

Abstract
Background
We analyzed whether co-occurring mutations influence the outcome of systemic therapy in ALK-rearranged non-small-cell lung cancer (NSCLC).
Patients and methods
ALK-rearranged stage IIIB/IV NSCLC patients were analyzed with next-generation sequencing and fluorescence in situ hybridization analyses on a centralized diagnostic platform. Median progression-free survival (PFS) and overall survival (OS) were determined in the total cohort and in treatment-related sub-cohorts. Cox regression analyses were carried out to exclude confounders.
Results
Among 216 patients with ALK-rearranged NSCLC, the frequency of pathogenic TP53 mutations was 23.8%, while other co-occurring mutations were rare events. In ALK/TP53 co-mutated patients, median PFS and OS were significantly lower compared with TP53 wildtype patients [PFS 3.9 months (95% CI: 2.4–5.6) versus 10.3 months (95% CI: 8.6–12.0), P < 0.001; OS 15.0 months (95% CI: 5.0–24.9) versus 50.0 months (95% CI: 22.9–77.1), P = 0.002]. This difference was confirmed in all treatment-related subgroups including chemotherapy only [PFS first-line chemotherapy 2.6 months (95% CI: 1.3–4.1) versus 6.2 months (95% CI: 1.8–10.5), P = 0.021; OS 2.0 months (95% CI: 0.0–4.6) versus 9.0 months (95% CI: 6.1–11.9), P = 0.035], crizotinib plus chemotherapy [PFS crizotinib 5.0 months (95% CI: 2.9–7.2) versus 14.0 months (95% CI: 8.0–20.1), P < 0.001; OS 17.0 months (95% CI: 6.7–27.3) versus not reached, P = 0.049] and crizotinib followed by next-generation ALK-inhibitor [PFS next-generation inhibitor 5.4 months (95% CI: 0.1–10.7) versus 9.9 months (95% CI: 6.4–13.5), P = 0.039; OS 7.0 months versus 50.0 months (95% CI: not reached), P = 0.001).
Conclusions
In ALK-rearranged NSCLC co-occurring TP53 mutations predict an unfavorable outcome of systemic therapy. Our observations encourage future research to understand the underlying molecular mechanisms and to improve treatment outcome of the ALK/TP53 co-mutated subgroup.
Keywords: ALK-rearranged NSCLC, sequential ALK-inhibitor therapy, TP53 mutation status
Key Message
About 25% of ALK-positive NSCLC patients harbor pathogenic TP53 mutations in pretreatment biopsies. ALK/TP53 co-mutated patients have a significantly worse PFS and OS compared with TP53 wildtype patients with chemotherapy, crizotinib as the only ALK-inhibitor and even with crizotinib followed by next-generation ALK inhibitors. New treatment strategies should be developed for these patients.
Introduction
ALK-positive non-small-cell lung cancer (NSCLC) is characterized by ALK gene rearrangements and an association with acinar histology, younger age and never-smoking status [1]. ALK rearrangements lead to constitutive activation of the encoded tyrosine kinase and downstream transforming signaling pathways [2]. Crizotinib, the first approved ALK-inhibitor, is superior to chemotherapy regarding overall response rate, progression-free survival (PFS), toxicity profile [3, 4] and overall survival (OS) [5–7]. Next-generation inhibitors with activity against ALK resistance mutations are in clinical evaluation and partly already approved [8–11]. An impressive OS was reported for sequential ALK-inhibitor therapy ranging from 45 to 89.6 months [12–14].
There are considerable differences in the clinical course of ALK-positive NSCLC patients treated with chemotherapy or ALK inhibitors [3, 4, 15, 16]. Genetic heterogeneity of ALK-positive tumors could explain this observation. We have molecularly analyzed 216 ALK-positive patients with advanced disease and hypothesized that co-occurring mutations might underlie these differences.
Patients and methods
Patients and samples
The study was carried out within the Network Genomic Medicine [17], which offers centralized molecular diagnostics at the University Hospital of Cologne for patients with lung cancer from 300 participating partners. The study was conducted in concordance with local ethical guidelines. Patients were treated with crizotinib, ceritinib, alectinib or brigatinib according to national guidelines or within clinical trials [PROFILE1005 (NCT00932451); PROFILE1007 (NCT00932893); CLDK378X2101 (NCT-1283516); ASCEND-5 (NCT01828112); ALTA (AP26113) (NCT02094573); ACCALIA (NCT01801111)].
