproteins (Figures 2 and 3). This is most likely due to the improved orientation of the CuAAC reaction that reduces background labeling but could also be a result of improved metabolic stability of the alkyne versus azide MCR. Notably, treatment of cells with Ac36AlkGlcNAc (2) allowed for the visualization of modified proteins in as little as 15 min and a large amount of labeling in only 1 h (Figure S3B), which could enable the specific identification of newly O-GlcNAcylated proteins in response to cellular stimuli (e.g., signaling or stress). Like Ac36AzGlcNAc (1), our new MCR cannot be metabolized to the UDP-6AlkGlcNAc by the canonical GlcNAc salvage pathway in mammalian cells, and we believe that it can be converted to 6AlkGlcNAc-1-phosphate by the action of the enzyme phosphoacetylglucosamine mutase, which we previously demonstrated can phosphorylate 6AzGlcNAc. It is also possible that there are uncharacterized small-molecule kinases that can generate these 1-phosphate sugars. In either case, a wide range of cell lines are strongly labeled by Ac36AlkGlcNAc (2) treatment, indicating that this MCR will be broadly applicable. By applying Ac36AlkGlcNAc (2) to MEF cells in an unbiased proteomics experiment, we were able to identify caspases-3 and −8 as being potentially O-GlcNAcylated (Figure 4A). A parallel proteomics experiment using our nonselective MCR, Ac4GlcNAlk,33 did not identify these caspases, demonstrating the utility of our new compound in discovery-based experiments. We then confirmed the MCR labeling of caspases-3 and −8, as well as caspase-9, in three different cancer cell types (Figure 6A). Notably, caspase-9 was not identified in our proteomics experiment, suggesting that applying Ac36AlkGlcNAc (2) in a quantitative proteomics experiment (e.g., SILAC labeling) may uncover additional interesting O-GlcNAcylated proteins. Critically, we were able to use chemoenzymatic labeling and/or WGA enrichment of O-GlcNAcylated proteins to independently confirm the endogenous O-GlcNAc modification of the caspases (Figures 5 and 6). This type of independent confirmation is a key first step after MCR-based identification of any O-GlcNAcylated protein, as treatment with metabolic reporters may alter the normal modification state of the cell.
We next chose to focus on caspase-8 given its key roles in development, immunology, and human disease.43 By taking advantage of chemoenzymatic labeling of endogenous O-GlcNAc modifications, we then showed that the O-GlcNAcylated fraction of caspase-8 is resistant to cleavage/activation upon induction of apoptosis by TNFα. It is possible that O-GlcNAcylation affects the ability of antibodies to recognize caspase-8; however, we observe a difference in the amount of cleaved caspase compared to full length and not an overall reduction in signal in the Western blot, so we think this is unlikely.
We also demonstrated that changing the levels of O-GlcNAcylation can affect the activation/cleavage of caspase-8 during the initiation of apoptotic signaling by TNFα. Using site-directed mutagenesis and chemoenzymatic detection, we found that the vast majority of O-GlcNAcylation on caspase-8 occurs at serine residues near the two cleavage sites required for full caspase-8 activation during apoptosis. We also demonstrated that O-GlcNAcylation of these sites inhibits their cleavage in vitro, which has been shown to be required for the full activation of caspase-8 during apoptosis,42 and we used caspase-8 mutants to indicate that O-GlcNAcylation of these sites can change the kinetics of caspase-8 cleavage/activation in living cells. However, we cannot completely rule out some differences in the cellular experiment, such as the activation of executioner caspases that then feed-back to activate our overexpressed caspase-8. We believe that this is certainly not the only O-GlcNAc modification that causes the phenotypic association between elevated O-GlcNAcylation levels and cell survival, but our results suggest that caspase-8 O-GlcNAcylation could contribute to the antiapoptotic function of O-GlcNAc in cells.
The exact stoichiometry and specific biological contexts where caspase-8 O-GlcNAcylation has a significant effect on overall apoptosis remain to be discovered, a line of investigation we are currently undertaking. Unfortunately, we have found that the generation of cell lines that stably express caspase-8 mutants, such as caspase-8(AAA), is difficult due to variable expression levels that result in autoactivation of apoptosis. Additionally, transient transfection of caspase-8 (as in Figure 9F) can be effected by the presence of OGT and OGA inhibitors in the media. To overcome these issues, we are currently exploring the use of CRISPR-Cas9 to introduce mutants into the caspase-8 gene at the chromosome level. Intriguingly, all of the apoptotic caspases in humans and mice, two out of the three caspases in Drosophila, and ced-3 in Caenorhabditis elegans have serine or threonine residues at or near their cleavage sites. However, the O-GlcNAcylation sites we identified here on caspase-8 are conserved only in mammals. Additionally, analysis of the known caspase cleavage sites in substrate proteins44 shows that serine or threonine is the second most favored amino acid at all of the surrounding eight residues. Many known caspase substrates have also been identified as being O-GlcNAcylated in a variety of proteomics experiments, although the specific sites of modification are largely unknown. Additionally, phosphorylation of serine/threonine residues near caspase cleavage sites has been shown to alter proteolysis rates.45–47 This raises the possibilities that O-GlcNAcylation of caspases is an evolutio-narily conserved mechanism for modulating caspase activation and that this modification may also protect caspase substrates from cleavage. We are currently exploring both of these avenues of research. In conclusion, our new MCR (Ac36AlkGlcNAc, 2) enabled the discovery of caspase O-GlcNAcylation, and our data show that this modification can inhibit caspase-8 cleavage/activation, which may play a role in the regulation of programmed cell death in development and disease.
