

Abstract
The recycling of ribosomal subunits after translation termination is critical for efficient gene expression. Tma64 (eIF2D), Tma20 (MCT-1), and Tma22 (DENR) function as 40S recycling factors in vitro, but it is unknown whether they perform this function in vivo. Ribosome profiling of tma deletion strains revealed 80S ribosomes queued behind the stop codon, consistent with a block in 40S recycling. We found that unrecycled ribosomes could reinitiate translation at AUG codons in the 3’UTR, as evidenced by peaks in the footprint data and 3’UTR reporter analysis. In vitro translation experiments using reporter mRNAs containing upstream ORFs (uORFs) further established that reinitiation increased in the absence of these proteins. In some cases, 40S ribosomes appeared to rejoin with 60S subunits and undergo an 80S reinitiation process in 3’UTRs. These results support a crucial role for Tma64, Tma20, and Tma22 in recycling 40S ribosomal subunits at stop codons and translation reinitiation.
Graphical Abstract
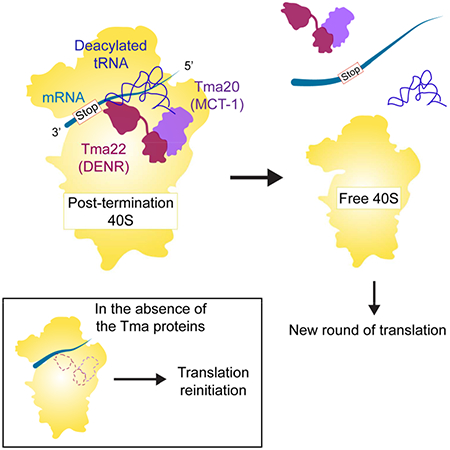
Young et al. (2018) show Tma64 (eIF2D) or the Tma20 (MCT-1)/Tma22 (DENR) complex removes 40S ribosomal subunits from mRNA after translation. Loss of 40S recycling in Tma mutants confers increased stop codon readthrough and reinitiation either by 40S subunits at AUG codons, or 80S ribosomes, on many mRNAs.
INTRODUCTION
Ribosome recycling is a critically important process that dissociates the post-termination ribosome complex into free ribosomal subunits, deacylated tRNA, and mRNA for use in further rounds of translation. Recycling begins after peptide release with the 80S ribosome positioned on a stop codon and bound to eukaryotic release factor 1 (eRF1) and the ATP-binding cassette protein Rli1 (yeast)/ABCE1 (mammals) (Becker et al., 2012; Shao et al., 2016). In vitro and in vivo studies have shown that Rli1/ABCE1 catalyzes the first stage of recycling, splitting the 80S ribosome into a free 60S subunit and a tRNA/mRNA bound 40S subunit (Pisarev et al., 2010; Shoemaker and Green, 2011; Young et al., 2015). The next step of recycling, dissociation of tRNA and mRNA from the 40S, has been reconstituted in vitro with the single protein ligatin/eIF2D, or the two interacting proteins MCT-1 (also known as MCTS1) and DENR, that are homologous to the N- and C-termini, respectively, of eIF2D (Skabkin et al., 2010). Earlier work reported that a subset of canonical initiation factors, including eIF1, eIF1A, eIF3 and eIF3j can also fulfill this function (Pisarev et al., 2007), suggesting that redundant mechanisms for 40S recycling may exist in the cell.
In addition to their observed 40S recycling role, eIF2D and MCT-1/DENR have also been shown in vitro to facilitate non-canonical initiation independent of the eIF2·GTP·Met-tRNAiMet ternary complex that normally brings Met-tRNAiMet to 40S subunits. eIF2D and MCT-1/DENR were found to promote recruitment of Met-tRNAiMet to complexes of the 40S ribosome and specialized mRNAs that had been assembled with the AUG directly in the P site in the absence of canonical initiation factors (Dmitriev et al., 2010; Skabkin et al., 2010).
Both DENR and MCT-1 have been shown to promote translation reinitiation downstream of uORFs with strong Kozak consensus sequences in Drosophila (Schleich et al., 2014) and in HeLa cells (Schleich et al., 2017). It was not determined whether the loss of reinitiation in the absence of these factors occurred due to a defect in Met-tRNAiMet recruitment to the newly scanning 40S (initiation defect) or because the deacylated tRNA at the P-site remained associated with the 40S subunit stalling the 40S at the uORF stop codon (recycling defect). The yeast homologues of eIF2D, MCT-1, and DENR have been independently identified as translation-machinery-associated (TMA) proteins Tma64, Tma20 and Tma22 (here denoted “Tma64/−20/−22”). Expression of human MCT-1 can rescue defects observed in the tma20 deletion strain, suggesting that MCT-1 and Tma20 are functional orthologues (Fleischer et al., 2006). Proper functioning of these factors has been shown to be important for human health, with mutations in DENR identified in patients with autism spectrum disorders (Haas et al., 2016; Neale et al., 2012), and MCT-1 is overexpressed in B- and T-cell lymphomas (Prosniak et al., 1998; Shi et al., 2003).
Further insight into the function of these factors can be discerned from sequence homology and structure. Both Tma64/eIF2D and Tma22/DENR contain a SUI1 (eIF1) domain at their C-terminus and were inhibited from binding a 40S subunit that had been preincubated with eIF1 (Skabkin et al., 2010). Recent cryo-EM and crystal structures of eIF2D and MCT-1/DENR bound to the ribosome (Lomakin et al., 2017; Weisser et al., 2017) have shown that these SUI1 domains adopt a similar fold to eIF1 and bind at the same location as eIF1 on the platform of the 40S subunit. The similarity between the SUI1 domain/40S binding interactions suggests that Tma64/eIF2D and Tma22/DENR may act like eIF1 in modulating tRNA access to the P-site (Hussain et al., 2014; Llacer et al., 2015; Rabl et al., 2011). Tma20/MCT-1 and the N-terminal part of Tma64/eIF2D bind below the 40S platform, and contacts the CCA tail of the initiator tRNA. Because no interactions were observed with the methionyl moiety (Weisser et al., 2017), it is conceivable that this interaction is not limited to the initiator tRNA, as would be the case in recycling. The generality of this interaction is consistent with the ability of eIF2D to operate with both the initiator and elongator tRNAs during non-canonical initiation complex formation (Dmitriev et al., 2010; Skabkin et al., 2010).
The binding site of Tma64/eIF2D or the complex of Tma20/MCT-1 and Tma22/DENR overlaps not only with the 40S binding site of eIF1 but also with those of the canonical initiation factor eIF2 that delivers Met-tRNAiMet to the 40S subunit. This overlap, and the interaction with tRNA, are consistent with either a role for Tma64/−20/−22 in substituting for eIF2, or in preventing 40S reinitiation by preventing binding of eIF2 and other canonical factors following dissociation of the 60S subunit. Consistent with these interpretations, Tma64/eIF2D occupies positions on the 40S ribosome where inter-subunit bridges form with the 60S subunit, presumably accounting for the strong ribosome anti-association activity of Tma64/eIF2D observed in vitro (Weisser et al., 2017). This activity could therefore enable Tma64/eIF2D to prevent reassociation of a 60S subunit to a post-termination 40S subunit at the stop codon, following Rli1/ABCE1-mediated 60S recycling, or premature joining of the 60S and 40S subunits prior to correct start site selection.
It therefore remains an open question whether Tma64 and Tma20/Tma22 function in initiation only, recycling only, or have distinct functions in both processes and potentially act between recycling and the next round of initiation. Here we show by ribosome profiling that loss of Tma64/−20/−22 leads to stalling of 80S ribosomes just upstream of a presumptive unrecycled 40S ribosome subunit at the stop codon. The absence of these factors also leads to increased translation of regions downstream of the stop codons, consistent with unrecycled 40S subunits forming competent pre-initiation complexes and scanning to a downstream start codon or, in other cases, first reassociating with 60S subunits and reinitiating translation as an 80S ribosome. These results show that the Tma proteins are critical factors that broadly promote the recycling of 40S ribosomal subunits at stop codons following Rli1/ABCE1-catalyzed dissociation of the 60S subunits and prevent translation reinitiation.
RESULTS
Deletion of TMA64 and TMA20 or TMA64 and TMA22 results in stalling of 80S ribosomes upstream of the stop codon and translation reinitiation in 3’UTRs
To determine if Tma64/−22/−20 are involved in ribosome recycling in vivo, we performed ribosome profiling on two double deletion strains: tma64Δ/tma20Δ and tma64Δ/tma22Δ (tmaΔΔ strains). We examined the tmaΔΔ strains to control for any redundancy between Tma64 and Tma20 or Tma22 (Fleischer et al., 2006; Gavin et al., 2002; Reinert et al., 2006). Possible redundancy of these factors is supported by observed negative genetic interactions between the TMA20 and TMA64 genes (Costanzo et al., 2010). Ribosome footprint levels in coding sequences (ORFs) were highly reproducible between biological replicates (Spearman’s R2 0.99 and 0.99, respectively for tma64Δ/tma20Δ and tma64Δ/tma22Δ)
Alignment of all genes at stop codons in WT cells revealed average ribosome density with strong 3-nt periodicity, corresponding to translation in a single reading frame, and a peak at the stop codon (Fig. 1A), consistent with existing results (Weinberg et al., 2016). Previously, we found that genetic depletion of Rli1 (rli1-d), which is responsible for dissociation of the 60S subunit from post-termination ribosomes in the first stage of recycling, resulted in a large increase in the stop codon peak (Fig. 1A inset) and the appearance of a smaller peak ~30 nt upstream (Fig. 1A). These data are consistent with a queued 80S ribosome positioned behind an 80S post-termination complex that is stalled at the stop codon (Young et al., 2015). Strikingly, the corresponding profiles for the two tmaΔΔ strains differ from both the WT and rli1-d profiles: there is no peak observed at the stop codon, but surprisingly, 80S ribosomes still accumulate ~30 nt upstream at levels higher than observed when Rli1 is depleted (Fig. 1A and B). These observations are consistent with unrecycled 40S subunits, which cannot be observed by 80S ribosome profiling, stalling at the stop codon and causing trailing 80S ribosomes to queue behind them.
Figure 1. Deletion of TMA64 and TMA20 or TMA64 and TMA22 results in stalling of the penultimate elongating ribosome and increased 3’UTR ribosome occupancy.