Fluorescence in situ hybridization
ALK, RET and ROS1 rearrangements were diagnosed using break-apart fluorescence in situ hybridization (FISH) [17]. MET and ERBB2 were tested for amplification as reported [18]. Details are described in supplementary Table S1, available at Annals of Oncology online.
Next-generation sequencing
Samples were analyzed with either a validated gene panel using AmpliSeq chemistry (Thermofisher, LUN3) comprising 102 amplicons of 14 different genes or a validated gene panel using GeneRead chemistry (Qiagen, LUN4), comprising 17 genes [19]. Details are described in supplementary Table S6, available at Annals of Oncology online. ALK variants were determined using the Archer® FusionPlex® Lung Kit and Archer Molecular Barcode (MBC) Adapters (both for Illumina) according to the manufacturer’s instructions.
Programmed death-ligand 1 immunohistochemistry
Programmed death-ligand 1 (PD-L1) immunohistochemistry was carried out on the Leica Bond platform using primary antibody clone 28-8 (Abcam, Cambridge, UK). Interpretation was done according to the Dako PD-L1 22C3 pharmDx guidelines, results were reported based on an integrated proportion score [20, 21].
Data collection
The Network Genomic Medicine database covers molecular diagnostics and basic demographic and clinical data. For treatment outcome medical records were reviewed. PFS was determined based on RECIST v1.1. Time of death was determined either via medical records or requests to local registry offices. OS was defined as the time from first diagnosis of stage IIIB/IV until death. For subjects alive at completion of this analysis, time to death was censored at the time of last contact.
Statistical analyses
Statistical analyses were carried out using IBM SPSS software 24 (IBM, Armonk, NY). Chi-squared and two-sided Fischer’s exact tests were used for analyzing qualitative variable characteristics in different groups. The Kaplan–Meier estimator was used to calculate OS and PFS. Two-sided log-rank tests were applied to compare differences between treatment groups. Cox proportional hazards model was used to adjust for potential confounders. P values <0.05 were considered statistically significant.
Results
Patient characteristics
Between January 2011 and December 2016, 423 ALK-positive patients were identified using FISH. From 289 patients with written informed consent, 53 had no stage IIIB/IV and 20 were lost to follow up. About 216 patients were eligible (Figure 1A). Median age, distribution of sex and histology are in line with earlier reports (Table 1) [3, 4]. Median follow-up was 34 months.
Figure 1.

(A) Flowsheet of molecular diagnostics. NGS, next-generation sequencing; FISH, fluorescence in situ hybridization; IHC, immunohistochemistry. (B) Allocation of patients to cohorts for evaluation of treatment-related OS. BSC, best supportive care; PFS, progression-free survival; OS, overall survival.
Table 1.
Baseline patient characteristics (n = 216)
| n | % | |
|---|---|---|
| Sex | 216 | |
| Male | 111 | 51.4 |
| Female | 105 | 48.6 |
| Age at diagnosis (years) | ||
| Mean | 58.09 | |
| Standard deviation | 14.52 | |
| Median | 58 (19–89) | |
| Histology | ||
| AD | 210 | 97.2 |
| Adenosquamous | 3 | 1.4 |
| w/o differentiation | 3 | 1.4 |
| Smoking history | ||
| Never | 86 | 46.3 |
| Former | 62 | 33.3 |
| Current | 38 | 20.4 |
| n/a | 30 | |
| ECOG performance status | ||
| 0 | 63 | 43.4 |
| 1 | 63 | 43.4 |
| 2 | 16 | 11.1 |
| 3 | 3 | 2.1 |
| n/a | 71 | |
| Tumor stage at diagnosis | ||
| I | 4 | 1.9 |
| II | 9 | 4.2 |
| IIIA | 14 | 6.5 |
| IIIB | 23 | 10.6 |
| IV | 166 | 76.8 |
w/o, without differentiation; n/a, not available; AD, adenocarcinoma.
From 147 (68%) patients’ tumors were analyzed by next-generation sequencing [LUN3 panel: 90 patients (61%); LUN4 panel: 57 patients (39%)]. Fifty patients (23%) were tested by additional single gene sequencing. Thirty-four (17%) of 197 patients were tested for PD-L1 expression, 135 (69%) received further FISH analyses. In 34 of 216 ALK-positive patients (16%) distribution of ALK variants was assessed by RNA sequencing (Figure 1A).