EXPERIMENTAL PROCEDURES
Synthesis of Known Small Molecules.
Known compounds alkyne- and azide-rhodamine (alk-rho and az-rho),48 UDP-GalNAz,49 Thiamet-G,9 Ac45SGlcNAc,50 Ac4GlcNAlk,34 Ac4ManNAlk,51 and Ac36AzGlcNAc (1)29 were synthesized according to literature procedures.
FLAG and WGA Enrichment of Caspases.
NIH3T3 cells stably expressing either FLAG-tagged caspase-3 or −8 were grown to 80% confluency before being treated with 20 μM Thiamet-G for 16 h. Cells were then harvested by trypsinization and washed two times with phosphate-buffered saline (PBS) (2 min, 2000g, 4 °C). Cell pellets were resuspended in 100 μL of 1% NP-40 lysis buffer [1% NP-40, 150 mM NaCl, 50 mM triethanolamine (TEA), pH 7.4] with Complete Mini, EDTA-free protease inhibitor cocktail tablets (Thermo Scientific) for 20 min and then centrifuged at 4 °C for 10 min at 10000g. The supernatant was collected, and the protein concentration was determined by BCA assay (Pierce, ThermoScientific). Total cell lysate (4 mg) was diluted to a final concentration of 1 mg/mL with 1% NP-40 buffer with Complete Mini, EDTA-free protease inhibitor cocktail tablets (Thermo Scientific). Diluted lysate was then precleared with Protein-G beads (30 μL, Pierce) prewashed three times with 1 mL of NP-40 buffer, for 1 h at 4 °C with full rotation. Beads were then pelleted by centrifugation (2 min, 2000g, 4 °C), and cleared supernatant was transferred to tubes containing EZview Red ANTI-FLAG M2 affinity beads (30 μL, Sigma), prewashed with cold NP-40 buffer two times followed by cold PBS two times. The samples were placed on a rotator for 2 h at 4 °C. Beads were collected by centrifugation at 2000g for 2 min at 4 °C, and the supernatant was carefully removed. Beads were subsequently washed three times by addition of 1 mL of cold PBS with rotation for 5 min before centrifuging for 2 min at 2000g. Protein was eluted by addition of 30 μL of 1% sodium dodecyl sulfate (SDS) buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) and boiling 5 min at 98 °C. The supernatant was allowed to cool to room temperature before being transferred to a new tube in which SDS concentration was diluted to 0.5% by addition of 1% NP-40 lysis buffer. The resulting diluted FLAG-enriched lysate was then transferred to new tubes containing prewashed (PBS 1×, cold NP-40 2×) sWGA agarose resin (60 μL, Vector Laboratories) and was incubated with rotation for 16 h at 4 °C. Following enrichment, resin was washed three times with PBS before proteins were eluted with 4% SDS buffer and boiling for 5 min at 98 °C. Samples were then analyzed by SDS-PAGE (Any Kd, Criterion Gel, Bio-Rad) followed by analysis by Western blotting.
FLAG-IP of Caspases.
NIH3T3 cells stably expressing either FLAG-tagged caspase-3 or −8 were grown to 80% confluency before being treated with 200 μM Ac4GlcNAc or Ac36AlkGlcNAc (2) for 16 h. Cells were then harvested by trypsinization and washed two times with PBS (2 min, 2000g, 4 °C). Cell pellets were resuspended in 100 μL of 1% NP-40 lysis buffer [1% NP-40, 150 mM NaCl, 50 mM TEA, pH 7.4] with Complete Mini, EDTA-free protease inhibitor cocktail tablets (Thermo Scientific) for 20 min and then centrifuged at 4 °C for 10 min at 10000g. The supernatant was collected and the protein concentration was determined by BCA assay (Pierce, ThermoScien-tific). Total cell lysate (2 mg) was diluted to a final concentration of 0.5 mg/mL with 1% NP-40 buffer with Complete Mini, EDTA-free protease inhibitor cocktail tablets (Thermo Scientific). Diluted lysate was then precleared with Protein-G beads (30 μL, Pierce) prewashed three times with 1 mL of NP-40 buffer, for 1 h at 4 °C with full rotation. Beads were then pelleted by centrifugation (2 min, 2000g, 4 °C), and cleared supernatant was transferred to tubes containing EZview Red ANTI-FLAG M2 affinity beads (30 μL, Sigma), prewashed with cold NP-40 buffer two times followed by cold PBS two times. The samples were placed on a rotator for 2 h at 4 °C. Beads were collected by centrifugation at 2000g for 2 min at 4 °C, and the supernatant was carefully removed. Beads were subsequently washed three times by addition of 1 mL of cold PBS with rotation for 5 min before centrifuging 2 min at 2000g. Protein was eluted by addition of 30 μL of 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) and boiling 5 min at 98 °C. Click chemistry with alkyne-biotin was performed in the presence of the FLAG beads assuming a volume of 60 μL. The click chemistry reaction was quenched by addition of 60 μL of 2× loading buffer and boiled 5 min at 98 °C. Samples were then analyzed by SDS-PAGE (Any Kd, Criterion Gel, Bio-Rad) followed by analysis by streptavidin or Western blotting.
