

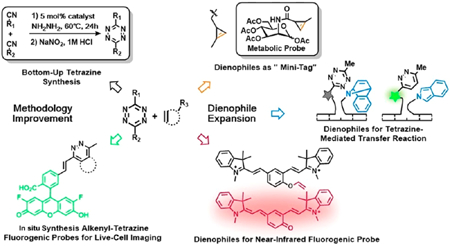
CONSPECTUS:
Bioorthogonal chemistry has found increased application in living systems over the past decade. In particular, tetrazine bioorthogonal chemistry has become a powerful tool for imaging, detection, and diagnostic purposes, as reflected in the increased number of examples reported in the literature. The popularity of tetrazine ligations are likely due to rapid and tunable kinetics, the existence of high quality fluorogenic probes, and the selectivity of reaction. In this Account, we summarize our recent efforts to advance tetrazine bioorthogonal chemistry through improvements in synthetic methodology, with an emphasis on developing new routes to tetrazines and expanding the range of useful dienophiles. These efforts have removed specific barriers that previously limited tetrazine ligations and have broadened their potential applications.
Among other advances, this Account describes how our group discovered new methodology for tetrazine synthesis by developing a Lewis acid-promoted, one-pot method for generating diverse symmetric and asymmetric alkyl tetrazines with functional substituents in satisfactory yields. We attached these tetrazines to microelectrodes and succeeded in controlling tetrazine ligation by changing the redox state of the reactants. Using this electrochemical control process, we were able to modify an electrode surface with redox probes and enzymes in a site-selective fashion.
This Account also describes how our group improved the ability of tetrazines to act as fluorogenic probes by developing a novel elimination-Heck cascade reaction to synthesize alkenyl tetrazine derivatives. In this approach, tetrazine was conjugated to fluorophores to produce strongly quenched probes that, after bioorthogonal reaction, are “turned on” to enhance fluorescence, in many cases by >100-fold. These probes have allowed no-wash fluorescence imaging in living cells and intact animals.
Finally, this Account reviews our efforts to expand the range of dienophile substrates to make tetrazine bioorthogonal chemistry compatible with specific biochemical and biomedical applications. We found that methylcyclopropene is sufficiently stable and reactive in the biological milieu to act as an efficient dienophile. The small size of the reactive tag minimizes steric hindrance, allowing cyclopropene to serve as a metabolic reporter group to reveal biological dynamics and function. We also used norbornadiene derivatives as strained dienophiles to undergo tetrazine-mediated transfer (TMT) reactions involving tetrazine ligation followed by a retro-Diels–Alder process. This TMT reaction generates a pair of nonligating products. Using nucleic acid-templated chemistry, we have combined the TMT reaction with our fluorogenic tetrazine probes to detect endogenous oncogenic microRNA at picomolar concentrations. In a further display of dienophile versatility, we used a novel vinyl ether to cage a near-infrared fluorophore in a nonfluorescent form. Then we opened the cage in a “click to release” tetrazine bioorthogonal reaction, restoring the fluorescent form of the fluorophore. Combining this label with a corresponding nucleic acid probe allowed fluorogenic detection of target mRNA.
In summary, this Account describes improvements in tetrazine and dienophile synthesis and application to advance tetrazine bioorthogonal chemistry. These advances have further enabled application of tetrazine ligation chemistry, not only in fundamental research but also in diagnostic studies.
Graphical Abstract
INTRODUCTION
Chemical biology is construed as a scientific research field aimed at developing and using chemical tools for biological studies.1 Numerous chemistries and technologies have emerged in recent decades to elucidate functions of biomolecules and probe physiological processes. For example, proteins of interest in live cells and transgenic animals can be visualized with high spatiotemporal resolution through fusion with various fluorescent proteins.2 The interaction between bioactive compounds and endogenous targets can be profiled using photoaffinity probes, which can facilitate drug development.3 These advancements are helping drive efforts by researchers to interrogate all classes of biomolecules in living animal models, ultimately for improving our understanding of biological processes and to develop diagnostics and therapeutics.4 In the early 2000s, the Bertozzi group reported a modified Staudinger reaction on cell surface.5 This pioneering work opened up a novel research field, bioorthogonal chemistry, in which unnatural partners can react efficiently, selectively and rapidly in the biological milieu in a nonintrusive manner.6 Since then, a variety of bioorthogonal chemistries have been developed and applied in numerous chemical biology studies.7
Tetrazine bioorthogonal reactions refer to inverse electron-demand Diels–Alder reactions between 1,2,4,5-tetrazine and diverse dienophiles. Tetrazine bioorthogonal chemistry was independently reported by two groups in 2008.8,9 The rapid kinetics of these reactions ensures efficient labeling even at the low concentrations typically found in vivo10 and has led to tetrazine bioorthogonal reactions becoming a commonly used bioconjugation tool.