For 175 patients (81%) follow-up data for OS were available including 7 patients (3.2%) treated with best supportive care. Thus, 168 patients (77.8%) were subdivided (Figure 1B) into cohort A including 42 patients (19.4%) treated with chemotherapy only, cohort B including 71 patients (33%) with crizotinib and chemotherapy, cohort C including 18 patients (8.3%) with first-line crizotinib and cohort D including 37 patients (17.1%) with ceritinib after crizotinib with or without chemotherapy. Supplementary Figure S2, available at Annals of Oncology online shows treatment sequences in cohort D.
From 41 patients (19%, cohort Z) no complete therapy data until death or final follow-up were available including 5 patients treated with alectinib and 2 with brigatinib.
Co-occurring mutations, PD-L1 status and ALK variants
Mutations in TP53 were the most frequent co-occurring mutations with 23.8% (34/143) of the tested patients. Among 36 TP53 mutations 34 were classified as nonfunctional [22], 2 were of unknown functional significance (supplementary Table S5, available at Annals of Oncology online). All other co-alterations occurred rarely with frequencies between 0.6% for BRAF (1/171), 0.6% for KRAS (1/174) and 3.6% (4/112) for low-level MET amplification (Figure 2A and supplementary Table S4, available at Annals of Oncology online). Four patients showed more than 1 co-occurring alteration (supplementary Figure S1, available at Annals of Oncology online).
Figure 2.
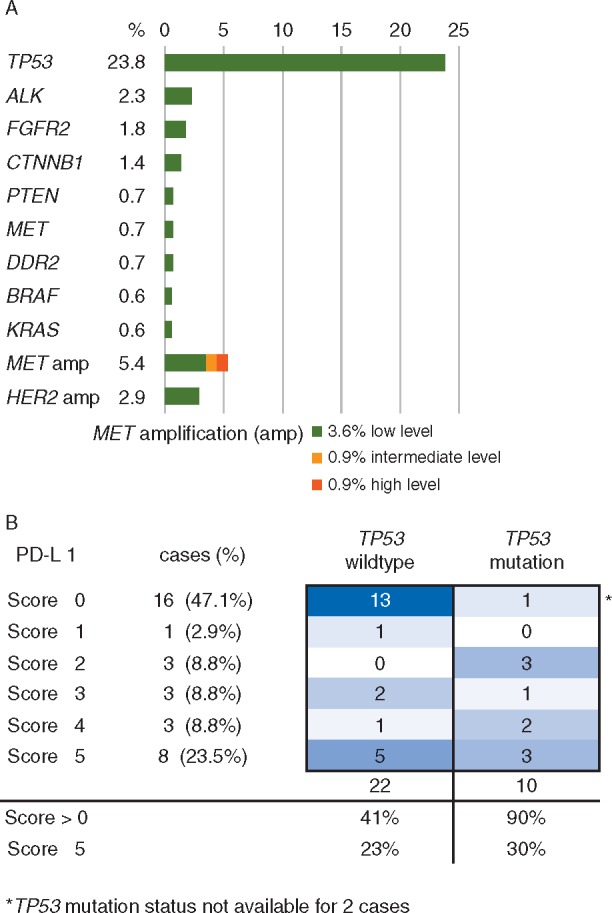
(A) Frequencies of co-occurring genetic aberrations in ALK-positive NSCLC patients. Results of NGS, single gene sequencing and FISH analysis in 197 ALK FISH-positive patients. (B) Correlation between PD-L1 positivity (expression score) and TP53 mutation status in 34 ALK-positive patients.
PD-L1 expression of tumor cells was assessed in 34 patients (supplementary Table S4, available at Annals of Oncology online). In eight patients (23.5%) the PD-L1 score [21] was 5, i.e. more than 50% of the tumor cells expressed PD-L1. PD-L1 positivity was significantly correlated with TP53 mutations [TP53 wildtype (wt): 41% PD-L1 positive/TP53 mutated: 90% PD-L1 positive; P = 0.009]. No significant difference was observed for high PD-L1 positivity (score 5) between wt and mutated TP53 (Figure 2B).
In 18 of 34 patients (53%) ALK variant 1 was found, in 14 patients (41%) variant 3a/b and in 2 patients (6%) variant 2 (supplementary Table S8, available at Annals of Oncology online).