Expression and Purification of GalT(Y289L).
pET23a GalT Y289L plasmid was provided by P. Qasba, National Cancer Institute, and was subsequently transformed into BL21 E. coli (Novagen). A 1 L culture containing ampicillin (100 μg mL−1) was inoculated with 10 mL of 50 mL of starter culture grown overnight at 37 °C. The 1 L culture was grown at 37 °C until an OD (A600) of 0.60 was obtained, at which time expression was induced with isopropyl β-D-1-thiogalacto-pyranoside (1 mM final concentration, 1000× stock in water) for 4 h at 37 °C. Cells were harvested by centrifugation (10 min, 4000g, 4 °C) and resuspended in 10 mL of suspension buffer (25% sucrose w/v in 1× PBS). Cells were lysed using sonication (30 s pulse, 30 s rest, 12 min at 4 °C). The resulting lysate was diluted to 80 mL in cold suspension buffer, and inclusion bodies were harvested by centrifugation (30 min, 15000g, 4 °C) and washed 8−10 times or until inclusion bodies turned white in color, by resuspension in 80 mL of suspension buffer followed by centrifugation. Inclusion bodies were then washed once with cold wash buffer (10 mM phosphate, pH 7.0) and harvested by centrifugation. Inclusion bodies were then resuspended in 14 mL of cold H2O, poured over solid guanidine HCl and Na2SO3 (resulting in 5 M GuHCl and 300 mM Na2SO3 final concentration), and vortexted vigorously, and the volume was adjusted to 25 mL with cold H2O. Freshly made NTSB solution (50 mM DTNB, 1 M Na2SO3 in water pH 8.0) was then added with vigorous vortexting to sulfenate free thiols. Completion of the reaction is indicated by a color change from dark orange to pale yellow. Protein was then precipitated with cold H2O (250 mL) and centrifuged immediately (30 min, 9900g, 4 °C). The protein pellet was washed three times by resuspension in cold H2O and centrifugation. The protein pellet was finally resuspended in 14 mL of cold H2O, poured over a solid guanidine HCl, vortexted, and then diluted to 25 mL with H2O (resulting in a 5 M final concentration). The protein solution was diluted to a final concentration of 1 mg mL−1 with a 5 M GuHCl solution (OD (A275) ≈ 2.0). This solution was diluted 10-fold into cold refolding buffer (5 mM EDTA, 4 mM cysteamine, 2 mM cystamine, 100 mM Tris base, pH 8.0) with gentle stirring. Protein was allowed to refold for 48 h at 4 °C without agitation. Refolding protein was dialyzed two times into cold H2O for 24 h and concentrated using centrifugal filters (30 kDa cutoff, Amicon Ultra, Millipore) to 3 mL. Buffer was exchanged once with 10 mL of reaction buffer (10 mM Tris base, pH 8.0), and final protein concentration was determined (OD (A275) ≈ 1.5). Purified protein was stored at 4 °C.
Chemoenzymatic Labeling.
Cells were collected by trypsinization and washed two times with PBS (2 min, 2000g, 4 °C). Cells were subsequently lysed by adding 13 μL of H2O, 25 μL of 0.05% SDS buffer (0.05% SDS, 5 mM MgCl2, 10 mM TEA, pH 7.4), and 1 μL of Benzonase (Sigma Ultra Pure) and incubated on ice for 30 min. Then, 100 μL of 4% SDS buffer was added (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4), the resuspended cell lysate was sonicated briefly in a bath sonicator, and cell debris was pelleted (10 min, 10000g, 15 °C). Protein concentration was determined by BCA Assay (Pierce, ThermoScientific), and the lysate was diluted to 1 μg μL−1 in 1% SDS chemoenzymatic buffer (1% SDS, 20 mM HEPES, pH 7.9). Proteins were precipitated by adding a 3× volume of methanol, a 0.75× volume of hloroform, and 2× volume of H2O followed by vortexing and centrifugation (5 min, 13000g, rt). The aqueous phase was discarded without disturbing the interface layer before adding a 2.5× volume of methanol, vortexing, and pelleting protein (5 min, 13000g, rt). The resulting protein pellet was allowed to air-dry for no longer than 20 min before being resuspended in 1% SDS chemoenzymatic buffer (1% SDS, 20 mM HEPES, pH 7.9). Protein concentration was normalized using the BCA Assay (Pierce, ThermoScientific) and diluted to 2.5 μg μL−1 in 1% SDS chemoenzymatic buffer. To start the chemoenzymatic transfer reaction, these reagents were added in the following order (for 100 μg of total protein): 49 μL of H2O, 80 μL of labeling buffer (2.5×; 5% NP40, 125 mM NaCl, 50 mM HEPES, pH 7.9), 55 μL of MnCl2 (100 mM in H2O), and 50 μL of UDP-GalNAz (0.5 mM in 10 mM HEPES, pH 7.9). This was mixed by pipeting. Finally, 7.5 μL of purified GalT Y289L (in 10 mM Tris pH 8.0) was added, and the reaction mixture was incubated for 16 h at 4 °C without agitation. Unreacted UDP-GalNAz was then removed by methanol−chloroform precipitation as described previously. Air-dried protein pellets were resuspended in 1% SDS CuAAC buffer (1% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) and subjected to CuAAC as detailed below.
Enrichment and Selective Elution of O-GlcNAcylated Proteins.