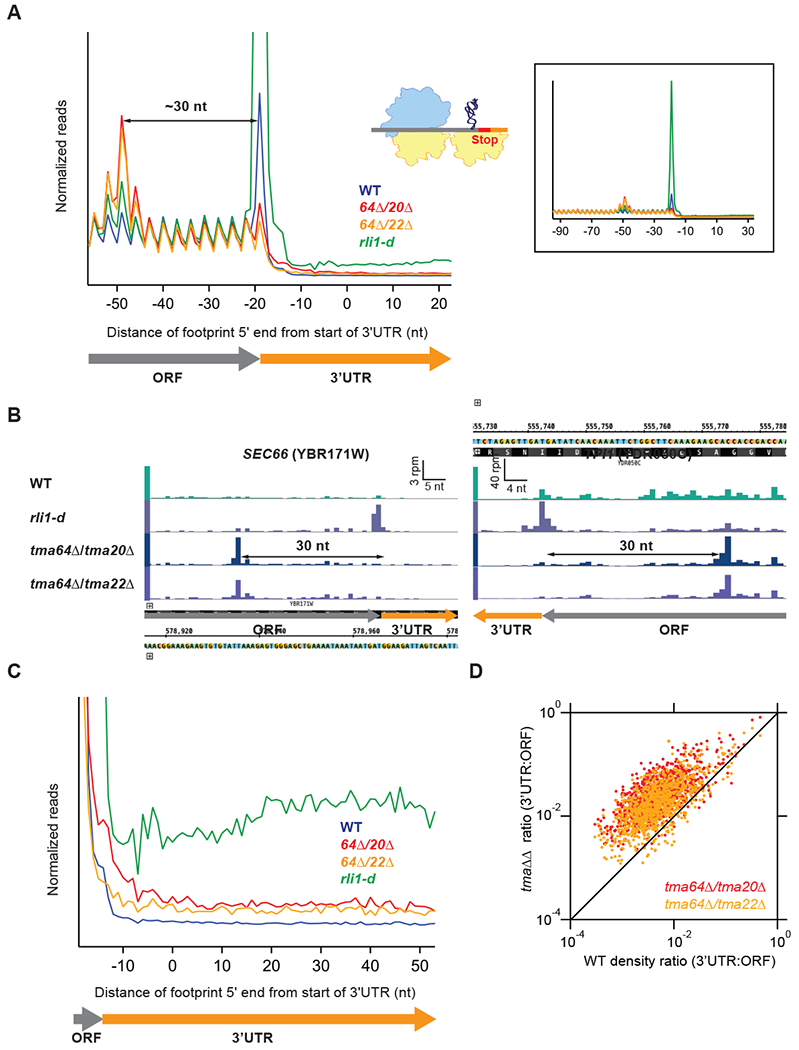
(A) Normalized average ribosome footprint occupancy (each gene weighted equally) from all genes aligned at their stop codons for WT, tma64Δ/tma20Δ, tma64Δ/tma22Δ, and rli1-d cells. Footprint 5’ ends are plotted. The schematic depicts a 40S subunit at the stop codon stalling and queuing of the penultimate elongating 80S ribosome.(Inset) Demagnified view of (A), showing full extent of ribosome stalling.
(B) Ribosome footprint profiles of genes SEC66 and TPI1 showing ribosome occupancy peaks 30 nt upstream of the stop codon that represent stalled elongating 80S ribosomes. Reads are shifted so peaks approximately correspond to ribosome A sites.
(C) Normalized average ribosome footprint occupancy for the first 60 nt of the 3’UTRs of all genes, magnified from (A).
(D) Ratio of footprint densities in 3’UTRs to the respective ORFs is plotted for tma64Δ/tma20Δ and tma64Δ/tma22Δ versus WT cells, for genes with >5 rpkm in ORFs and > 0.5 rpkm in 3’UTRs. Each point represents the data for 1 gene, revealing extensive ribosome occupancy in the 3’UTRs of most genes.
While the largest penultimate peak is observed ~30 nt upstream of the stop codon in the tma double deletions, smaller ribosome occupancy peaks are also observed ~27 and ~33 nt upstream of the stop codon (Fig. 1A). These peaks could arise from differences in the conformation of the post-termination 40S complex (Archer et al., 2016) or sequence-dependent accessibility of the mRNA to RNase 1 digestion (see variation between examples, Fig. 1B and Suppl. Fig. 1A).
Ribosome occupancies are elevated in the 3’UTRs of the two tma double deletion (tmaΔΔ) strains, although at a lower level than observed in the rli1-d strain (Fig. 1C). The distribution of 3’UTR ribosomes in the tmaΔΔ strains do not exhibit a 3-nt periodicity in the 3’UTR, unlike a strain expressing a suppressor tRNA, (Suppl. Fig. 1B inset) (Guydosh and Green, 2014), suggesting that the 3’UTR ribosomes in the tmaΔΔ strains do not occupy a single reading frame. A comparison of the ratio between ribosome density in the 3’UTR vs. ORF for each transcript revealed that an increase in 3’UTR density occurred on most genes (Fig. 1D). These data were similar in both replicates and highly significant (Suppl. Fig. 1 C-E). These data are therefore consistent with the notion that Tma64/−22/−20 normally block reinitiation in multiple reading frames by promoting recycling of 40S post-termination complexes (post-TCs).
Ribosome occupancy peaks are observed at 3’UTR AUG codons consistent with 40S reinitiation
To evaluate potential mechanisms for translation reinitiation in the tmaΔΔ strains, we more carefully examined the ribosome occupancy across 3’UTRs by searching for peaks of ribosome density at AUG codons. The presence of such peaks would be consistent with a mechanism where Tma64/−22/−20 prevent canonical initiation factors from binding to 40S post-TCs at stop codons with subsequent scanning and reinitiation (Weinberg et al., 2016). We found many examples where peaks appeared at 3’UTR AUG codons in the tmaΔΔ strains but rarely in WT and rli1-d strains, illustrated for the RPL14A and RPS30B genes in Fig. 2A. To determine whether this trend occurred broadly, we averaged ribosome density around all 3’UTR AUG codons and found an obvious density peak in the tmaΔΔ strains 3-4 fold above the background density (Fig. 2B, left panel). In WT cells, we also observed peaks on AUGs, though at a much lower absolute level. This implies that loss of these factors strongly enhances a low-level reinitiation process present in the WT strain. The pronounced AUG density peak was observed in both replicates for each tmaΔΔ strain (Suppl. Fig. 1F left panel).
Figure 2. Ribosome occupancy peaks are observed at 3’UTR AUG codons consistent with 40S reinitiation.
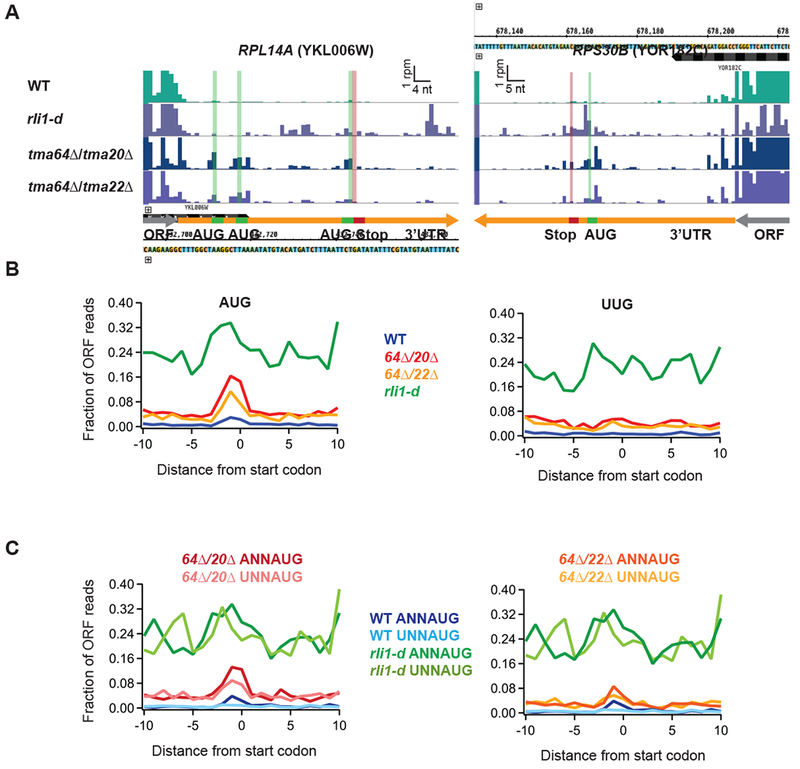
(A) Ribosome footprint profiles of the 3’UTRs of genes RPL14A and RPS30B showing ribosome occupancy peaks at 3’UTR AUG codons. Green line and bars represent AUG start codon(s) and red lines and bars represent stop codons in the same frame. Gene annotations are drawn to correspond to data for ribosome P sites (AUG codons) or A sites (stop codons). Note that the 3rd AUG and stop codon for RPL14A are slightly offset for visibility.
(B) Average fraction of ribosome occupancy in a window surrounding 3’UTR AUG codons (left) and 3’UTR near-cognate UUG codons (right) normalized to ORF ribosome occupancy level (all frames included). Data shown for WT, tma64Δ/tma20Δ, tma64Δ/tma22Δ, and rli1-d cells. Reads are shifted so peaks approximately correspond to ribosome P sites.
(C) Average ribosome occupancy on AUG codons, as for B (left), but data are filtered according to start codon context: ANNAUG strong context and UNNAUG weak context for tma64Δ/tma20Δ (left) and tma64Δ/tma22Δ (right). Reads are shifted so peaks approximately correspond to ribosome P sites.
Similar peaks were not observed in 3’UTRs at the near-cognate UUG codons (Fig. 2B, right panel), suggesting that the AUG peaks were created by 40S preinitiation complexes that select start codons by a high-fidelity scanning process. This was also true in both replicates for each tmaΔΔ strain (Suppl. Fig. 1F, right panel). As a control, we performed this analysis on data obtained from the rli1-d strain and did not see evidence for a strong initiation peak above the elevated background level (Fig. 2B), consistent with non-AUG dependent 80S reinitiation occurring on depletion of Rli1 (Young et al., 2015).
To test further whether these 3’UTR 80S peaks adhered to known rules of start codon selection, we next identified AUG codons in known good and poor sequence contexts. In general, the presence of an adenine base at positions −3 nt upstream of AUG is preferred relative to uracil, among genes in Saccharomyces species (Shabalina et al., 2004). We therefore averaged the ribosome density around AUG codons preceded by either an A or U at the −3 position (Fig. 2C). For both tmaΔΔ strains, good contexts showed higher occupancy than poor contexts, consistent with canonical 40S initiation.
Direct detection of epitope-tagged 3’UTR translation products is consistent with both 40S and 80S reinitiation in the absence of Tma64/−20/−22
To determine more directly if 40S reinitiation takes place in the 3’UTRs of the tmaΔΔ strains, we designed reporter constructs to assay for polypeptides predicted to arise from initiation at 3’UTR AUG codons in 6 candidate genes. The coding sequences for 13 tandem Myc epitopes were inserted immediately upstream of the first in-frame stop codon encountered downstream of a 3’UTR AUG codon exhibiting a peak of ribosome occupancy in tmaΔΔ cells, and thus predicted to be the site of 40S reinitiation. For genes IPPI and RPL27A, two or three different reading frames were tested, respectively, by tagging the corresponding ORFs in separate strains, for a total of nine distinct Myc13-tagged 3’UTR reporters (Suppl. Fig. 2A-F). To improve the sensitivity of detection, these reporter genes were cloned into a high copy vector and transformed into the WT and tma64Δtma20Δ strains.
Western analysis revealed low-molecular weight Myc13-tagged polypeptides expressed in the tagged tmaΔΔ strains at levels much greater than observed in the corresponding tagged WT strains, for seven of the nine tagged 3’UTR ORF reporters derived from six different genes (Fig. 3A and Suppl. Fig. 3A; bands marked with cyan dots). Variations in abundance among the reporter polypeptides could derive from sequence-dependent variability in protein stability or could reflect underlying variations in levels of reinitiation for different genes. Myc13-tagged polypeptides were expressed from both 3’UTR ORFs tagged for IPP1, one of three tagged ORFs tested for RPL27A, and the single tagged ORFs constructed for RPS27A, RPS30B, RPS26B, and RPLI4A. We showed previously that the Myc13 tag migrates anomalously at ~35 kD, such that Myc13 tagging of Ost4, a yeast protein of only 36 amino acids, yields a tagged product that migrates aberrantly at ~40 kD rather than its ~25 kD molecular weight (Young et al. 2015). The seven Myc13-tagged products expressed in the tmaΔΔ cells co-migrate with Ost4-myc, consistent with the expression of short 3’UTR-encoded peptides that provides direct evidence for reinitiation of translation in the absence of Tma64/−20/−22. The expression in WT cells of less abundant, co-migrating Myc-tagged products from the RPL27A, RPS30B, RPL14A, and IPP1_tag2 reporters (Fig. 3A and Suppl. Fig. 3A) suggests that low-level reinitiation occurs in the 3’UTRs of these four genes even in the presence of Tma64/−20/−22.