PFS dependent on therapy and TP53 mutations
PFS was assessed in 157 patients with first-line chemotherapy (140 patients cohorts A–D plus 17 patients cohort Z, see Figure 1B), thereof 149 patients with platinum-based chemotherapy (109 resp. 103 PD at data cutoff), for crizotinib after chemotherapy in 112 patients (cohorts B–D and partly Z; 73 PD at data cutoff), for crizotinib first line in 24 patients (cohorts C and D and partly Z; 12 PD at data cutoff) and for ceritinib after crizotinib with or without chemotherapy in 43 patients (cohorts D and partly Z; 28 PD at data cutoff). PFS of next-generation ALK inhibitors was calculated in 50 patients combined for ceritinib, alectinib (5 patients, 4 PD at data cutoff) and brigatinib (2 patients, 1 PD at data cutoff) because of the small patient number. PFS for first-line chemotherapy [5.4 months (95% CI: 3.7–7.1)] and for the subgroup of first-line platinum-based chemotherapy [5.5 months (95% CI: 3.8–7.2)] was inferior to PFS for first-line crizotinib [12.3 months (95% CI: 0.0–34.9); P = 0.001] and for crizotinib after chemotherapy [9.4 months (95% CI: 6.4–12.4); P < 0.001]. PFS for next-generation ALK inhibitors as sequential therapy after crizotinib was 7.0 months [(95% CI: 5.4–8.6); P = 0.449].
TP53 mutations were a negative prognostic factor for PFS regardless of systemic therapy. Median PFS with first-line chemotherapy was 2.6 months (95% CI: 1.3–4.1) with mutated TP53 (n = 27) and 6.2 months (95% CI: 1.8–10.5) with TP53 wt (n = 75) (P = 0.021). For crizotinib first-line median PFS was 5.5 months only (95% CI: 0.0–10.9) with mutated TP53 (n = 3) versus 29.9 months (95% CI: 0.0–63.9) with TP53 wt (n = 15) (P = 0.007). Similarly, for crizotinib after chemotherapy median PFS was 5.0 months (95% CI: 2.3–7.8) with mutated TP53 (n = 19) versus 14.0 months (95% CI: 9.5–18.6) with TP53 wt (n = 56) (P = 0.004). Regardless of treatment line, TP53 mutation status segregated the median PFS of crizotinib-treated patients in an unfavorable TP53-mutated group [n = 22; 5.0 months (95% CI: 2.9–7.2)] and a favorable TP53 wt group (n = 71; 14.0 months (95% CI: 8.0–20.1); P < 0.001]. Also, median PFS with next-generation ALK inhibitors after crizotinib was worse in patients with mutated TP53 [n = 11; 5.4 months (95% CI: 0.1–10.7)] compared with TP53 wt [n = 22, 9.9 months (95% CI: 6.4–13.5); P = 0.039]. In total, PFS of TP53 co-mutated patients was 3.9 months [n = 60 (95% CI: 2.4–5.6)] and 10.3 months in TP53 wt patients [n = 168 (95% CI: 8.6–12.0)] regardless of treatment (P < 0.001) (Figure 3A and supplementary Table S2, available at Annals of Oncology online).
Figure 3.

(A) PFS with different systemic treatments dependent on TP53 mutation status. Kaplan–Meier blots for the total cohort (n = 228), for chemotherapy (n = 102), for crizotinib (n = 93) and for next-generation ALK inhibitors (n = 33). (B) OS in the treatment-related cohorts dependent on TP53 mutation status. Kaplan–Meier blots for the total cohort (n = 143), for chemotherapy (n = 22), for crizotinib (n = 63) and for ceritinib (n = 24).
The ALK variant 3a/b subgroup (cohorts A–D, n = 20) showed a nonsignificant trend toward better PFS with 11.9 months (95% CI: 0.9–23.1) versus variant 1 (n = 31) with 7.9 months (95% CI: 1.6–14.4) (P = 0.285). TP53 mutations were negative predictive in both variant subgroups (n = 30; P = 0.001) with a strong trend in variant 1 [2.6 month (95% CI: 0.0–10.9) versus 15.9 months (95% CI: 1.4–30.6); P = 0.068] and reaching statistical significance in variant 3a/b (P = 0.022). Cox regression suggested a negative impact of TP53 mutations on PFS regardless of ALK variants (n = 30; P = 0.002) (supplementary Tables S8 and S9, available at Annals of Oncology online).