Cell lysates (∼1 mg at 1 mg mL−1) that had been labeled either chemoenzymatically or with a metabolic chemical reporter were subjected to CuAAC conditions with freshly made cocktail [azide- or alkyne-azo-biotin (100 μM, 5 mM stock solution in DMSO) (Click Chemistry Tools); tris(2-carboxyethyl)phosphine hydrochloride (1 mM, 50 mM freshly prepared stock solution in water); tris[(1-benzyl-1-H-1,2,3-triazol-4-yl)methyl]amine (100 μM, 10 mM stock solution in DMSO); CuSO4·5H2O (1 mM, 50 mM freshly prepared stock solution in water)]. After 1 h, the proteins were precipitated by addition of 4 volumes of ice-cold methanol and incubation at −20 °C for 2 h. Precipitated proteins were collected by centrifugation (30 min, 5000g, 4 °C) and washed three times with ice-cold methanol, with resuspension of the pellet each time. The pellet was then air-dried for 15 min and then resuspended in 800 μL of resuspension buffer (6 M urea, 2 M thiourea, 10 mM HEPES, pH 8.0) by bath sonication. Samples were then transferred to 2 mL dolphin-nosed tubes containing streptavidin beads (25 μL of a 50% slurry per sample, Thermo), washed two times with 1 mL of PBS and one time with 1 mL of resuspension buffer, and resuspended in resuspension buffer (200 μL). Samples were then incubated on a rotator for 2 h. Beads were washed two times with resuspension buffer, two times in PBS (1 mL), and two times with 1% SDS in PBS buffer. Beads were then incubated in 25 μL of sodium dithionite solution (1% SDS, 25 mM sodium dithionite) for 30 min at room temperature to elute captured proteins. The beads were centrifuged for 2 min at 2000g, and the eluent was collected. The elution step was repeated, and the eluents were combined. Proteins were then precipitated in ice cold methanol (1 mL) overnight in −20 °C. Protein was collected by centrifugation (10 min, 10000g, 4 °C), the pellet was allowed to air-dry for 5 min, and then 30 μL of 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) was added to each sample. The mixture was sonicated in a bath sonicator to ensure complete dissolution, and 30 μL of 2× loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol) was then added. Samples were then boiled for 5 min at 98 °C before being separated by SDS-PAGE (4−20% Tris-Glycine Gel, Bio-Rad).
Endogenous Caspase-8 Chemoenzymatic/Biotin IP Following Induction of Apoptosis.
HeLa cells were plated at 2 × 106 cells in a 150 mm dish 48 h prior to activation. Apoptosis was then induced by treating with 10 ng mL−1 TNFα and 1 μg mL−1 cycloheximide (CHX) for 6 h. Cells in the media were combined with cells that were collected by trypsinization before they were pelleted by centrifugation (6 min, 2000g, 4 °C) and washed with PBS (1 mL) three times. Cells were subsequently lysed by adding 26 μL of H2O, 50 μL of 0.05% SDS buffer (0.05% SDS, 5 mM MgCl2, 10 mM TEA, pH 7.4) containing Z-VAD-FMK (Enzo Life Sciences), and 1 μL of Benzonase (Sigma Ultra Pure) and incubated on ice for 30 min. Then, 200 μL of 4% SDS buffer was added (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4), the resuspended cell lysate was sonicated briefly in a bath sonicator, and cell debris was pelleted (10 min, 10000g, 15 °C). Precipitated protein was subjected to chemoenzymatic transfer followed by click chemistry with alkyne-azo-biotin before being subjected to streptavidin enrichment, SDS-PAGE, and Western blotting as previously described.
Caspase Activation Kinetics.
HeLa cells were treated at 70−75% confluency with either 100 μM 5SGlcNAc, 5 μM Thiamet-G (1000× stocks in DMSO), or DMSO vehicle for 16 h. Apoptosis was then induced by treating with 10 ng mL−1 TNFα and 1 μg mL−1 CHX for either 0, 1, 2, 3, 4, or 6 h. Cells in the media were combined with cells that were collected by trypsinization. The cells were then pelleted by centrifugation (6 min, 2000g, 4 °C) and washed with PBS (1 mL) three times. Cell pellets were then resuspended in 50 μL of 1% NP-40 lysis buffer [1% NP-40, 150 mM NaCl, 50 mM TEA, pH 7.4] with Complete Mini protease inhibitor cocktail (Roche Biosciences) and Z-VAD-FMK (Enzo Life Sciences) for 15 min and then centrifuged (10 min, 10000g, 4 °C). The supernatant (soluble cell lysate) was collected, and the protein concentration was determined by BCA assay (Pierce, ThermoScientific). Samples were prepared at a protein concentration of 2 mg mL−1 with 4× loading buffer (8% SDS, 40% glycerol, 0.4% bromophenol blue, 2.8% B-mercaptoethanol, 200 mM Tris-HCl, pH 6.8) and boiled at 5 min at 98 °C. A 40 μg amount of protein was then loaded per lane for SDS-PAGE separation (Criterion TGX 4−20%, Bio-Rad).
Caspase Wild-Type vs Mutant Chemoenzymatic/Biotin IP.
H1299 cells at 90% confluency were cotransfected with pCDNA caspase-8 catalytically dead, wild-type (HA-tagged, 20 μg) and either mutant (FLAG-tagged, 20 μg), (S386,387A) mutant (FLAG-tagged, 20 μg), or (S375A) mutant (FLAG-tagged, 20 μg) DNA using Liptofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. Cells were harvested 48 h post-transfection and subsequently lysed using SDS-lysis before being subjected to chemoenzymatic transfer according to protocols described above. Following chemo-enzymatic transfer, protein was precipitated by MeOH/CHCl3 and resuspended in 1% SDS buffer. CuAAC was preformed as described previously with alkyne-azo-biotin. Following CuAAC, enrichment was conducted according to protocols previously described. Following the final elution step, both eluents were combined, and proteins were then precipitated in ice cold methanol (1 mL) overnight at −20 °C. Protein was collected by centrifugation (10 min, 10000g, 4 °C), and the pellet was allowed to air-dry for 5 min, at which time 20 μL of 4% SDS buffer (4% SDS, 150 mM NaCl, 50 mM TEA, pH 7.4) was added to each sample. The mixture was sonicated in a bath sonicator to ensure complete dissolution, and 20 μL of 2× loading buffer (20% glycerol, 0.2% bromophenol blue, 1.4% β-mercaptoethanol) was then added. Samples were boiled for 5 min at 98 °C before being subjected to SDS-PAGE and Western blotting as described above. Plasmids were available upon request.