Figure 3. Detection of epitope-tagged 3’UTR translation products that result from 40S and 80S reinitiation in the tma deletion strains.
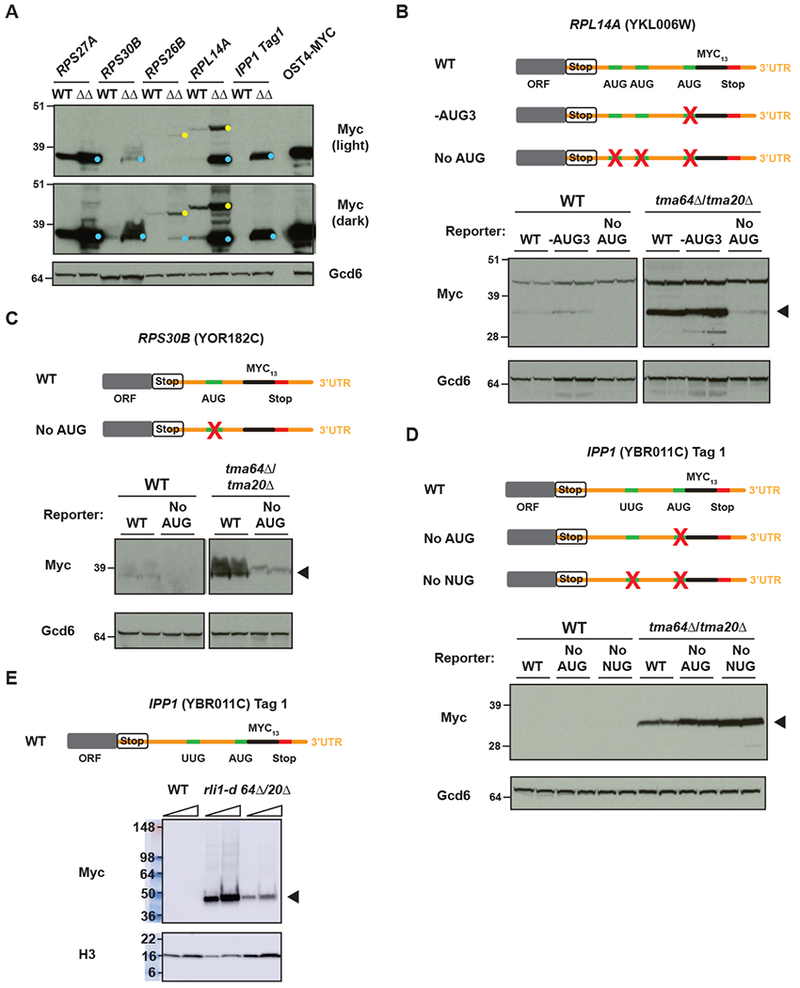
(A) WCEs from WT or tma64Δ/tma20Δ cells expressing reporters were subjected to western analysis using antibodies against c-Myc (upper blots) or Gcd6 or Histone H3 (control, lower blots). The YDY231 strain, which carries a C-terminally myc13-tagged OST4 gene, was included as a size marker for small 3’UTR reinitiation products. 3’UTR reinitation products are marked with cyan dots and readthrough/frameshift products are marked with yellow dots.
(B)-(E) Mutations of the AUG codons in the 3’UTRs of RPL14A (B) and RPS30B (C) resulted in loss of the 3’UTR reinitiation product (arrowheads), supporting a 40S reinitiation model. A larger readthrough product is observed for RPL14A running above the reinitiation product (B). (D) Mutation of the AUG and a near-cognate UUG in the IPP1_tag 1 reporter did not result in loss of the 3’UTR reinitation product, suggesting that it is the result of 80S reinitiation. (The relatively greater abundance of the products of the two mutant reporters might result from increased protein stability owing to altered amino acid sequences). (E) Expression of the IPP_tag 1 product was observed in the rli1-d strain confirming that 80S reinitiation is possible on this transcript and could account for the AUG-independent reinitiation observed in the tma64Δ/tma20Δ strain. For westerns shown in all panels, at least 2 biological replicates were used.
For the RPL14A and RPS26B 3’UTR reporters, slower migrating Myc13-tagged products were also detected, and are expressed at higher levels in tmaΔΔ versus WT strains (Fig. 3A, yellow dots). These products are consistent with readthrough of the main ORF stop codons in tmaΔΔ cells, consistent with previous findings of increased readthrough in tma20Δ cells (Fleischer et al 2006). For the RPS26B reporter, a −1 frameshift would also be required to place the 3’UTR Myc13 coding sequences in the same (“zero”) reading frame of the main ORF. The fact that the putative read-through product for RPL14A migrates more slowly than that for RPS26B (Fig. 3A) can be explained by noting that Rpl14a is 1716 Da larger than Rps26b. The putative readthrough and reinitiation products expressed from the RPL14A reporter were observed at comparable levels in both tmaΔΔ strains (Suppl. Fig. 3B).
To test the theory that the observed reinitiation products expressed from 3’UTR reporters result from 40S reinitiation, the predicted AUG start codons, and in some cases alternative AUG or near-cognate UUG start codons, located upstream of the Myc13 coding sequences were mutated to non-cognate AAA triplets (noted in Suppl Fig. 2A-F). Failure to express the putative reinitiation products from the mutated reporter genes would support canonical 40S reinitiation occurring in these 3’UTRs. Mutation of all three in-frame AUG codons upstream of the Myc13 coding sequences in the RPL14A reporter essentially eliminated expression of the putative ~50 kDa reinitiation product without affecting expression of the larger putative read-through product (Fig. 3B, No AUG), whereas mutating the third AUG codon alone had no effect on expression of either tagged polypeptide (Fig. 3B, -AUG3). These results suggest that one or both of the first two AUGs serve as start codons for reinitiation in the tmaΔΔ strains (Fig. 2A, left). Mutation of the single AUG predicted to serve as the start codon for reinitiation at RPS30B on the basis of 80S densities (Fig. 2A, right) also dramatically reduced expression of the putative reinitiation product from this 3’UTR reporter (Fig. 3C), suggesting that most of the reinitiation occurs at this AUG (Fig. 3C). Examination of darker exposures of the blots from WT cells for these two reporters indicate that the same AUG mutations eliminate or reduce expression of the reinitiation products that are produced from the WT versions of these reporters in cells containing Tma64/−20/−22 (Suppl. Fig. 4A-B). Measurement of reporter mRNA levels by qRT-PCR (Suppl. Fig. 4C) revealed that the levels of WT and “No AUG” reporter mRNA levels were similar to each other in both WT and tma64Δ/tma20Δ cells for the RPL14A and RPS30B reporters, indicating that the decrease in reinitation products expressed from the “No AUG” variants results from loss of 40S reinitiation, not a decrease in reporter mRNA levels. Taken together, the presence of 80S ribosome profiling peaks on 3’UTR AUG codons, and the dependence on AUG codons for reinitiation in some 3’UTRs indicates that 40S reinitiation is a prominent mechanism for starting translation in 3’UTRs in the absence of Tma64/−20/−22.
While a 40S reinitiation mechanism accounts for the above-described reporter peptides, it offers only a partial explanation for reinitiation on other reporters. In the case of RPS27A, mutating the single AUG codon predicted to serve as its reinitiation start codon (Suppl. Fig. 2D) conferred a significant, albeit partial reduction in expression of the putative reinitiation product by ~50% (Suppl. Fig. 4D). Similarly, expression of the putative reinitiation product of the IPP1_tag 2 reporter was reduced by only ~50% on mutating the AUG codon predicted to be its reinitiation start site; although a somewhat greater reduction occurred when an upstream in-frame UUG and downstream in-frame AUG were also mutated (Suppl. Fig. 4E), which might indicate that reinitiation can occur at more than one AUG or near-cognate start codon in this gene.
For the remaining three 3’UTR reporters whose expression is elevated in tma64Δ/tma20Δ cells, IPP1_tag 1, RPS26B, and RPL27A_tag 3, 40S reinitiation makes only a minor contribution to reporter protein expression, as we observed little or no reduction in reporter expression on mutating in-frame upstream start (AUG or UUG) codons (Fig. 3D and Suppl. Fig. 4F-G). As observed above for other reporters, measurement of the IPP1_tag 1 transcript level by qRT-PCR (Suppl. Fig. 4C) showed that simultaneous mutation of upstream AUG and UUG codons did not affect mRNA abundance. An alternative mechanism to account for these last observations is that loss of Tma64/−20/−22 can result in the 40S subunit remaining at the stop codon for an extended period of time allowing 60S subunit reassociation to occur, which might be favored by loss of the reported anti-association activity of Tma64/−20/−22 (Weisser et al., 2017). These reassociated post-termination 80S ribosomes would then migrate into the 3’UTR and undergo 80S reinitiation as previously observed in cells depleted of Rli1 (Young et al. 2015). To test this, we introduced the IPP1_tag 1 reporter into the rli1-d mutant and cultured the cells under non-permissive conditions. Remarkably, depletion of Rli1 in the rli1-d strain increased expression of a Myc13-tagged product that co-migrates with the tagged polypeptide observed in the tma64Δtma20Δ double mutant (Fig. 3E). This finding confirms that 80S reinitiation is possible on this transcript and could account for the AUG-independent reinitiation observed for this and other 3’UTR reporters in cells lacking Tma64/−20.
Further support for the conclusion that eliminating the Tma proteins can evoke non-canonical 80S reinitiation came from analyzing four previously described reporters that express Myc13-tagged polypeptides encoded in the 3’UTRs of SED1, YDR524C, YMR122W-A, or CWP2, which were all shown to be expressed on Rli1 depletion in rli1-d cells via 80S reinitiation (Young et al., 2015). Expression of these reporter polypeptides also occured in cells lacking Tma64/−20 or Tma64/−22 (Suppl. Fig. 5). Thus, it appears that elimination of Tma64/−20 or Tma64/−22 evokes a combination of 40S and 80S reinitiation triggered by accumulation of 40S post-TC complexes at main ORF stop codons.
Absence of Tma64/Tma20 or Tma64/Tma22 enhances reinitiation in cell extracts
To test further our conclusion that loss of Tma64/−20/−22 leads to increased reinitiation downstream of stop codons, we used a cell free expression system from yeast to translate a series of firefly luciferase (Fluc) reinitiation reporters. These reporters were composed of a short 5’ uORF followed by (or slightly overlapping with) the Fluc coding sequence (Schematics in Fig. 4A). In all cases, Fluc was out-of-frame with the uORFs. The reporter constructs were transcribed in vitro, and the capped and polyadenylated mRNAs were added to yeast lysates prepared from WT and each tmaΔΔ strain, and the resulting reactions were monitored for luminescence. The Vmax was calculated for each reaction and normalized to the Vmax of a Fluc reporter with no uORF to account for batch to batch experimental variation (Suppl Fig. 6A). These normalized rates were then divided by the rate obtained in WT extract (Fig. 4A left).
Figure 4. Loss of Tma64 and Tma22 or Tma20 enhance reinitiation in cell extracts.