Within the last 10 years, tetrazine bioorthogonal chemistry has been applied to diverse areas of chemical biology.11 Weissleder and co-workers have elegantly implemented this chemistry for various biological labeling and imaging studies.10 Other pioneering groups have implemented metabolic probes for various biomolecules.12–15 The research groups of Chin and others have extensively studied protein dynamics and functions using genetic code expansion strategies.16–19 The Fox laboratory has been pushing the envelope of dienophile trans-cyclooctene (TCO) reactivity, while maintaining stability in vivo.20 The potential for biomaterial formation and nano-particle decoration has also been demonstrated.21,22 Theoretical calculations of tetrazine bioorthogonal reactions by the Houk group promise to guide further expansion of applications.23
Development of versatile tetrazines and dienophiles to expand the bioorthogonal chemistry tool kit for different research purposes continues to be important. Here we recount our advances in improving tetrazine synthetic methodology24,25 and in discovering new dienophiles26–29 and associated tetrazine bioorthogonal reaction types.27 We also describe how the resulting expansion tetrazine bioorthogonal chemistry has been exploited by our groups and others for biological applications.30–32
IMPROVEMENTS IN TETRAZINE SYNTHETIC METHODOLOGY
The first synthesis of 1,2,4,5-tetrazine can be traced back to late 19th century.33,34 In the classical route, known as the Pinner synthesis, tetrazine is produced in a two-step procedure: condensation of aromatic nitriles with hydrazine to form dihydrotetrazines, followed by oxidation. However, this approach can be applied only to synthesis of aromatic tetrazines. Alkyl tetrazines with functional groups are usually difficult to obtain or are generated in low yield. Therefore, we aimed to develop a straightforward route to broaden access to a diverse range of tetrazines. Previous studies had shown that a transition metal can promote nucleophilic attack of nitrile by hydrazine.35 We hypothesized that coordination with transition metals could activate alkyl nitriles for attack by hydrazine, enabling the synthesis of the corresponding tetrazine. By screening a series of Lewis acid metals, we found that divalent nickel and zinc salts can efficiently catalyze formation of an amidrazone intermediate, generating 3,6-dibenzyl-1,2,4,5-tetrazine product 1 in up to 95% yield (Figure 1A).24 This synthesis cannot be achieved using the classical Pinner synthesis.36 Gently tuning the ratio between the two starting nitriles in the presence of 5% catalyst loading allows the generation of unsymmetric tetrazines in moderate to good yield (Figure 1B). The resulting unsymmetric tetrazines 2–7, bearing amino, hydroxyl, or carboxyl functional groups, can be used for future derivatization. We found that our Lewis acid promoted one-pot synthesis strategy can significantly increase the yield of monosubstituted tetrazines by using formamidine salts as starting material (Figure 1C). This methodology has allowed several groups to reliably obtain tetrazine derivatives with novel structures, which have been endowed with new functions to drive exploration of intracellular no-wash fluorogenic imaging,37 as well as pretargeted 18F and 11C positron emission tomography.38,39
Figure 1.

(A) Optimized reaction condition of one-pot tetrazine synthesis. (B) Representative substrate scope. Reaction yield shown under each product. (C) Synthesis of monosubstituted tetrazine.
Despite its advantages, metal catalyzed de novo tetrazine synthesis has limitations. It involves anhydrous hydrazine, which is not sold in Europe or China because of safety concerns. In addition, since hydrazine is a good nucleophile and reductant, it can undergo side reactions with some functional groups under heating. We sought to overcome this problem by preparing a tetrazine building block which could be attached to functional molecules easily and efficiently via a mild reaction.25 Tetrazine is one of the most electron-poor aromatic systems,34 meaning that it can function as an extremely strong electron-withdrawing group. This electron deficiency can also be utilized as an electron acceptor for molecular electronics, nonlinear optics and fluorogenic probe design.40 Based on this, we hypothesized that we could use Heck coupling, which functions well with electron-deficient vinyl reactants, to append tetrazines onto various molecules.