Peptide Synthesis.
All solid-phase peptide syntheses were conducted manually on unprotected Rink amide ChemMatrix resin (PCAS BioMatrix) with an estimated loading of 0.6 mmol g−1. Protected O-GlcNAcylated serine was prepared as previously described.11,52 Commercially available N-Fmoc and side-chain-protected amino acids (10 equiv, Advanced ChemTech) were activated for 20 min with HBTU (10 equiv, Novabiochem) and N,N-diisopropylethylamine (DIEA) (20 equiv, Sigma) and then coupled to the resin for 1 h, bubbling with N2 to mix. Reaction completion was checked using the Kaiser test. Briefly, a small amount of resin was incubated with equal volumes of 5% w/v ninhydrin in EtOH, 80% w/v phenol in EtOH, and 20 μM KCN in pyridine and heated to 99 °C for 5 min in a sealed tube. If necessary, a second coupling was conducted with 10 equiv of amino acid, 10 equiv of HOBt (Novabiochem), and 15 equiv of N,N′-dicyclohexyl-carbodiimide (DCC) (Sigma) for 2 h, with N2 mixing. After successful coupling, the terminal Fmoc group was removed with 20% v/v piperidine in dimethylformamide (DMF) for 5 min with N2 mixing and then for an addition 15 min with fresh 20% piperidine in DMF. When peptides were completed, the final Fmoc group was removed as described above, and the N-terminal amine was acetylated with 5 equiv each of pyridine and acetic anhydride in DMF. Peptides were then cleaved from the resin by incubating in cleavage cocktail (95:2.5:2.5 TFA/H2O/triisopropylsilane) for 3.5 h at room temperature. The peptides were then diluted ∼1/10 in cold diethyl ether and precipitated overnight (−80 °C). The resulting suspensions were centrifuged (30 min, 5000g, 4 °C), and the pellets were resuspended in fresh Et2O and centrifuged again (30 min, 5000g, 4 °C). The pellets were then resuspended in H2O, flash frozen, and lyophillized. At this point, glycopeptides 2 and 4 were deprotected with hydrazine hydrate (80% v/v in MeOH) for 2 h, followed by concentration under vacuum. Crude lyophillized material was purified by RP-HPLC (0−50% B gradient over 60 min; A = 0.1% trifluoroacetic acid in H2O; B = 0.1% trifluoroacetic acid, 10% H2O, 90% acetonitrile) over a C18 semipreparative column (Vydac). Purified peptides were then characterized by ESI-MS (Agilent 6100 series Quadrupole LC-MS): 1, calcd = 720.3 Da (M + H+), obsd = 720.2 Da (M + H+); 2, calcd = 923.4 Da (M + H+), obsd = 923.3 Da (M + H+); 3, calcd = 835.4 Da (M + H+), obsd = 835.2 Da (M + H+); 4, calcd = 1241.5 Da (M + H+), obsd = 1241.2 Da (M + H+); 5, calcd = 704.3 Da (M + H+), obsd = 704.2 Da (M + H+); 6, calcd = 819.3 Da (M + H+), obsd = 819.2 Da (M + H+); 7, calcd = 803.4 Da (M + H+), obsd = 803.4 Da (M + H+); 8, calcd = 1033.5 Da (M + H+), obsd = 1033.2 Da (M + H+).
In Vitro Caspase-8 Cleavage Assay.
Peptides 1−8 (1000× stock in DMSO) were added to 4 units of recombinant caspase-8 (Enzo Life Sciences) in 100 μL of general caspase activity buffer (20 mM HEPES, 100 mM NaCl, 0.1% CHAPS, 10% sucrose, 1 mM EDTA, 10 mM DTT, pH 7.2) to give a final concentration of 2 mM. The mixtures were incubated overnight at 37 °C and subjected to HPLC analysis by RP-HPLC. Purified peptides were then characterized by ESI-MS (Agilent 6200 series Quadrupole LC-MS): 1 uncleaved, calcd = 720.3 Da (M + H+), obsd = 720.2 Da (M + H+); 1 cleaved, calcd = 505.2 Da (M + H+), obsd = 505.3 Da (M + H+); 2 uncleaved, calcd = 923.4 Da (M + H+), obsd = 923.2 Da (M + H+); 2 cleaved, calcd = 505.2 Da (M + H+), obsd = 505.2 Da (M + H+); 3 uncleaved, calcd = 835.4 Da (M + H+), obsd = 835.2 Da (M + H+); 3 cleaved, calcd = 549.2 Da (M + H+), obsd = 549.2 Da (M + H+); 4 uncleaved, calcd = 1241.5 Da (M + H+), obsd = 1241.5 Da (M - H+); 4 cleaved, calcd = 549.2 Da (M + H+), obsd = 549.2 Da (M + H+); 5 uncleaved, calcd = 704.3 Da (M + H+), obsd = 704.2 (M + H+); 5 cleaved, calcd = 505.2 Da (M + H+), obsd = 505.2 Da (M + H+); 6 uncleaved, calcd = 819.3 Da (M + H+2), obsd = 819.2 Da (M + H+); 6 cleaved, calcd = 505.2 Da (M + H+), obsd = 505.2 Da (M + H+) 7 uncleaved, calcd = 803.4 Da (M + H+), obsd = 803.4 (M + H+); 7 uncleaved, calcd = 803.4 Da (M + H+), obsd = 803.3 Da (M + H+); 7 cleaved, calcd = 548.2 Da (M + H+), obsd = 548.2 Da (M + H+); 8 uncleaved, calcd = 1033.5 Da (M + H+), obsd = 1033.2 Da (M + H+); 8 cleaved, calcd = 549.2 Da (M + H+), obsd = 549.2 Da (M + H+).