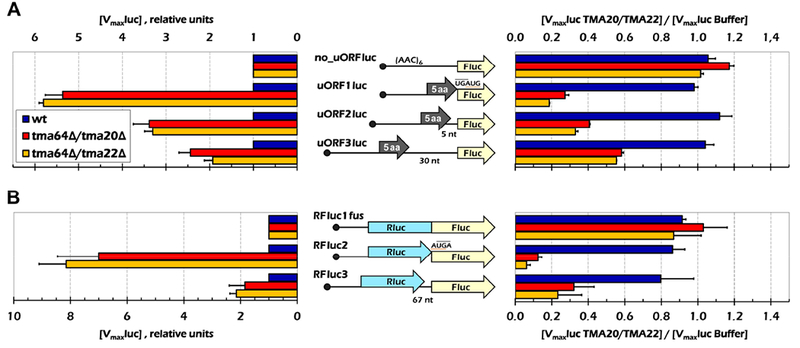
(A) In vitro synthesized uORF-containing mRNAs (schematically shown in the middle) were translated in cell lysates prepared form three yeast strains in the absence (left panel) or in the presence (right panel) of the recombinant Tma20/Tma22 dimer. The luciferase activities were continuously measured and maximal rates of the product accumulation were calculated. In the left panel, the values were normalized to that obtained for the no_uORFluc construct in the same lysate and shown as a ratio to the rates in the WT lysate. On the right, luciferase activity was measured in the presence and absence of recombinant Tma protein. Luciferase levels in the presence of Tma factor were normalized to levels observed upon addition of buffer. Error bars, mean ±SD. (B) The same assay as in (A) was performed on bicistronic Rluc-Fluc mRNA constructs in the yeast lysates. A similar trend was observed, showing that these factors also function after long ORFs in this assay. For all panels, 3 replicates each containing 2 or more mRNA preparations were used for each experiment.
The presence of a uORF would be expected to greatly diminish the signal from Fluc because most ribosomes would translate the uORF and then be recycled at the uORF stop codon, without ever reaching the Fluc start codon. As expected, compared to the control mRNA lacking a uORF (no_uORFluc), all of the uORF-containing constructs exhibited considerably lower Fluc production in the WT extract (Suppl Fig. 6A, blue bars, compare no_uORFluc to other constructs). We assume that this low-level Fluc production indicates that the majority of scanning ribosomes translate the uORFs in these mRNAs, rather than bypass them by leaky scanning, and then fail to reinitate at the Fluc AUG codon. The low level of reinitiation is consistent with the relatively inefficient “canonical” mechanism of 40S reinitiation (Kozak, 2002; Mueller and Hinnebusch, 1986; Williams et al., 1988).
In accordance with a role for Tma64/−20/−22 in promoting ribosome recycling and suppressing reinitiation downstream, Fluc expression was elevated to varying extents in the lysates prepared from both tmaΔΔ strains compared to WT for all of the mRNAs harboring a uORF (Suppl. Fig. 6A; summarized in Fig. 4A, left panel). These results are difficult to reconcile with a stimulatory role for Tma64/−20/−22 in delivery of Met-tRNAiMet to the Fluc start codon during reinitiation, in which case the absence of these factors would decrease Fluc expression from uORF-containing mRNAs. Rather, the increased Fluc expression we observed is more consistent with the idea that impaired recycling in the tmaΔΔ extracts preserves 40S post-termination complexes at the uORF stop codons that can resume scanning and reinitiate at the Fluc coding sequences.
The increase in Fluc expression was greatest, ~6-fold, when the stop codon of a 5-codon uORF partially overlaps the AUG of the main ORF (UGAUG in construct uORF1luc). Strikingly, the effect of eliminating Tma64/−22/−20 was progressively diminished on increasing the intercistronic distance from 0 to either 5 nt or 30 nt (Fig. 4A, left, uORF1luc vs. uORF2luc and uORF3luc). This was counterintuitive since canonical reinitiation efficiency, as on GCN4 mRNA, is known to increase with increasing distance between the uORF and main ORF (Grant et al., 1994; Kozak, 1987), as the greater distance provides increased opportunity for the recruitment of eIF2-Met-tRNAiMet-GTP. Indeed, we observed this trend in the WT extract (Suppl Fig. 6A, cf. uOR1Fluc, uORF2luc, uORF3luc). One way to explain why the opposite trend has occurred in the tmaΔΔ strain is that an additional activity of Tma64/−22/−20 is to “protect” unrecycled 40S subunits from binding eIF3 (Lomakin et al., 2017) and acquiring eIF2-Met-tRNAiMet-GTP. A longer distance from the uORF to the Fluc ORF would give additional time for the Tma proteins to gradually dissociate and lose their ability to block reinitiation; accordingly, the absence of Tma proteins in tmaΔΔ extracts confers a progressively smaller stimulation of reinitiation as the uORF-Fluc spacing increases.
To confirm that the increased reinitiation for the uORF-containing mRNAs in the tmaΔΔ extracts resulted from the absence of the Tma proteins, bacterially expressed Tma20/Tma22 dimer was purified and added to the lysates prior to incubation with the reinitiation reporters (Fig. 4A, right panel). Adding Tma20/Tma22 to the WT extract had a negligible effect on Fluc production for all mRNAs, but reduced Fluc production in both mutant lysates (Fig. 4A, right), to an extent that mirrored the increased Fluc production observed in the unsupplemented mutant lysates (Fig. 4A, left). These are the results expected if adding purified Tma20/Tma22 fully complements the absence of these factors in the mutant extract.
It was shown previously that expression of human MCT-1 complemented translation-related defects in a tma20Δ yeast strain (Fleischer et al., 2006). To investigate this complementation further, we purified recombinant human MCT-1 protein and MCT-1/DENR dimer, both expressed in bacteria, and purified human eIF2D from HeLa cells. Remarkably, addition of MCT-1 alone strongly suppressed the high-level Fluc production given by uORF1luc mRNA in the tma64Δ/tma20Δ lysate but not in the tma64Δ/tma22Δ lysate (Suppl Fig. 6B), indicating efficient complementation of the increased reinitiation conferred by tma20Δ in the double mutant lysate. By contrast, the purified MCT-1/DENR dimer greatly reduced reinitiation in both mutant extracts, indicating complementation of tma20Δ or tma22Δ in the respective double mutants. Human eIF2D demonstrated similar, albeit less efficient, complementation of both mutant extracts, indicating that eIF2D can functionally substitute for yeast Tma64, and that the activities of Tma64 and Tma20/Tma22 are at least partially redundant. We further analyzed the effects of human MCT-1/DENR dimer on translation of the full set of mRNA constructs (Suppl. Fig. 6C) and observed suppression of reinitiation highly similar to that given by purified Tma20/Tma22 dimer (Fig. 4A, right).
Finally, we applied the same approach to examine the effects of eliminating Tma64/−22/−20 on translation of bicistronic reporter mRNAs harboring a full-length coding sequence for Renilla luciferase (Rluc) positioned upstream of the Fluc coding sequence (Fig. 4B). In the control construct, Rluc and Fluc cistrons were fused in the same reading frame without an intervening stop codon (RFluc1fus construct), while in the other two mRNAs Rluc overlapped Fluc by one 1 nt (AUGA; RFluc2), or the two coding sequences were separated by 67 nt of unrelated vector sequence (RFluc3). Fluc expression from mRNAs RFluc2 and RFluc3 was normalized to that of RFluc1fus in each extract, and results from the mutant extracts were expressed relative to those for the WT extract. Consistent with our previous observations, Fluc production was higher in both mutant extracts compared to WT for constructs RFluc2 and RFluc3, in which Fluc production depends on reinitiation following termination at the Rluc stop codon, but not for the RFluc1fus control mRNA (Fig. 4B, left, RFluc2 & RFluc3 vs. RFluc1fus). Also consistent with the results above, it appeared that the stimulation of reinitiation in the mutant extracts was diminished by increasing the intercistronic separation between Rluc and Fluc in RFluc3 versus RFluc2 (Fig. 4B, left). Finally, addition of recombinant Tma20/Tma22 complemented the elevated reinitiation observed for RFluc2 and RFluc3 in both mutant extracts to an extent that mirrored the increased reinitiation seen in the unsupplemented mutant extracts (Fig. 4B, right). In summary, we have reconstituted on reporter mRNAs in yeast extracts lacking Tma64/Tma20 the increased frequency of reinitiation following termination that we observed by ribosome profiling in the 3’UTRs of native mRNAs in the corresponding tmaΔΔ mutant cells.
The canonical initiation factor eIF1/SUI1 may not play a role in recycling of post-termination 40S ribosomes in vivo
Since it has been observed that 40S subunits can be recycled in a mammalian reconstituted system by the canonical initiation factors eIF1, eIF1A, eIF3 and eIF3j (Pisarev et al., 2007), we wondered whether, in addition to the Tma64/−20/−22 pathway, a redundant 40S recycling pathway utilizing eIF1 existed in the cell. Therefore, we also performed ribosome profiling on a sui1-L96P strain, encoding a mutant variant of eIF1, to test if eIF1 is involved in 40S recycling (As SUI1 is essential, a mutant was employed to partially abrogate its function). It has been previously shown that this mutation reduces the fidelity of start codon selection by weakening the binding of eIF1 to multiple components of the 43S pre-initiation complex (PIC) (Martin-Marcos et al., 2011; Obayashi et al., 2017). Dissociation of eIF1 from the PIC is a key step required for start codon selection during scanning, such that mutations that reduce eIF1 affinity for the PIC increase the probability of inappropriate eIF1 dissociation and attendant translation initiation at near-cognate start codons (the Sui− phenotype) (Hinnebusch, 2014). WT (SUI1) and sui1-L96P alleles of eIF1 were transformed into WT and tma64Δ/tma20Δ strains where the native eIF1 gene had been placed under the control of a GAL promoter and could therefore be repressed with glucose as a carbon source.
The eIF1 AUG start codon is in poor context (C at the −3 position), which enables translational autoregulation of the SUI1 gene. Low eIF1 levels, or Sui− mutations like sui1-L96P that weaken eIF1 binding to the PIC (Obayashi et al., 2017), decrease leaky scanning past the suboptimal SUI1 AUG codon and thus increase translation of SUI1 mRNA (Martin-Marcos et al., 2011). Consistent with these previous results, we observed increased ribosome occupancy on the SUI1 transcript in the sui1-L96P mutant relative to the WT SUI1 strain (Fig. 5A). Increased ribosome occupancies were also observed in upstream ORFs located in 5’UTRs that initiate with UUG in the sui1-L96P strain (e.g. within the YRO2 5’UTR, Fig. 5B), consistent with the relaxed discrimination against near-cognate start codons expected for the sui1-L96P strain.
Figure 5. The canonical initiation factor eIF1/SUI1 may not play a role in recycling of post-termination 40S ribosomes.
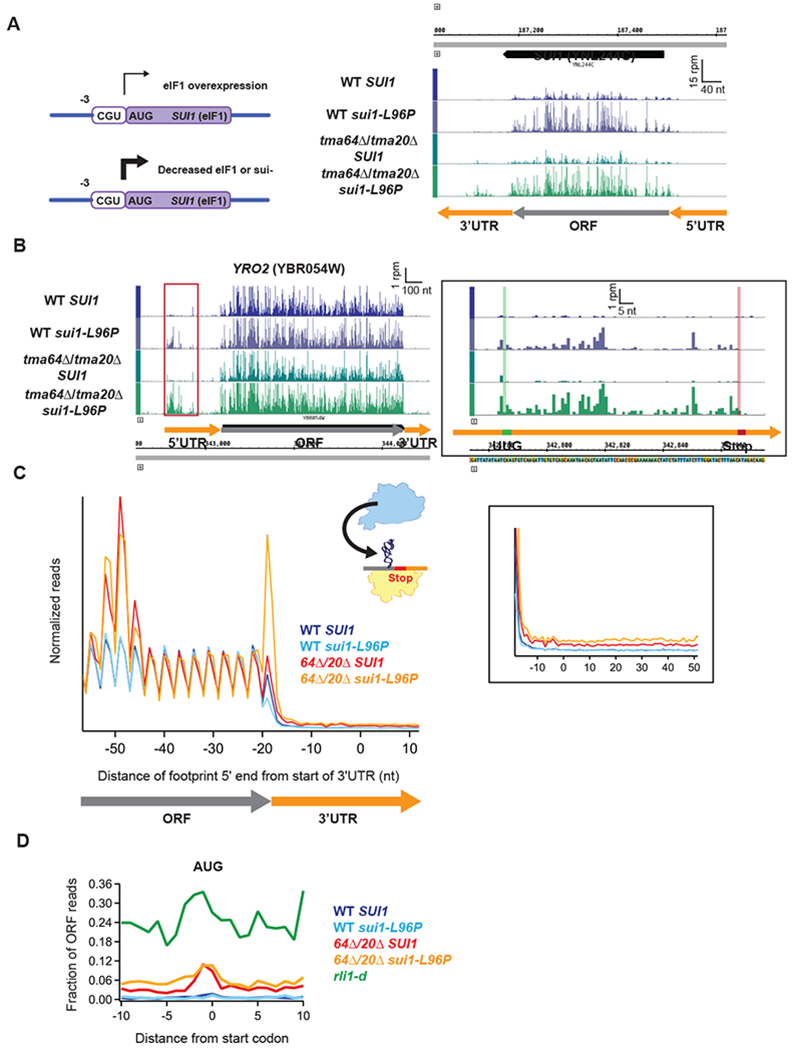
(A) Diagram showing the translational autoregulation of the SUI1 gene (left). The eIF1 AUG start codon is in poor context (C at the −3 position), making it sensitive to changes in the expression level of eIF1. (Right) Genome view of the SUI1 gene showing increased ribosome occupancy in the strains carrying the sui1-L96P allele, confirming the expected Sui− phenotype. Gene annotations correspond to ribosome P sites (AUG codons) or A sites (stop codons).