OS dependent on therapy and TP53 mutations
OS was assessed for 168 patients in cohorts A–D (Figure 1B). Median OS with chemotherapy only (cohort A, n = 42, 31 events at data cutoff) was with 9.0 months (95% CI: 5.0–12.9) inferior to all other cohorts treated with ALK inhibitors: cohort B (n = 71, 32 events) 31.0 months (95% CI: 0.4–61.6); P < 0.001, cohort C (n = 18, 2 events) median not reached (P = 0.001), cohort D (n = 37, 20 events) 45.0 months (95% CI: 32.3–57.7); P < 0.001. OS of patients treated with crizotinib starting from the first dose (n = 89; cohorts B + C) was 17.0 months (95% CI: 10.6–23.9).
TP53 mutations were a strong negative predictor for median OS in all cohorts. Median OS of mutated TP53 patients (n = 34) was 15.0 months (95% CI: 5.0–24.9) compared with 50.0 months (95% CI: 22.9–77.1) for TP53 wt patients (n = 109) (P = 0.002). With chemotherapy only (cohort A), the median OS in TP53-mutated patients (n = 7) was 2.0 months (95% CI: 0.0–4.6) compared with 9.0 months (95% CI: 6.1–11.9) in TP53 wt patients (n = 15) (P = 0.035). For crizotinib-treated patients (cohorts B + C), OS for TP53-mutated patients (n = 13) was 17.0 months (95% CI: 6.7–27.3) compared with TP53 wt patients (n = 50) for whom the median OS was not reached (P = 0.049). Also for patients treated with ceritinib after crizotinib (cohort D), a striking difference in OS was observed with 7.0 months only (95% CI: not reached) for TP53 mutated patients (n = 4) and 50.0 months (95% CI: not reached) (P = 0.001) for TP53 wt patients (n = 20).
Within the TP53-mutated patient cohort, median OS with chemotherapy only (n = 7) was 2.0 months (95% CI: 0.0–4.6) and thus inferior to ALK-inhibitor treatment (n = 15) with 17.0 months (95% CI: 4.6–29.4) (P = 0.025). For TP53 wt patients treated with chemotherapy only (n = 15), the median OS was 9.0 months (95% CI: 6.1–11.9) compared with a median OS of 50.0 months (95% CI: 22.3–77.7) (P < 0.001) for TP53 wt patients treated with ALK inhibitors with or without chemotherapy (n = 54) (Figure 3B and supplementary Table S3, available at Annals of Oncology online).
In univariate analysis including age, sex, smoking history, current smoker status, Eastern Cooperative Oncology Group (ECOG) performance status, number of brain metastases, number of treatment lines before crizotinib or ceritinib and TP53 mutation status only current smoker status and TP53 mutations were significant negative prognostic factors for OS (P = 0.016 and P = 0.002, respectively). In multivariate Cox regression analysis only TP53 mutation remained an independent negative prognostic factor (P = 0.004) (supplementary Table S7, available at Annals of Oncology online).
Patients with ALK variant 3a/b (n = 14) had a nonsignificant better OS of 50 months (95% CI: 0.0–108.9) compared with variant 1 (n = 18) with 29.0 months (95% CI: 9.4–48.6) (P = 0.815). TP53 mutations were prognostic negative in both variant subgroups reaching statistical significance in variant 1 (P = 0.032) (supplementary Table S8, available at Annals of Oncology online).
Discussion
We show that in ALK-positive NSCLC TP53 mutations separate roughly one quarter of patients with a substantially worse outcome. PFS and OS were inferior compared with TP53 wt patients treated with chemotherapy and ALK inhibitors.
TP53 alterations may damage tumor suppressor functions as loss of function mutations or trigger inhibition of apoptosis and genomic instability as gain of function mutations [23]. Thus, a negative prognostic impact of TP53 mutations in cancer has been postulated and preclinical observations support this hypothesis [24]. While in unselected NSCLC such a negative prognostic impact has not been proven unequivocally [25–29], it has been reported in numerous reports for EGFR-mutated NSCLC treated with EGFR inhibitors. These results, however, only partly reached statistical significance [30–34]. In ALK-positive lung cancer, TP53 mutations so far have not been described as significant negative prognostic factors.