Calcium Phosphate Transfection.
One day before transfection, 5 × 105 target cells were plated. One hour before transfection, media was exchanged for fresh media. To an Eppendorf tube were added water (398 μL, autoclaved), 2 M CaCl2 (62 μL, filter sterilized), and 40 μL of DNA (500 ng/μL, 20 μg total, phenol/chloroform extracted, EtOH precipitated). The solution was mixed by pipeting; then the DNA solution was added dropwise to a Falcon tube containing 500 μL of 2× HBS (500 mM HEPES, 1.5 mM Na2HPO4, 280 mM NaCl, 10 mM KCl, 12 mM dextrose, pH 7.05, filter sterilized) while pumping bubbles. The resulting solution was added dropwise to target cells while swirling slowly. A fine black precipitate was observed after 30 min, and the media was opaque in color. Seven hours after transfection, media was exchanged for fresh media.
Caspase-8 Knockout with CRISPR.
Expression vector pSpCas9-(BB)-2A-Puro (PX459) (Addgene no. 48139) was digested with BbsI, and a pair of annealed oligonucleotides corresponding to the first exon of human caspase-8 (CACCGCTCTTCCGAATTAATAGAC and AAACGTCTATTAATTCGGAAGAGC)53 was ligated into the guide RNA to generate SpCas9-Casp8, which was confirmed by sequencing (Laragen, Culver City, CA, USA). HeLa cells were then transiently transfected with SpCas9-Casp8 using calcium phosphate transfection methods as described above. Forty-eight hours post-transfection, 1 μg mL−1 puromycin was added to the media for the next 72 h. Caspase-8 knockout cells were then further selected for by inducing apoptosis with 10 ng mL−1 TNF-alpha and 1 μg mL−1 cycloheximide for 24 h. Following clonal selection using cloning cylinders, Western blot analysis showed five out of five selected colonies had no detectable caspase-8. Colony number 1 was selected for sequencing and further experiments. A 2 kb fragment of genomic caspase-8 DNA was amplified using PCR with primers CTTTGTGACATG-GTACCTGGC and GTGTCCGATAATAGGAGATGTT. An additional round of PCR was then used to introduce attB recombination sites enabling Gateway cloning (ThermoFisher) into donor vector pDONR201 according to the manufacturer’s protocol. E. coli (DH5α, ThermoFisher) was then transformed with these plasmids and grown on antibiotic-containing agar plates. Twenty colonies were then selected for sequencing (Laragen).
Caspase-8 Overexpression Co-IP.
HeLa caspsae-8 CRISPR cells at 90% confluency were cotransfected with pCDNA caspase-8 catalytically dead, wild-type (HA-tagged, 20 μg), and wild-type (FLAG-tagged, 20 μg) DNA using Liptofectamine 2000 (Invitrogen), according to the manufacturer’s protocol). Forty-eight hours post-transfection, cells were harvested by trypsinization and then lysed by resuspension in 150 μL of lysis buffer with 1 mM phenyl-methanesulfonyl fluoride (PMSF, 200 mM stock in isopropyl alcohol) (10× lysis buffer, Cell Signaling) followed by sonication (5 s on, 5 s rest, three times). Protein concentration was determined using the BCA assay (Pierce, ThermoScientific), and 200 μg of lysate was diluted to 1 μg μL−1 in lysis buffer. Lysate was then precleared with 30 μL of magnetic protein-G beads (Cell Signaling, #8740) for 1 h at 4 °C with end-over-end rotation. Inputs were generated at 2 μg μL−1 using 4× loading buffer (8% SDS, 40% glycerol, 0.4% bromophenol blue, 2.8% B-mercaptoethanol, 200 mM Tris-HCl, pH 6.8) before boiling for 5 min at 98 °C. Magnetic beads were captured using a magnetic rack, and precleared lysate was transferred to a new, precooled tube. Either FLAG primary antibody (1:50 dilution, Cell Signaling, #2368) or HA primary antibody (1:50 dilution, Cell Signaling, #3724) was added to the precleared lysate. The appropriate isotype control was used at the same protein concentration as the primary antibody (rabbit isotype control, Cell Signaling, #3900). A 30 μL amount of magnetic protein-G beads was then added and incubated overnight with end-over-end rotation at 4 °C. Protein-G beads were then washed five times with 500 μL of lysis buffer containing 1 mM PMSF. Beads were then pelleted gently by centrifugation (3 min, 2000g, 4 °C) before 3× loading buffer (Cell Signaling, #7722) was added and samples were boiled for 5 min at 98 °C. After removal of magnetic beads, samples were analyzed by SDS-PAGE (4−20% Tris-Glycine Gel, Bio-Rad) and analyzed by Western blotting.
Caspase-8 Overexpression Co-IP with Inhibitor Treatment.
HeLa caspase-8 CRISPR cells at 90% confluency were cotransfected with pCDNA caspase-8 catalytically dead, wild-type (HA-tagged, 10 μg) and wild-type (FLAG-tagged, 10 μg) DNA using Liptofectamine 2000 (Invitrogen), according to the manufacturer’s protocol. Seven hours following transfection cells were replated at 7.5 × 105 per 10 cm dish for treatment. The following day, cells were treated with either 5SGlcNAc for 16 h (200 μM, 1000× stock in DMSO), Thiamet-G for 20 h (20 μM, 1000× stock in DMSO), or DMSO vehicle. Following treatment, cells were harvested and IP was conducted according to the protocol described above.