(B) Genome view of YRO2 showing increased ribosome occupancy in the 5’UTRs of the strains carrying the sui1-L96P allele. (Inset) Close-up of red box showing a UUG-initiated uORF. Reads are shifted so peaks approximately correspond to ribosome P sites.
(C) Normalized average ribosome footprint occupancy (each gene weighted equally) from all genes aligned at their stop codons for WT SUI1, WT sui1-L96P, tma64Δ/tma20Δ SUI1, and tma64Δ/tma20Δ sui1-L96P cells. The schematic depicts reassociation of a 60S subunit to a 40S subunit stalled at the stop codon. Footprint 5’ ends are plotted. (Inset) Zoomed-in view of the 3’UTR.
(D) Average fraction of ribosome occupancy in a window surrounding 3’UTR AUG codons normalized to ORF ribosome occupancy level (all frames included). Reads are shifted so peaks approximately correspond to P sites.
If eIF1 plays a role in 40S subunit recycling, we reasoned that the sui1-L96P eIF1 mutant might exhibit recycling defects similar to those observed in the tma double deletion strains. Ribosome profiling of the sui1-L96P mutant (grown in glucose medium) showed no stalling of the penultimate ribosome (Fig. 5C) and no increase in 3’UTR ribosome occupancy (Fig. 5C inset) observed in the tmaΔΔ strain, consistent with eIF1 playing no role in post-termination 40S recycling. As expected, given the low level of ribosomes observed in the 3’UTRs of the sui1-L96P mutant, no ribosome occupancy peak is observed at 3’UTR AUG codons either in the sui1-L96P or WT (SUI1) strain (Fig. 5D).
To investigate whether there is redundancy between Tma64/20/22 and eIF1, we created a triple mutant comprised of a tma64Δ/tma20Δ strain harboring the galactose-repressible native SUI1 gene and carrying the sui1-L96P allele. As observed for the sui1-L96P allele in the WT strain, increased ribosome occupancy was observed on the SUI1 transcript and on 5’UTR uORFs that initiate at UUG, consistent with a similar defect in translation initiation fidelity (Fig. 5A and 5B).
Surprisingly, the tma64Δ/tma20Δ strain carrying sui1-L96P showed strong 80S peaks at main ORF stop codons and increased 3’UTR ribosome occupancies genomewide relative to the tma64Δ/tma20Δ SUI1 strain (Fig. 5C). The 80S accumulation at stop codons observed in the triple mutant was unexpected given its absence in the tmaΔΔ mutant strain (Fig. 1A above). One possible explanation for both the accumulation of 80S complexes at stop codons and increased 3’UTR occupancies would be that 60S subunits can rejoin the unrecycled 40S ribosomes stalled at main ORF stop codons and the resulting 80S complexes would engage in non-canonical 80S reinitiation in 3’UTRs. Supporting this hypothesis, the sui1-L96P mutation leads to lower polysomes, which can be expected to increase the fraction of free 60S subunits in the cell available to reassociate with stalled 40S post-TCs. Moreover, comparing the fraction of 80S footprints that accumulate on 3’UTR AUG codons between the tma64Δ/tma20Δ/SUI1 double and tma64Δ/tma20Δ/sui1-L96P triple mutant reveals that the additional 3’UTR reads in the triple mutant do not increase the observed peak on AUG codons and instead generally increase reads throughout the 3’UTR (Fig. 5D, red vs. orange). As a result, the profile for the triple mutant in Fig. 5D appears to be intermediate between those of the tma64Δ/tma20Δ double mutant and the rli1-d strain (Fig. 5D, red vs. green). Thus, although the eIF1-L96P substitution increases ribosome occupancies in 3’UTRs, the increased 80S footprint densities do not appear to result from increased 40S reinitiation at AUG codons in the manner expected if eIF1 functioned redundantly with Tma64/−20/−22 in recycling 40S post-TCs.
Discussion
With a combined approach of in vivo ribosome profiling and analysis of 3’UTR peptide reporters in mutants lacking Tma64/eIF2D, Tma20/MCT-1, and Tma22/DENR, and in vitro analysis of reinitiation with luciferase-based reporters in mutant cell extracts, we have shown that elimination of these proteins from yeast results in reinitiation of translation downstream of stop codons and production of short peptides encoded in 3’UTRs. We therefore conclude that these factors promote ribosome recycling and prevent translation of 3’UTRs (Fig. 6).
Figure 6. Schematic model depicting the fate of post-TCs in Rli1-depleted cells and tmaΔΔ strains.
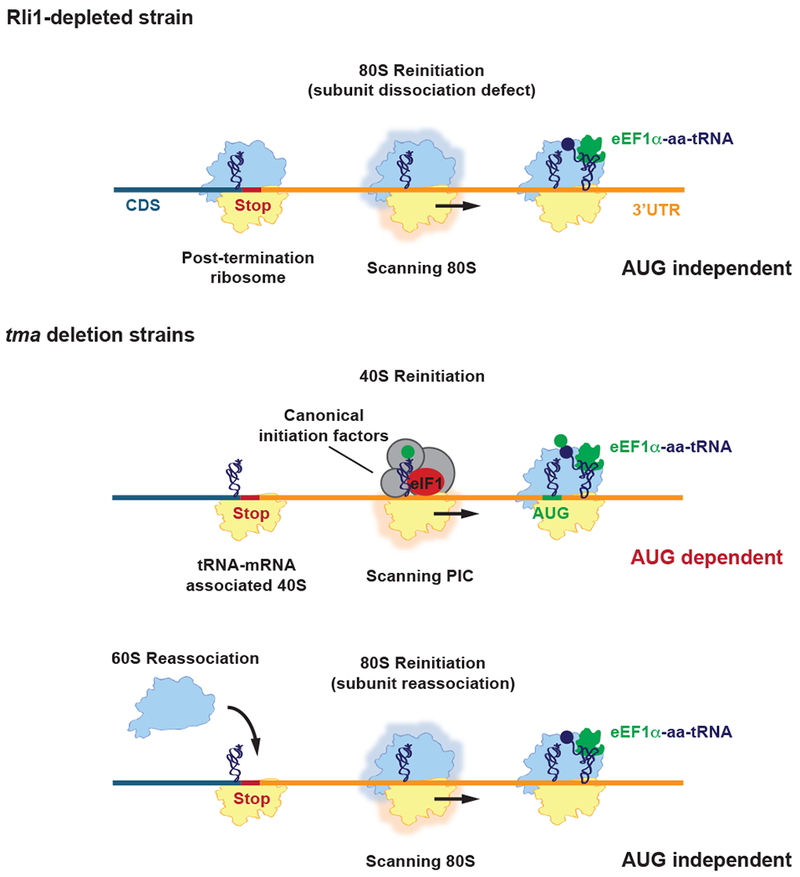
In Rli1-depleted cells, most post-TCs are not recycled, migrate a short distance from the stop codon, and reinitiate translation in an 80S AUG-independent manner, producing short 3’UTR encoded polypeptides (top row). In the tmaΔΔ strains, 60S subunit dissociation occurs, leaving a 40S post-TC stalled at the stop codon. The 40S post-TC can either form a 40S preinitiation complex (PIC) and scan into the 3’UTR, reinitiating translation at a canonical AUG start codon (middle row), or alternatively, reassociate with a 60S subunit and reinitiate translation in an 80S AUG-independent manner similar to what is observed in Rli1-depleted cells (bottom row).
Furthermore, our data offer some insight on the underlying mechanism of this recycling function. Most strikingly, we found that deletion of these “Tma” factors reduced the 80S footprint peak (Fig. 1A-B) at stop codons but evoked accumulation of 80S ribosomes upstream at the position expected from queuing of elongating ribosomes behind a stalled, un-recycled termination complex. These dual phenotypes would be expected for the loss of a factor specifically required for recycling the 40S subunit following removal of the 60S subunit by eRF1/Rli1. We therefore propose that elimination of Tma64/−20/−22 results in accumulation of unrecycled 40S subunits (not observable by 80S ribosome profiling methods) at stop codons. This model is bolstered by the observation of prominent 80S ribosome footprint peaks at 3’UTR AUG codons in the tmaΔΔ mutant strains (Fig. 2B). Ribosome profiling density from 80S ribosome footprints is known to exhibit peaks at AUG codons at the start of coding sequences (Weinberg et al., 2016). This signal therefore serves as a hallmark of the canonical 40S scanning process, whereby a 40S ribosome scans along the mRNA until finding a start codon and then recruits a 60S subunit to form an 80S initiation complex. We therefore conclude that the unrecycled 40S ribosomes at stop codons can also scan downstream 3’UTR sequences until reaching an AUG start codon and thereupon assemble an 80S initiation complex. Supporting this interpretation, the size of these peaks is modulated by the upstream sequence context (Fig. 2C) in a manner consistent with known models for 40S initiation (Shabalina et al., 2004). This model is further strengthened by our in vivo reporter data showing that peptides are generated from these 3’UTR sequences (Fig. 3A); and that in numerous cases removal of in-frame downstream AUG codons reduced or eliminated expression of the 3’UTR reporter peptides (Fig. 3B-C, Suppl Fig. 4D-E). Our in vitro translation data (Fig. 4) are also consistent with reinitiation at downstream AUG codons by 40S subunits normally recycled by the “Tma” proteins, as translation of AUG-initated Fluc coding sequences was enhanced in tmaΔΔ extracts for reporter mRNAs harboring uORFs or an upstream Rluc cistron. Thus, in aggregate, our results strongly support the view that the absence of the “Tma” factors leads to accumulation of unrecycled 40S ribosomes on stop codons, followed by reinitiation of downstream translation, in many cases, via a scanning mechanism resulting in AUG recognition (Fig. 6). This view implies that the “Tma” proteins promote recycling of the post-TC 40S subunit and may also impede reinitiation by occluding binding sites for eIF2 or other canonical initiation factors (e.g. eIF1 or eIF3) on the 40S and thereby prevent reassembly of an initiation complex.