The outcome in our treatment-related subgroups independently of TP53 status confirmed what has been described in clinical trials [3, 4, 16] and registry analyses [6, 7, 12–14, 35]: superiority of ALK inhibitors over chemotherapy in terms of PFS and superiority of sequential ALK-inhibitor therapy compared with crizotinib monotherapy in terms of OS. Our results additionally show that TP53 mutations represent the by far most frequent co-occurring mutations in ALK-positive NSCLC. By comparison, we found other co-mutations with a frequency of below 4% only; among them rarely those with actionable mutations like BRAFV600, high-level MET amplification or activating KRAS mutation.
Most important, our results suggest that about one-fourth of ALK-positive patients do not substantially benefit from recent progress of targeted therapy. As a limitation, concerning the use of next-generation ALK inhibitors statistically valid OS data could be assessed only for ceritinib. Future studies will have to prove, whether our findings can be confirmed for other next-generation ALK inhibitors.
In many cancer types, TP53 mutations were shown to be associated with higher genetic instability [24]. Accordingly, we could recently show that early TP53 mutations can lead to chromosomal instability in ALK-positive NSCLC [36]. It is tempting to speculate that a higher mutational burden might lead to a better efficacy of immune checkpoint inhibitors. Of note, the proportion of patients with PD-L1 positive tumor cells is enriched in our TP53-mutated group, although the number of patients is rather small.
Recently, in post ALK-inhibitor treatment biopsies it was shown that the type of ALK variant influences the development of ALK-inhibitor resistance mutations. In particular, EML4-ALK variant 3 was correlated with the development of ALK G1202R resistance mutation and a better PFS under treatment with the third-generation ALK-inhibitor lorlatinib, but not with first- and second-generation ALK inhibitors [37]. Similarly, in our pretreatment biopsies we saw a nearly equal distribution between ALK variants 1 and 3a/b and no significant influence of first- and second-generation ALK inhibitors on PFS. TP53 mutations were negative prognostic in terms of PFS and OS in both variant subgroups. Based on the low patient number, which limits our conclusions, significance was only partly reached. It remains to be elucidated whether TP53 mutation status and ALK variant status are independent prognostic factors.
In summary, we here describe TP53 mutations as the first pretreatment biomarker in ALK-positive NSCLC identifying patients with a substantially worse outcome from therapy. In future clinical trials stratification of this patient subgroup should be considered and new treatment strategies investigated to improve the outcome of ALK/TP53 co-mutated patients.
Funding
None declared.
Disclosure
AK: Advisory Role for BMS, AbbVie, Novartis; CA: Research Funding from Roche; MS: Honoraria, Advisory Role and Travel Expenses from Novartis, BMS, Takeda, Boehringer Ingelheim; SM-B: Honoraria and Advisory Role from AstraZeneca, BMS, Novartis; DS: BB Biotech AG; RR: Advisory Role and Travel Expenses from Boehringer Ingelheim, Lilly; SM: Honoraria, Advisory Role and Research Funding from Novartis, Pfizer, Roche, Boehringer Ingelheim; LN: Honoraria, Advisory Role, Research Funding and Travel Expenses from Pfizer, Celgene, Novartis, Roche, BMS, Boehringer Ingelheim, MSD; JF: Honoraria from BMS and AstraZeneca; FK: Honoraria and Advisory Role from Takeda, AbbVie, BMS, MSD, Roche, Novartis, Celgene; MS: Honoraria, Advisory Role and Travel Expenses from Pfizer, BMS, Roche, MSD, Lilly, Celgene, AstraZeneca; SK: Honoraria, Advisory Role and Travel Expenses from Roche, Novartis, Chugai, AstraZeneca, Boehringer Ingelheim; DK: Advisory Role and Travel Expenses from Roche, Boehringer Ingelheim, Novartis, Grifols; JB: Honoraria, Advisory Role and Travel Expenses from Boehringer Ingelheim, BMS, AstraZeneca, Novartis, MSD, Pfizer, Roche, Chugai; BS: Honoraria from Boehringer Ingelheim, BMS, Roche; JB: Honoraria from Amgen, Janssen; MS: Honoraria and Advisory Role from Pfizer, Roche, Novartis, Takeda; K-OK: Advisory Role and Research Funding from MSD, BMS, Roche, Pfizer, Boehringer Ingelheim; RT: Advisory Role and Research Funding from Roche, J&J, Novartis, AstraZeneca, Bayer, New Oncology AG, Clovis, Boehringer Ingelheim, Merck, MSD, Lilly, Sanofi-Aventis, Daiichi-Sankyo, Puma; TZ: Honoraria and Advisory Role from Roche, Novartis, Lilly, Pfizer, Merck; AMS: Honoraria, Advisory Role, Research Funding and Travel Expenses from BMS, Roche; RB: Honoraria from Pfizer, Novartis; JW: Advisory Role, Research Funding and Travel Expenses from AbbVie, AstraZeneca, BMS, Boehringer Ingelheim, Chugai, Ignyta, Lilly, MSD, Novartis, Pfizer, Roche. All remaining authors have declared no conflicts of interest.