Caspase Wild-Type vs Mutant Autoactivation.
Caspase-8 knockout HeLa cells were transiently transfected using Lipofectamine 2000 according to the manufacturer’s protocol with 19 μg of a pRetroX-Tet-on empty vector (Clontech) and 1 μg of a pcDNA3 vector (ThermoFisher) containing either caspase-8, caspase-8(AAA), or caspase-8(WWW). Media was exchanged after 6 h of transfection, and this time was defined as 0 h after transfection. After the indicated lengths of time, cells in the media were combined with cells that were collected by trypsinization. The cells were then pelleted by centrifugation (6 min, 2000g, 4 °C) and washed with PBS (1 mL) three times. Cell pellets were then resuspended in 50 μL of 1% NP-40 lysis buffer [1% NP-40, 150 mM NaCl, 50 mM TEA, pH 7.4] with Complete Mini protease inhibitor cocktail (Roche Biosciences) and Z-VAD-FMK (Enzo Life Sciences) for 15 min and then centrifuged (10 min, 10000g, 4 °C). The supernatant (soluble cell lysate) was collected, and the protein concentration was determined by BCA assay (Pierce, ThermoScientific). Samples were prepared at a protein concentration of 3 mg mL−1 with 4× loading buffer (8% SDS, 40% glycerol, 0.4% bromophenol blue, 2.8% B-mercaptoethanol, 200 mM Tris-HCl, pH 6.8) and boiled at 5 min at 98 °C. A 40 μg amount of protein was then loaded per lane for SDS-PAGE separation (Criterion TGX 4−20%, Bio-Rad).
Supplementary Material
ACKNOWLEDGMENTS
We thank Mary O’Reilly (O’Reilly Science Art) for preparation of the table of contents figure and Henrik Molina (Rockefeller University) for assistance with the proteomics analysis. K.N.C. is a fellow of the National Science Foundation Graduate Research Fellowship Program (DGE-0937362). This research was supported by the National Science Foundation (CHE-1506503 to M.R.P.), the Damon Runyon Cancer Research Foundation (DDR-19–12 to M.R.P.), Susan G. Komen for the Cure (CCR14299333 to M.R.P.), the American Cancer Society (RSG-14–225-01-CCG to M.R.P.), and in part by the National Cancer Institute of the U.S. National Institutes of Health (CCSG P30CA014089). LC-MS/MS proteomic analysis was performed at the Rockefeller University Proteomics Resource Center.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b02213.
Supporting figures, experimental methods, and NMR characterization (PDF)
Proteomic data tables (XLS)
REFERENCES
- (1).Zachara NE; Hart GW Chem. Rev 2002, 102, 431. [DOI] [PubMed] [Google Scholar]
- (2).Bond MR; Hanover JA J. Cell Biol 2015, 208, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vocadlo DJ Curr. Opin. Chem. Biol 2012, 16, 488. [DOI] [PubMed] [Google Scholar]
- (4).Shafi R; Iyer SP; Ellies LG; O’Donnell N; Marek KW; Chui D; Hart GW; Marth JD Proc. Natl. Acad. Sci. U. S. A 2000, 97, 5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).O’Donnell N; Zachara NE; Hart GW; Marth JD Mol. Cell. Biol 2004, 24, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yang YR; Song M; Lee H; Jeon Y; Choi E-J; Jang H-J; Moon HY; Byun H-Y; Kim E-K; Kim DH; Lee MN; Koh A; Ghim J; Choi JH; Lee-Kwon W; Kim KT; Ryu SH; Suh P-G Aging Cell 2012, 1, 439. [DOI] [PubMed] [Google Scholar]
- (7).Sinclair DAR; Syrzycka M; Macauley MS; Rastgardani T; Komljenovic I; Vocadlo DJ; Brock HW; Honda BM Proc. Natl. Acad. Sci. U. S. A 2009, 106, 13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ma Z; Vosseller K Amino Acids 2013, 45, 719. [DOI] [PubMed] [Google Scholar]
- (9).Yuzwa SA; Macauley MS; Heinonen JE; Shan X; Dennis RJ; He Y; Whitworth GE; Stubbs KA; McEachern EJ; Davies GJ; Vocadlo DJ Nat. Chem. Biol 2008, 4, 483. [DOI] [PubMed] [Google Scholar]
- (10).Yuzwa SA; Shan X; Macauley MS; Clark T; Skorobogatko Y; Vosseller K; Vocadlo DJ Nat. Chem. Biol 2012, 8, 393. [DOI] [PubMed] [Google Scholar]
- (11).Marotta NP; Cherwien CA; Abeywardana T; Pratt MR ChemBioChem 2012, 13, 2665. [DOI] [PubMed] [Google Scholar]
- (12).Marotta NP; Lin YH; Lewis YE; Ambroso MR; Zaro BW; Roth MT; Arnold DB; Langen R; Pratt MR Nat. Chem 2015, 7, 913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Darley-Usmar VM; Ball LE; Chatham JC J. Mol. Cell. Cardiol 2012, 52, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zachara NE Am. J. Physiol Heart Circ 2012, 302, H1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Groves JA; Lee A; Yildirir G; Zachara NE Cell Stress Chaperones 2013, 18, 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ma Z; Vocadlo DJ; Vosseller KJ Biol. Chem 2013, 288, 15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zachara N; O’Donnell N; Cheung W; Mercer J; Marth J; Hart GJ Biol. Chem 2004, 279, 30133. [DOI] [PubMed] [Google Scholar]