Beyond a role in promoting reinitiation of post-termination 40S ribosomes at AUG codons, our in vivo reporter data also support a parallel 80S reinitiation mechanism occurring by an AUG-independent mechanism (Fig. 6). For some of our in vivo 3’UTR reporters, we found that eliminating AUG codons (and even in-frame near-cognate codons), had no effect on expression of the 3’UTR reporter peptides, suggesting that ribosomes reinitiate translation without the need for an AUG (or near-cognate) start codon. We have previously shown that Rli1 depletion results in widespread 80S reinitiation because unrecycled 80S subunits accumulate on stop codons in the absence of the 60S-dissociation activity of Rli1. These unrecycled 80S ribosomes presumably then “scan” into the 3’UTR and reinitiate in an AUG-independent manner and produce short 3’UTR-encoded peptides. Indeed, we observed 3’UTR reporter peptides (Suppl. Fig. 5) for several genes in tmaΔΔ cells shown previously to be produced at high levels in rli1-d cells via non-canonical 80S reinitiation (Young et al., 2015). To explain these cases, an appealing model is that the stalled 40S subunit is able to rejoin with a free 60S subunit and immediately scan downstream into the 3’UTR and carry out non-canonical 80S reinitiation. While such an 80S reinitiation mechanism is prevented during normal initiation in the 5’UTR by the anti-60S association activity of several canonical initiation factors that are bound to the 40S ribosome, here many of these factors are likely to be initially absent after 60S removal by eRF1/Rli1, offering a window of opportunity for the 60S to rejoin. It is possible that Rli1 would remain bound to the 40S ribosome at the stop codon, as observed in a recent structure obtained in the presence of AMP-PNP (Heuer et al., 2017), and thereby provide anti-association activity. In this case, a 60S reassociation model would require the release of Rli1 prior to 80S reinitiation. In WT cells, the “Tma” factors may therefore play a role in preventing 60S reassociation in the moments immediately after 60S dissociation. Recent structures of eIF2D or MCT-1/DENR bound to 40S subunits (Lomakin et al., 2017; Weisser et al., 2017) provide the structural basis for this anti-association activity. It appears that this sort of 80S reinitiation mechanism is increased in our triple mutant strain where both TMA64 and TMA20 are deleted and eIF1 is mutated. The addition of the eIF1 mutation to the tmaΔΔ strain may increase the propensity for 80S reinitiation because the ability of eIF1 to promote the competing pathway of 40S reinitiation is impaired and the number of free 60S subunits is overall higher in this strain owing to reduced canonical initiation.
While many mechanistic questions remain to be answered, our study offers compelling evidence that eIF2D/Tma64, MCT-1/Tma20, and DENR/Tma22 play a role in promoting 40S ribosome recycling and impeding reinitiation in 3’UTRs by distinct mechanisms. This finding therefore suggests new avenues for understanding how mutation of these factors or altered expression levels in humans is associated with diseases such as autism and B- and T-cell lymphoma. Similarly, changes in recycling at 5’UTR uORF stop codons in cells from these patients may also affect reinitiation at the main start codon of open reading frames and alter the expression of genes involved in neuronal migration in autism, and oncogenes or tumor suppressor genes involved in lymphoma. The role of unregulated reinitiation on non-coding sequences could therefore be an important avenue for further investigation in studies of disease etiology in these patients.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nicholas Guydosh (nicholas.guydosh@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All Saccharomyces cerevisiae strains used in this study are derived from the BY4741 background. They were maintained on either YPD plates or SC-Leu and SC-Ura plates for transformants. Cultures were grown at 30°C. Escherichia coli DH5a transformants were maintained on LB + 100 μg/ml Ampicillin plates. Cultures were grown at 37°C.
METHOD DETAILS
Yeast Strain Constructions
Yeast strains used in this study are listed in Table S1. The primers used for strain construction and verification are listed in Table S2.
The tmaΔΔ strains YDY10 (tma64Δ/tma20Δ) and YDY12 (tma64Δ/tma22Δ) were constructed in two steps. Firstly, the tma64Δ::kanMX4 deletion allele in F1946 (n4051) was converted to tma64Δ::hphMX4, by transformation with BamHI/SpeI digested pAG32 (carrying hphMX4) followed by selection on YPD containing 300 μg/mL Hygromycin B, to produce strain FZY793. TMA20 and TMA22 were then independently deleted in strain FZY793 by transformation with the tma20Δ::kanMX4 and tma22Δ::kanMX4 alleles PCR-amplified from yeast strains F1950 (n328) and F1948 (n6812), respectively, selecting transformants on YPD containing 200 μg/mL G418, to produce strains YDY10 and YDY12.
Strains YDY14 and YDY26 were constructed by transforming WT strain BY4741 and YDY10 (tma64Δ/tma20Δ) with the PGAL1 promoter sequence, PCR-amplified from plasmid pFA6a-His3MX6-PGAL1 using the primers PGAL1-SUI1f and PGAL1-SUI1r that appended sequences homologous to the desired integration site at the start of the SUI1 gene, and selecting on SCGAL-His.
WT and tma64Δ/tma20Δ strains harboring MYC13 tags in the 3’UTRs of candidate genes were constructed by transforming BY4741 and YDY10 with fragments containing MYC13 and HIS3 coding sequences, PCR-amplified from plasmid pFA6a-13myc-His3MX6 using the appropriate primers with 5’ sequences homologous to the desired integration sites within the candidate gene 3’UTRs, and selecting on SC-His. PCR amplification from chromosomal DNA and DNA sequencing of the amplified fragment was used to confirm the correct integration events.
Western analysis
MYC13 epitope tags were inserted just upstream of 3’UTR stop codons where an inframe, upstream AUG codon showed ribosome occupancy peaks in six candidate genes. These tagged genes were cloned onto high copy plasmids. The reporter constructs were transformed into WT and tma64Δ/tma20Δ strains and the transformed strains were grown to log-phase before preparation of WCEs.
Plasmid constructions
Plasmids used in this study are listed in Supplemental Tables S3 and S4. The primers used for plasmid construction and verification are listed in Table S2.
WT high copy plasmids containing MYC13-tagged candidate genes were constructed by PCR amplifying fragments from MYC13-tagged WT and tma64Δ/tma20Δ strains using primers that introduced SphI and XbaI restriction sites at either end, and inserting the resulting fragments between the SphI and XbaI sites of YEplac195. The cloned fragments contain the region 5’ of the candidate gene, so that the candidate gene is expressed from its endogenous promoter, the coding sequence for the candidate gene, the MYC13-tagged 3’UTR, and the ADH1 terminator sequence inserted by the MYC13 tagging cassette. Mutagenesis of the WT high copy plasmids to remove AUG start codons and near-cognate start codons was carried out using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent 210518).
To prepare TMA20 and TMA22 encoding plasmids for expression in E.coli, cDNAs corresponding to the TMA20 and TMA22 coding regions were amplified from total cDNA of BY4741 yeast strain by PCR with primer pairs TMA20for and TMA20rev, TMA22for and TMA22rev, respectively, and inserted into pET11c (Novagen) at NdeI and BamHI restriction sites to yield pET11c-TMA20 and pET11c-TMA22 expressing untagged versions of the corresponding proteins.
Ribosome footprint profiling
Preparation of Ribosome Footprint Libraries
Ribosome profiling was performed as previously described in (Guydosh and Green, 2014; Young et al., 2015).
BY4741, YDY10 (tma64Δ/tma20Δ), and YDY12 (tma64Δ/tma22Δ) were grown in YPD to an OD600 of 0.6, fast filtered and frozen in liquid nitrogen. Cells were lysed in a freezer mill in the presence of lysis buffer (20 mM Tris [pH8], 140 mM KCl, 1.5 mM MgCl2, 1% Triton X-100) containing 0.1 mg/ml CHX. Lysates were digested with 15U of RNase I (Ambion; AM2294) per OD for 1h at room temperature (25 °C), and separated on a 10-50% sucrose gradient to isolate monosomes. RNA was purified from monosome fractions using hot phenol chloroform, and 25-34 nt ribosome footprints were size selected from a 15% TBE-Urea gel. The purified RNA fragments were dephosphorylated using PNK (NEB; M0201L), and ligated to universal miRNA cloning linker (NEB; S1315S) using truncated T4 RNA ligase 2 (NEB; M0242L). The ligated RNA footprints were size selected on a 10% TBE-Urea gel, and reverse transcribed using Superscript III (Invitrogen; 18080044). The reverse transcribed footprints were again size selected on a 10% TBE-Urea gel, and circularized using CircLigase ssDNA Ligase (Epicentre; CL4111K). Ribosomal RNA footprints were removed from the circularized libraries by oligonucleotide subtraction hybridization. The subtracted libraries were amplified by PCR to add unique 6 nt indexes for each library and common Illumina primer and flow cell binding regions. Libraries were assessed and quantified by BioAnalyzer using the High Sensitivity DNA Kit (Agilent 5067-4626). Sequencing was performed on an Illumina HiSeq2000 or 2500 machine at the NHLBI DNA Sequencing and Genomics Core at NIH (Bethesda, MD).
YDY14 (PGAL1-SUI1) and YDY26 (tma64Δ/tma22Δ PGAL1-SUI1) transformed with pJCB101 (sc SUI1) and pPMB03 (sc sui1-L96P) were grown in SC-Leu to an OD600 of 0.6, fast filtered and frozen in liquid nitrogen. Libraries were constructed as described above.
Data shown from the SUP4-o and rli1-d strains are identical to those previously published (Guydosh and Green, 2014; Young et al., 2015).
Read Preparation and Sequence Alignment
The R64-1-1 S288C reference genome assembly (SacCer3) was used for all analysis (Saccharomyces Genome Database Project). De-multiplexed sequences were first processed in some cases to remove low quality reads (any position with Phred score <20 or assigned N). Following a search for the linker (footprints), contaminating ladder oligos were removed and alignment to the RNA gene database (http://downloads.yeastgenome.org/sequence/S288C_reference/rna/archive/rna_coding_R64-1-1_20110203.fasta.gz) was performed to remove noncoding contaminants. Remaining reads were then aligned to the genome and those that did not align were aligned to a database of annotated splice junctions. Footprint reads left over at this stage were then trimmed of consecutive 3’ As and realigned to the genome and splice junction database. Matches in these various alignment rounds were pooled for calculation of ribosome occupancy.
All reads that aligned to multiple coding sequences were discarded. Read occupancy was set by giving one count per read 5’ end and in some cases shifted to align with various active sites in the ribosome (i.e. start of the A site) as described below. Read counts were then normalized to units of reads per million (rpm) (by dividing by the total number of million mapped reads in a sample). Alignments were performed with Bowtie 1.1.2 or 1.01 (Langmead et al., 2009) and included the parameters: - y -a -m 1 --best --strata. Read lengths were assessed with the FastQC software (Babraham Bioinformatics). Two mismatches were permitted in the alignments to coding sequences. All other analysis software used Biopython 1.58 or 1.63. Unless noted otherwise, data from biological replicate samples were pooled for all analysis. Python code for the three basic analyses (Gene average, gene quantitation, and position average plots) is provided (Supplementary file). Statistics showing the number of reads passing each step of processing are given in Table S5.
Analysis of Aligned Reads
In general, ORFs marked dubious or those that overlapped with other transcripts were ignored in the analysis. Annotations for 3’ UTRs (Nagalakshmi et al., 2008) that used coordinates from the R64-1-1 genome assembly were downloaded from Saccharomyces Genome Database Project.
Gene-wide occupancy (quantitation) was quantitated into density units of reads per kilobase per million mapped reads (rpkm) by taking reads mapping to an annotated sequence and dividing by the gene length in kilobases. For gene quantitation analyses, 3’ UTRs were extended 25 nt downstream from their annotated endpoints in order to ensure all ribosomes that partially protected poly(A) sequences were accounted for. Reads that mapped in the first or last 15 nt of genes were left out to eliminate artefacts associated with initiating and terminating ribosomes. To be included in the analysis, a threshold of 5 rpkm was required of ORF reads and 0.5 rpkm for 3’UTR reads. Reads were shifted by 13 nt to correspond to the center of the P site.