Supplementary Material
{kind=link}
{kind=link}
Footnotes
Data presented in part at the European Society of Medical Oncology Annual Meeting 2017, Madrid, Spain (abstract #3757) and the German Society of Hematology/Oncology Annual Meeting 2017, Stuttgart, Germany (abstract A-908-0026-00596).
References
- 1. Inamura K, Takeuchi K, Togashi Y. et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol 2009; 22(4): 508–515. [DOI] [PubMed] [Google Scholar]
- 2. Wellstein A, Toretsky JA.. Hunting ALK to feed targeted cancer therapy. Nat Med 2011; 17(3): 290–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shaw AT, Kim D-W, Nakagawa K. et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med 2013; 368(25): 2385–2394. [DOI] [PubMed] [Google Scholar]
- 4. Solomon BJ, Mok T, Kim D-W. et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 2014; 371(23): 2167–2177. [DOI] [PubMed] [Google Scholar]
- 5. Clinical Lung Cancer Genome Project (CLCGP); Network Genomic Medicine (NGM). A genomics-based classification of human lung tumors. Sci Transl Med 2013; 5(209): 209ra153.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barlesi F, Mazieres J, Merlio JP. et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016; 387(10026): 1415–1426. [DOI] [PubMed] [Google Scholar]
- 7. Kris MG, Johnson BE, Berry LD. et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014; 311(19): 1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gadgeel SM. Sequencing of ALK inhibitors in ALK+ non-small cell lung cancer. Curr Treat Options Oncol 2017; 18(6): 36.. [DOI] [PubMed] [Google Scholar]
- 9. Lin JJ, Riely GJ, Shaw AT.. Targeting ALK: precision medicine takes on drug resistance. Cancer Discov 2017; 7(2): 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Markham A. Brigatinib: first global approval. Drugs 2017; 77(10): 1131–1135. [DOI] [PubMed] [Google Scholar]
- 11. Mok TSK, Crino L, Felip E. et al. The accelerated path of ceritinib: translating pre-clinical development into clinical efficacy. Cancer Treat Rev 2017; 55: 181–189. [DOI] [PubMed] [Google Scholar]
- 12. Duruisseaux M, Besse B, Cadranel J. et al. Overall survival with crizotinib and next-generation ALK inhibitors in ALK-positive non-small-cell lung cancer (IFCT-1302 CLINALK): a French nationwide cohort retrospective study. Oncotarget 2017; 8(13): 21903–21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gainor JF, Tan DS, De Pas T. et al. Progression-free and overall survival in ALK-positive NSCLC patients treated with sequential crizotinib and ceritinib. Clin Cancer Res 2015; 21(12): 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito K, Hataji O, Fujimoto H. et al. Sequential use of ALK inhibitors: an optional approach. J Thorac Oncol 2016; 11(12): e153–e154. [DOI] [PubMed] [Google Scholar]
- 15. Peters S, Camidge DR, Shaw AT. et al. Alectinib versus crizotinib in untreated ALK-positive non–small-cell lung cancer. N Engl J Med 2017; 377(9): 829–838. [DOI] [PubMed] [Google Scholar]
- 16. Soria JC, Tan DSW, Chiari R. et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 2017; 389(10072): 917–929. [DOI] [PubMed] [Google Scholar]
- 17. Heydt C, Kostenko A, Merkelbach-Bruse S. et al. ALK evaluation in the world of multiplex testing: Network Genomic Medicine (NGM): the Cologne model for implementing personalised oncology. Ann Oncol 2016; 27(suppl_3): iii25–iii34. [DOI] [PubMed] [Google Scholar]