- (18).Yi W; Clark PM; Mason DE; Keenan MC; Hill C; Goddard WA; Peters EC; Driggers EM; Hsieh-Wilson LC Science 2012, 337, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Rao X; Duan X; Mao W; Li X; Li Z; Li Q; Zheng Z; Xu H; Chen M; Wang PG; Wang Y; Shen B; Yi W Nat. Commun 2015, 6, 8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Riedl SJ; Shi Y Nat. Rev. Mol. Cell Biol 2004, 5, 897. [DOI] [PubMed] [Google Scholar]
- (21).Elmore S Toxicol. Pathol 2007, 35, 495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li J; Yuan J Oncogene 2008, 27, 6194. [DOI] [PubMed] [Google Scholar]
- (23).Fuchs Y; Steller H Nat. Rev. Mol. Cell Biol 2015, 16, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dix M; Simon G; Cravatt B Cell 2008, 134, 679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mahrus S; Trinidad J; Barkan D; Sali A; Burlingame A; Wells J Cell 2008, 134, 866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Crawford ED; Wells JA Annu. Rev. Biochem 2011, 80, 1055. [DOI] [PubMed] [Google Scholar]
- (27).Pop C; Salvesen GS J. Biol. Chem 2009, 284, 21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Stennicke HR; Renatus M; Meldal M; Salvesen GS Biochem. J 2000, 350, 563. [PMC free article] [PubMed] [Google Scholar]
- (29).Chuh KN; Zaro BW; Piller F; Piller V; Pratt MR J. Am. Chem. Soc 2014, 136, 12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Speers A; Cravatt B Chem. Biol 2004, 11, 53510.1016/j.chembiol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- (31).Mayer A; Gloster TM; Chou WK; Vocadlo DJ; Tanner ME Bioorg. Med. Chem. Lett 2011, 21, 1199. [DOI] [PubMed] [Google Scholar]
- (32).Kim EJ; Bond MR; Nam G; Hanover JA Bull. Korean Chem. Soc 2017, 38, 264. [Google Scholar]
- (33).Zaro BW; Yang Y-Y; Hang HC; Pratt MR Proc. Natl. Acad. Sci. U. S. A 2011, 108, 8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Uttamapinant C; Tangpeerachaikul A; Grecian S; Clarke S; Singh U; Slade P; Gee KR; Ting AY Angew. Chem., Int. Ed 2012, 51, 5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Clark PM; Dweck JF; Mason DE; Hart CR; Buck SB; Peters EC; Agnew BJ; Hsieh-Wilson LC J. Am. Chem. Soc 2008, 130, 11576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chang DW; Xing Z; Capacio VL; Peter ME; Yang X EMBO J 2003, 22, 4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gloster TM Curr. Opin. Struct. Biol 2014, 28C, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Liu X; Li L; Wang Y; Yan H; Ma X; Wang PG; Zhang L FASEB J 2014, 28, 3362. [DOI] [PubMed] [Google Scholar]
- (39).Pathak S; Alonso J; Schimpl M; Rafie K; Blair DE; Borodkin VS; Schüttelkopf AW; Albarbarawi O; van Aalten DMF Nat. Struct. Mol. Biol 2015, 22, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Boatright KM; Renatus M; Scott FL; Sperandio S; Shin H; Pedersen IM; Ricci JE; Edris WA; Sutherlin DP; Green DR; Salvesen GS Mol. Cell 2003, 11, 529. [DOI] [PubMed] [Google Scholar]
- (41).Pop C; Fitzgerald P; Green DR; Salvesen GS Biochemistry 2007, 46, 4398. [DOI] [PubMed] [Google Scholar]
- (42).Oberst A; Pop C; Tremblay AG; Blais V; Denault J-B; Salvesen GS; Green DR J. Biol. Chem 2010, 285, 16632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Salvesen GS; Walsh CM Semin. Immunol 2014, 26, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Fridman A; Pak I; Butts BD; Hoek M; Nicholson DW; Mehmet H Apoptosis 2013, 18, 369. [DOI] [PubMed] [Google Scholar]
- (45).Tözsér J; Bagossi P; Zahuczky G; Specht SI; Majerova E; Copeland TD Biochem. J 2003, 372, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Degli Esposti M.; Ferry G; Masdehors P; Boutin JA; Hickman JA; Dive CJ Biol. Chem 2003, 278, 15749. [DOI] [PubMed] [Google Scholar]
- (47).Dix MM; Simon GM; Wang C; Okerberg E; Patricelli MP; Cravatt BF Cell 2012, 150, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Charron G; Zhang MM; Yount JS; Wilson J; Raghavan AS; Shamir E; Hang HC J. Am. Chem. Soc 2009, 131, 4967. [DOI] [PubMed] [Google Scholar]
- (49).Hang HC; Yu C; Kato DL; Bertozzi CR Proc. Natl. Acad. Sci. U. S. A 2003, 100, 14846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Gloster TM; Zandberg WF; Heinonen JE; Shen DL; Deng L; Vocadlo DJ Nat. Chem. Biol 2011, 7, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Hsu T-L; Hanson SR; Kishikawa K; Wang S-K; Sawa M; Wong C-H Proc. Natl. Acad. Sci. U. S. A 2007, 104, 2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mitchell SA; Pratt MR; Hruby VJ; Polt RJ Org. Chem 2001, 66, 2327. [DOI] [PubMed] [Google Scholar]
- (53).Kranz D; Boutros M EMBO J 2014, 33, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.