Gene-average plots (“metagene”) were constructed by excluding genes with features that were smaller than the window size (300 nt upstream of ORF and 100 nt of downstream region). In all cases, a minimal threshold of 5 rpkm was required from the ORF reads and genes were equally weighted according to reads in the coding sequence.
Position-average plots (“metacodon”) were similarly created by averaging together (with equal weight) reads in a window about every occurrence of a particular motif in a 3’UTR. Genes with no reads in the 3’ UTR were excluded from these analyses. Reads were normalized to the total reads present in the ORF and only genes exceeding a threshold of 5 rpkm were used. Reads were shifted by 12 nt to correspond to the start of the A site.
Biochemical Techniques
Western analysis was conducted using a mouse monoclonal antibody for c-myc (Roche; 1167203001), a rabbit polyclonal antibody for GCD6 (Bushman et al., 1993), and a rabbit polyclonal antibody for Histone H3 (Abcam; Ab1791).
Real-time quantitative RT-PCR (qRT-PCR) analysis of high copy reporter constructs
The BY4741(WT) and tma64Δ/tma20Δ strains were transformed with the RPL14A, RPS30B, and IPP1_tag1 WT and “No AUG/NUG” reporters and grown in triplicate in SC-Ura medium to an OD600 of 0.8-1 and snap frozen on dry ice.
RNA isolation
Cell pellets were resuspended in 400 μl of AE Buffer (50 mM sodium acetate, 10 mM EDTA, pH 5.2). 40 μl of 10% SDS (1/10 vol.) was added to each sample followed by 500 μl of pre-heated phenol equilibrated with AE Buffer. Samples were mixed by vortexing and incubated at 65°C for 5 minutes. After incubation, the samples were rapidly cooled in a dry-ice/ethanol bath and spun at 8000 rpm for 10 minutes at room temperature. The bottom phenol phase was removed and the above phenol extraction was repeated. After the second spin the aqueous phase was transferred to new tubes and extracted twice with Phenol:Chloroform:Isoamyl Alcohol (25:24:1) and once with Chloroform:Isoamyl Alcohol (24:1). The aqueous phase was transferred to new tubes and precipitated overnight at −20°C with 3M sodium acetate, pH 5.2 (1/10 vol.) and ethanol (2.5 vol.). RNA was pelleted by spinning at 4°C for 10 minutes at top speed in a microcentrifuge. The RNA pellets were washed with 70% ethanol, air dried, and resuspended in RNAse-free water. Genomic DNA contamination was removed from the RNA samples using RNase-free DNaseI (Roche; 04-716-728-001) according to the manufacturer’s instructions. For each sample, 5.5 μg of total RNA was digested with 1U of RNase-free DNase in a 10 μl reaction.
Real-time quantitative RT-PCR (qRT-PCR)
The level of mRNA for the RPL14A, RPS30B, and IPP1_tag1 reporters in the BY4741 and tma64Δ/tma20Δ strains, relative to the amount of actin (ACT1) mRNA, was quantified by qRT-PCR analysis. cDNA was synthesized from 1 μg of RNA using Superscript™ III First-Strand Synthesis SuperMix (Invitrogen; 11752-050) according to the manufacturer’s protocol. The synthesized first-strand cDNA was diluted 1:10, and 2 μl of the diluted cDNA was used for subsequent real-time PCR amplification using the Stratagene MX3000P and Brilliant III SYBR® Green QPCR Master Mix (Stratagene; 600882) according to the manufacturer’s instructions. The primers used in the qRT-PCR analysis for the reporters are listed in Table S2. The primers were designed to span from the main ORF to the beginning of the 3’UTR Myc13-tag for each reporter to ensure that full-length reporter mRNAs were being quantified. The real-time PCR reactions were carried out in triplicate for each cDNA sample to obtain average Ct values. Fold change was calculated using the 2−ΔΔCt method as described in (Schmittgen and Livak, 2008).
Preparation of capped polyadenylated reporter mRNAs
For synthesis of polyadenylated mRNAs encoding the firefly and Renilla luciferases, 50T-tailed PCR products were used as templates (Dmitriev et al., 2007), as described below. The templates for mRNAs of uORFluc series were prepared on the basis of plasmid pSV40-Fluc (Dmitriev et al., 2007) by using uORF0, uORF1, uORF2 or uORF3 as a T7-promoter containing forward primer, and FLA1 as the reverse one. For RFluc1fus and RFluc2, the corresponding plasmids pRFluc1fus and pRFluc2 gifted by I.Osterman (MSU) were used, while RFluc3 was prepared on the basis of pRF plasmid (Dmitriev et al., 2007); in these three cases primers RFluc1 and FLA2 were taken for generation of the templates. For transcription, RiboMAX kit (Promega) was utilized. The resulting transcripts were precipitated with 2M LiCl, then capped with Vaccinia Capping System (NEB) and precipitated with LiCl once again. All mRNA transcripts were checked for integrity by denaturing urea polyacrylamide gel electrophoresis.
In vitro translation in the yeast cell-free system
For in vitro translation reactions we used a self-made yeast cell-free system, as described in (Prokhorova et al., 2016). Yeast cultures were grown in 0.3l of YPD in 1l flasks at 30°C, 180 rpm, to OD600 = 1.2 and harvested by centrifugation at 3000g, 5 min, RT. Cell were washed with mQ H2O in 50 ml Falcon tubes and centrifuged at 2300g, 5 min, RT. This usually produced ~2 g of cells that were resuspended in 20 ml (10 ml per 1 g of cells) of Resuspension buffer (1M sorbitol, 2mM EDTA, 14 mM β-mercaptoethanol) and incubated for 30 min at RT, rotating manually from time to time. After centrifugation (2300g, 5 min, RT), the pellet was resuspended in 10 ml of 1M sorbitol and 100 μl lyticase (10 000 U/ml) were added and incubated for 45 min at RT, rotating periodically. Then spheroplasts were pelleted by centrifugation (2300g, 5 min, RT), put into ice, resuspended in 2 ml of ice-cold Buffer A (30 mM HEPES-KOH, pH 7.4, 100 mM KOAc, 2 mM Mg(OAc)2, 2 mM DTT) and disrupted in 2 ml Dounce homogenizer (Kimble Chase #885300-0002 , pestle B, 50 strokes). The lysate was transferred to 1.5 ml tubes and centrifuged at 20000g, 15 min at 3°C, the supernatant was supplemented with creatine phosphokinase (10 mg/ml) to final concentration 50 μg/ml and stored at −80°C. Translation reactions were performed in a total volume of 15 μl, containing 7.5 μl of the extract, translation buffer (25 mM Hepes-KOH pH 7.4, 2 mM DTT, 3 mM Mg(OAc)2, 12 mM creatine phosphate, 1 mM ATP, 0.4 mM GTP, 126 mM KOAc, and 50 μM of each amino acid), 3 u of RiboLock RNase inhibitor (Thermo Fisher Scientific), 1 mM D-luciferin and 0.12 pmol mRNA. In experiments with recombinant proteins, 1 μl of protein solution diluted in buffer A100 (20 mM Tris-HCl, pH 7.4, 100 mM KCl, 10% glycerol, 1 mM DTT), or just the buffer, were added, as indicated. Translation mixtures were incubated in a white 384-well plate (F-bottom, non-binding polystyrol, Grenier GR-781904), and covered by a PCR plate seal at 25 °C in the TECAN Infinite 200Pro reader with continuous measurement of the luciferase activity (integration time 3 s). After 1 h of incubation, luciferase accumulation rates were calculated (as an increment of light intensity units per second) and the maximum rates (Vmax) were found (usually at a time point nearly 15 min of incubation). These Vmax were taken for further analysis, as described in the text. All the experiments were repeated with two independently prepared batches of the extracts and at least two preparations of mRNAs (totally at least 3 experiments), each in triplicate. The values were normalized as indicated in the main text, and SDs were calculated.
Recombinant proteins expression and purification
E.coli strain Rosetta pLacI-RARE was used to express recombinant untagged proteins. Protein dimer components were expressed separately and then combined at the stage of cell lysates for further purification. Cells were grown in LB medium containing 50 μg/ml ampicillin and 34 μg/ml chloramphenicol. For yeast TMA20 and TMA22 proteins, the culture was initially grown at 37°C and when the OD600 reached 0.6–0.8, it was cooled down to 25°C and subsequently induced with 0.3 mM IPTG for 5 h at 25°C. For human DENR, the expression was induced at OD600 0.6–0.8 by addition of IPTG to the final concentration of 0.5 mM. Cells were incubated for additional 3 h at 37°C before harvesting. To express MCT-1, cells were initially grown at 37°C to the OD600 0.8–1.0, then cell culture was cooled down to 18°C and IPTG was added to the final concentration of 0.5 mM. Cells were grown overnight at 18°C and then harvested by centrifugation. Cells were resuspended in the buffer (50 mM Tris-HCl, pH 6.8, 200 mM NaCl, 10 mM β-mercaptoethanol, 0.1 mM PMSF) and sonicated. Cellular debris was removed by centrifugation and supernatants containing TMA20 and TMA22, or MCT-1 and DENR were mixed together and loaded on the S-Sepharose column equilibrated with Buffer A (50 mM Tris–HCl, pH 6.8, 0.1 mM EDTA, 10 mM β- mercaptoethanol) containing 50 mM NaCl. TMA20/TMA22 or MCT-1/DENR dimers were eluted from the column by linear gradient of NaCl (50–1000 mM) in Buffer A. Fractions containing both proteins were combined and then diluted tenfold with Buffer A and loaded on the Heparin-Sepharose column. The dimers were eluted as described above. Finally, the dimers were purified on a Superdex 75 column (GE Healthcare) equilibrated with Buffer B (20 mM Tris-HCl, pH 7.4, 100 mM KCl, 10 mM NH4OAc, 5 % glycerol, 2 mM DTT). TMA20/TMA22 and MCT-1/DENR dimers were concentrated to 250 μM, frozen in liquid nitrogen in small aliquots and stored at −80°C. Individual MCT-1 protein was purified by the same protocol.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of immunoblot bands was performed using ImageJ (Version 2.00-rc-43/1.51p). Graphs show a minimum of 4 biological and technical repeats. Error bars are +/− SEM. Statistical significance was calculated using 2-tailed students T-tests. For blots where quantitation was not performed, at least two individual replicates were performed to verify the result was reproducible. Graphs were made with Igor Pro 7.00 (Wavemetrics). Boxplot analysis performed and created by Rstudio ver. 1.1.447.