- 18. Schildhaus HU, Heukamp LC, Merkelbach-Bruse S. et al. Definition of a fluorescence in-situ hybridization score identifies high- and low-level FGFR1 amplification types in squamous cell lung cancer. Mod Pathol 2012; 25(11): 1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koitzsch U, Heydt C, Attig H. et al. Use of the GeneReader NGS System in a clinical pathology laboratory: a comparative study. J Clin Pathol 2017; 70(8): 725–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Scheel AH, Baenfer G, Baretton G. et al. Interlaboratory concordance of PD-L1 immunohistochemistry for non-small-cell lung cancer. Histopathology 2018; 72(3): 449–459. [DOI] [PubMed] [Google Scholar]
- 21. Scheel AH, Dietel M, Heukamp LC. et al. Harmonized PD-L1 immunohistochemistry for pulmonary squamous-cell and adenocarcinomas. Mod Pathol 2016; 29(10): 1165–1172. [DOI] [PubMed] [Google Scholar]
- 22. Kato S, Han SY, Liu W. et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci USA 2003; 100(14): 8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brosh R, Rotter V.. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer 2009; 9(10): 701–713. [DOI] [PubMed] [Google Scholar]
- 24. Hainaut P, Pfeifer GP.. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med 2016; 6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ahrendt SA, Hu Y, Buta M. et al. p53 mutations and survival in stage I non-small-cell lung cancer: results of a prospective study. J Natl Cancer Inst 2003; 95(13): 961–970. [DOI] [PubMed] [Google Scholar]
- 26. Govindan R, Weber J.. TP53 mutations and lung cancer: not all mutations are created equal. Clin Cancer Res 2014; 20(17): 4419–4421. [DOI] [PubMed] [Google Scholar]
- 27. Kosaka T, Yatabe Y, Onozato R. et al. Prognostic implication of EGFR, KRAS, and TP53 gene mutations in a large cohort of Japanese patients with surgically treated lung adenocarcinoma. J Thorac Oncol 2009; 4(1): 22–29. [DOI] [PubMed] [Google Scholar]
- 28. Rosell R, Monzo M, Pifarre A. et al. Molecular staging of non-small cell lung cancer according to K-ras genotypes. Clin Cancer Res 1996; 2(6): 1083–1086. [PubMed] [Google Scholar]
- 29. Tsao MS, Aviel-Ronen S, Ding K. et al. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. J Clin Oncol 2007; 25(33): 5240–5247. [DOI] [PubMed] [Google Scholar]
- 30. Canale M, Petracci E, Delmonte A. et al. Impact of TP53 mutations on outcome in EGFR-mutated patients treated with first-line tyrosine kinase inhibitors. Clin Cancer Res 2017; 23(9): 2195–2202. [DOI] [PubMed] [Google Scholar]
- 31. Labbe C, Cabanero M, Korpanty GJ. et al. Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer 2017; 111: 23–29. [DOI] [PubMed] [Google Scholar]
- 32. Yu HA, Jordan E, Ni A. et al. Concurrent genetic alterations identified by next-generation sequencing in pre-treatment, metastatic EGFR-mutant lung cancers. J Clin Oncol 2016; 34: 9053–9053. [Google Scholar]
- 33. Aisner DL, Sholl LM, Berry LD. et al. The impact of smoking and TP53 mutations in lung adenocarcinoma patients with targetable mutations—The Lung Cancer Mutation Consortium (LCMC2). Clin Cancer Res 2018; 24(5): 1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. VanderLaan PA, Rangachari D, Mockus SM. et al. Mutations in TP53, PIK3CA, PTEN and other genes in EGFR mutated lung cancers: correlation with clinical outcomes. Lung Cancer 2017; 106: 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kostenko A, Michels SYF, Fassunke J. et al. Survival following implementation of next-generation sequencing in routine diagnostics of advanced lung cancer: results of the German Network Genomic Medicine. J Clin Oncol 2016; 34: 9085–9085. [Google Scholar]
- 36. Alidousty C, Baar T, Martelotto LG. et al. Genetic instability and recurrent MYC amplification in ALK-translocated NSCLC: a central role of TP53 mutations. J Pathol 2018; 246: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lin JJ, Zhu VW, Yoda S. et al. Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. J Clin Oncol 2018; 36: 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.