DATA AND SOFTWARE AVAILABILTY
Custom software is available as a supplementary file. Raw and analyzed data have been deposited in the NCBI GEO database under the accession number GSE108942. Reviewers may access the data with this token: mtibemqajdqrbkd
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Monoclonal Antibody c-Myc (9E10) | Roche | 1167203001 |
| Rabbit Polyclonal Antibody GCD6 | Covance (CM002) | (Bushman et al., 1993) |
| Rabbit Polyclonal Anti-Histone H3 Antibody | Abcam | Ab1791 |
| Bacterial and Virus Strains | ||
| DH5alpha | NEB | C2987H |
| Rosetta pLacI-RARE | Novagen | 71404 |
| Biological Samples | ||
| N/A | N/A | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma | C7698 |
| RNase I | Ambion | AM2294 |
| PNK | NEB | M0201L |
| Universal miRNA cloning linker | NEB | S1315S |
| T4 RNA ligase 2, truncated | NEB | M0242L |
| Superscript III Reverse Transcriptase | Invitrogen | 18080044 |
| Lyticase | Sigma | L2524 |
| Creatine kinase | Sigma | 000000010127566001 |
| Creatine phosphate | Sigma | 10621714001 |
| RiboLock RNase Inhibitor | Thermo Fisher Scientific | EO0381 |
| D-luciferin (Beetle Luciferin, Potassium Salt) | Promega | E1602 |
| DNase I recombinant, RNase-free | Roche | 04-716-728-001 |
| SuperScript III First-Strand Synthesis SuperMix for qRT-PCR | Invitrogen | 11752-050 |
| Critical Commercial Assays | ||
| QuikChange Lightning Site-Directed Mutagenesis Kit | Agilent Technologies | 210518 |
| CircLigase ssDNA Ligase Kit | Epicentre | CL4115K |
| Agilent High Sensitivity DNA Kit | Agilent | 5067-4626 |
| RiboMAX Large Scale RNA Production Systems | Promega | P1300 |
| Vaccinia Capping System | NEB | M2080S |
| Brilliant III Ultra-Fast SYBR Green QPCR Master Mix | Agilent Technologies | 600882 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE108942 |
| Experimental Models: Cell Lines | ||
| N/A | N/A | N/A |
| Experimental Models: Organisms/Strains | ||
| A full list of yeast strains used in this study is provided in Table S1. | This paper | N/A |
| Oligonucleotides | ||
| A full list of oligonucleotides used in this study is provided in Table S2. | This paper | N/A |
| Recombinant DNA | ||
| A full list of plasmids and high copy reporter plasmids used in this study are provided in Tables S3 and S4. | This paper | N/A |
| Software and Algorithms | ||
| Igor Pro Version 7.00 | Wavemetrics | 15-500 |
| ImageJ Version 2.00-rc-43/1.51p | https://imagej.nih.gov/ij/ | N/A |
| Position average and metagene | This paper | makeposavg makeavggene |
| Bowtie | Github | (Langmead et al., 2009) |
| Other | ||
Supplementary Material
Table S2. Oligonucleotides used in this study, related to STAR Methods
Data File 1. Source code for python routines, related to STAR Methods.
Highlights.
Tma64 (eIF2D) and Tma20 (MCT-1)/Tma22 (DENR) recycle 40S subunits at stop codons
An unrecycled 40S can reinitiate translation at AUGs after uORFs or in the 3 ’UTR
Unrecycled 40S subunits can reassociate with the 60S and undergo 80S reinitiation
Tma64 (eIF2D) and Tma20 (MCT-1)/Tma22 (DENR) may block 60S or eIF binding to the 40S
ACKNOWLEDGMENTS
We thank Ilya Osterman for RFluc1fus and RFluc2 constructs. We are grateful to Ivan Kulakovskiy, Andrey Lando and Pavel Sinitcyn for their assistance in data analysis, and Dmitri Andreev and Ilya Terenin for discussing results. We are grateful for feedback from Tom Dever and Jon Lorsch, and other members of the Hinnebusch, Dever, and Guydosh labs.
FUNDING
The work was supported by a grant of the Russian Federation (14.W03.31.0012 to S.E.D.). The experiments with MCT-1/DENR proteins were supported by the Russian Science Foundation (RSF 14-50-00060 to S.E.D.). This research was supported by the Intramural Research Programs of the NIH, the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD, HD001004 to A.G.H.) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK, DK075132 to N.R.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Archer SK, Shirokikh NE, Beilharz TH, and Preiss T (2016). Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature 535, 570–574. [DOI] [PubMed] [Google Scholar]
- Becker T, Franckenberg S, Wickles S, Shoemaker CJ, Anger AM, Armache JP, Sieber H, Ungewickell C, Berninghausen O, Daberkow I, et al. (2012). Structural basis of highly conserved ribosome recycling in eukaryotes and archaea. Nature 482, 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman JL, Foiani M, Cigan AM, Paddon CJ, and Hinnebusch AG (1993). Guanine nucleotide exchange factor for eukaryotic translation initiation factor 2 in Saccharomyces cerevisiae: interactions between the essential subunits GCD2, GCD6, and GCD7 and the regulatory subunit GCN3. Mol Cell Biol 13, 4618–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. (2010). The genetic landscape of a cell. Science 327, 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev SE, Andreev DE, Terenin IM, Olovnikov IA, Prassolov VS, Merrick WC, and Shatsky IN (2007). Efficient translation initiation directed by the 900-nucleotide-long and GC-rich 5’ untranslated region of the human retrotransposon LINE-1 mRNA is strictly cap dependent rather than internal ribosome entry site mediated. Mol Cell Biol 27, 4685–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev SE, Terenin IM, Andreev DE, Ivanov PA, Dunaevsky JE, Merrick WC, and Shatsky IN (2010). GTP-independent tRNA delivery to the ribosomal P-site by a novel eukaryotic translation factor. J Biol Chem 285, 26779–26787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer TC, Weaver CM, McAfee KJ, Jennings JL, and Link AJ (2006). Systematic identification and functional screens of uncharacterized proteins associated with eukaryotic ribosomal complexes. Genes Dev 20, 1294–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. (2002). Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415, 141–147. [DOI] [PubMed] [Google Scholar]
- Gietz RD, and Sugino A (1988). New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, and McCusker JH (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15, 1541–1553. [DOI] [PubMed] [Google Scholar]
- Grant CM, Miller PF, and Hinnebusch AG (1994). Requirements for intercistronic distance and level of eukaryotic initiation factor 2 activity in reinitiation on GCN4 mRNA vary with the downstream cistron. Mol Cell Biol 14, 2616–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guydosh NR, and Green R (2014). Dom34 rescues ribosomes in 3’ untranslated regions. Cell 156, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas MA, Ngo L, Li SS, Schleich S, Qu Z, Vanyai HK, Cullen HD, Cardona-Alberich A, Gladwyn-Ng IE, Pagnamenta AT, et al. (2016). De Novo Mutations in DENR Disrupt Neuronal Development and Link Congenital Neurological Disorders to Faulty mRNA Translation Re-initiation. Cell Rep 15, 2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer A, Gerovac M, Schmidt C, Trowitzsch S, Preis A , Kotter P, Berninghausen O, Becker T, Beckmann R, and Tampe R (2017). Structure of the 40S-ABCE1 post-splitting complex in ribosome recycling and translation initiation. Nat Struct Mol Biol 24, 453–460. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG (2014). The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem 83, 779–812. [DOI] [PubMed] [Google Scholar]
- Hussain T, Llacer JL, Fernandez IS, Munoz A, Martin-Marcos P, Savva CG, Lorsch JR, Hinnebusch AG, and Ramakrishnan V (2014). Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell 159, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1987). Effects of intercistronic length on the efficiency of reinitiation by eucaryotic ribosomes. Mol Cell Biol 7, 3438–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (2002). Pushing the limits of the scanning mechanism for initiation of translation. Gene 299, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGrandeur T, and Parker R (1999). The cis acting sequences responsible for the differential decay of the unstable MFA2 and stable PGK1 transcripts in yeast include the context of the translational start codon. RNA 5, 420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memoryefficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llacer JL, Hussain T, Marler L, Aitken CE, Thakur A, Lorsch JR, Hinnebusch AG, and Ramakrishnan V (2015). Conformational Differences between Open and Closed States of the Eukaryotic Translation Initiation Complex. Mol Cell 59, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomakin IB, Stolboushkina EA, Vaidya AT, Zhao C, Garber MB, Dmitriev SE, and Steitz TA (2017). Crystal Structure of the Human Ribosome in Complex with DENR-MCT-1. Cell Rep 20, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Martin-Marcos P, Cheung YN, and Hinnebusch AG (2011). Functional elements in initiation factors 1, 1A, and 2beta discriminate against poor AUG context and non-AUG start codons. Mol Cell Biol 31, 4814–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PP, and Hinnebusch AG (1986). Multiple upstream AUG codons mediate translational control of GCN4. Cell 45, 201–207. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, and Snyder M (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi E, Luna RE, Nagata T, Martin-Marcos P, Hiraishi H, Singh CR, Erzberger JP, Zhang F, Arthanari H, Morris J, et al. (2017). Molecular Landscape of the Ribosome Pre-initiation Complex during mRNA Scanning: Structural Role for eIF3c and Its Control by eIF5. Cell Rep 18, 2651–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Hellen CU, and Pestova TV (2007). Recycling of eukaryotic posttermination ribosomal complexes. Cell 131, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisarev AV, Skabkin MA, Pisareva VP, Skabkina OV, Rakotondrafara AM, Hentze MW, Hellen CU, and Pestova TV (2010). The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol Cell 37, 196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokhorova IV, Akulich KA, Makeeva DS, Osterman IA, Skvortsov DA, Sergiev PV, Dontsova OA, Yusupova G, Yusupov MM, and Dmitriev SE (2016). Amicoumacin A induces cancer cell death by targeting the eukaryotic ribosome. Sci Rep 6, 27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosniak M, Dierov J, Okami K, Tilton B, Jameson B, Sawaya BE, and Gartenhaus RB (1998). A novel candidate oncogene, MCT-1, is involved in cell cycle progression. Cancer Res 58, 4233–4237. [PubMed] [Google Scholar]
- Rabl J, Leibundgut M, Ataide SF, Haag A, and Ban N (2011). Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331, 730–736. [DOI] [PubMed] [Google Scholar]
- Reinert LS, Shi B, Nandi S, Mazan-Mamczarz K, Vitolo M, Bachman KE, He H, and Gartenhaus RB (2006). MCT-1 protein interacts with the cap complex and modulates messenger RNA translational profiles. Cancer Res 66, 8994–9001. [DOI] [PubMed] [Google Scholar]
- Schleich S, Acevedo JM, Clemm von Hohenberg K, and Teleman AA (2017). Identification of transcripts with short stuORFs as targets for DENR*MCTS1-dependent translation in human cells. Sci Rep 7, 3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleich S, Strassburger K, Janiesch PC, Koledachkina T, Miller KK, Haneke K, Cheng YS, Kuechler K, Stoecklin G, Duncan KE, et al. (2014). DENR-MCT-1 promotes translation re-initiation downstream of uORFs to control tissue growth. Nature 512, 208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, and Livak KJ (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Schwartz DC, and Parker R (1999). Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol 19, 5247–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina SA, Ogurtsov AY, Rogozin IB, Koonin EV, and Lipman DJ (2004). Comparative analysis of orthologous eukaryotic mRNAs: potential hidden functional signals. Nucleic Acids Res 32, 1774–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Murray J, Brown A, Taunton J, Ramakrishnan V, and Hegde RS (2016). Decoding Mammalian Ribosome-mRNA States by Translational GTPase Complexes. Cell 167, 1229–1240 e1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, Hsu HL, Evens AM, Gordon LI, and Gartenhaus RB (2003). Expression of the candidate MCT-1 oncogene in B- and T-cell lymphoid malignancies. Blood 102, 297–302. [DOI] [PubMed] [Google Scholar]
- Shoemaker CJ, and Green R (2011). Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc Natl Acad Sci U S A 108, E1392–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skabkin MA, Skabkina OV, Dhote V, Komar AA, Hellen CU, and Pestova TV (2010). Activities of Ligatin and MCT-1/DENR in eukaryotic translation initiation and ribosomal recycling. Genes Dev 24, 1787–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, and Bartel DP (2016). Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep 14, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisser M, Schafer T, Leibundgut M, Bohringer D, Aylett CHS, and Ban N (2017). Structural and Functional Insights into Human Re-initiation Complexes. Mol Cell 67, 447–456 e447. [DOI] [PubMed] [Google Scholar]
- Williams NP, Mueller PP, and Hinnebusch AG (1988). The positive regulatory function of the 5’-proximal open reading frames in GCN4 mRNA can be mimicked by heterologous, short coding sequences. Mol Cell Biol 8, 3827–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DJ, Guydosh NR, Zhang F, Hinnebusch AG, and Green R (2015). Rli1/ABCE1 Recycles Terminating Ribosomes and Controls Translation Reinitiation in 3’UTRs In Vivo. Cell 162, 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Oligonucleotides used in this study, related to STAR Methods
Data File 1. Source code for python routines, related to STAR Methods.