We initiated this work by synthesizing the Heck coupling reactant 3-methyl-6-vinyl-1,2,4,5-tetrazine 8. We found that 8 was extremely volatile during rotary evaporation and difficult to isolate. In contrast, the tetrazine mesylate precursor 9 was a stable pink powder. Thus, we used 9 as a precursor, and explored a one-pot elimination-Heck cascade coupling with aryl halide. Screening reaction conditions led us to identify 3% [Pd2(dba)3] and 12% iron complex ligand 10 as the best combination; microwave irradiation for 30 min at 50 °C smoothly generated Heck product 11 in nearly quantitative yield (Figure 2A). Interestingly, conventional heating gave little to no product. This mild coupling strategy is compatible with different substituents at position 3 on tetrazine (12–17), and it is capable of appending tetrazines to a variety of functional skeletons (Figure 2B). We generated 24 π-conjugated tetrazines in good yield, including derivatives of deoxyuridine 18, amino acids 19 and 20, as well as long conjugated double-coupling products 21. We believe this practical synthesis protocol can contribute to the design and production of chemical biology probes and conjugated materials.
Figure 2.

(A) In situ elimination–Heck cascade reaction. (B) Representative substrate scope. The number of bromide equivalents, reaction time, and yield are shown under each product.
DEVELOPMENT OF FULL-SPECTRUM FLUOROGENIC PROBES
Fluorescence labeling is one of the most commonly used strategies for visualizing the spatiotemporal dynamics of biological molecules. The standard approach is to use a constitutively fluorescent molecule and to wash the labeled sample extensively to remove excess dye that contributes to background. However, a more effective approach is to use fluorogenic probes: the fluorescence of the probe is quenched under normal conditions and is activated only when the probe engages in a specific reaction or enters a specific environment. This can result in strong signal with minimal background, which is extremely useful for intracellular and in vivo imaging.41 Numerous bioorthogonal fluorogenic probes have been developed in recent years. Early tetrazine fluorogenic probes contained a flexible aliphatic linker, resulting in a moderate turn-on ratio, which refers to the ratio of probe fluorescence when activated to probe fluorescence when quenched.42 With our tetrazine synthetic methodology in hand, we aimed to design a new generation of tetrazine fluorogenic probes.25
Since the double bond generated by the Heck reaction is the ideal bridge for energy transfer, we hypothesized that a tetrazine acceptor could efficiently quench the fluorescence of donor fluorophores based on the “through bond energy transfer” (TBET) mechanism.43 Therefore, we coupled tetrazine with various popular fluorophore skeletons, generating 2′,7′-difluorofluorescein (Oregon Green) probe 22, BODIPY probe 23, and tetramethylrhodamine probe 24 in yields of 53%–83% (Figure 3A–C). To our delight, all three probes were stable and highly quenched in solution. After reacting with dienophiles, both green emitting probes, 22 and 23, exhibited a turn-on ratio > 100. The greatest ratio of 400 was observed with the bioorthogonal reaction between Oregon Green probe 22 and TCO. The rhodamine probe 24, whose emission was red-shifted (569–573 nm), showed a turn-on ratio up to 76-fold (Figure 3C). Following this work, several groups have developed a wide range of fluorogenic cassettes.44,45 The suitability of our fluorogenic probes for live-cell imaging was demonstrated in a labeling experiment with TCO-decorated antibodies (Figure 3D). The in vivo application of alkenyltetrazine fluorogenic probes was elegantly demonstrated by the Bertozzi group through systemic imaging of zebrafish glycans.15
Figure 3.

(A–C) Fluorogenic reaction of 22–24 with dienophiles. (D) Live-cell imaging of LS174T cells using fluorogenic probe 22. Left: cells were pretargeted with TCO-decorated A33 antibodies. Right: cells were treated with unmodified antibodies. (E) Structure of cyclopropene 25.
We subsequently turned our attention to designing near-infrared (NIR) fluorogenic probes28 offering imaging possibilities deeper in tissue with less interference from tissue autofluorescence. However, the existing tetrazine fluorogenic probes relied on TBET or Förster resonance energy transfer (FRET) to achieve quenching: the electronic excited state energy of the donor (fluorophores) transfers to the acceptor (tetrazine) through bond (TBET) or space (FRET). Tetrazines have an inherent absorbance around 500–550 nm. Thus, farred and NIR fluorophores with relatively longer emission wavelengths are challenging to quench. Therefore, we decided to design a tetrazine near-infrared fluorogenic probe through a different quenching mechanism: internal charge transfer (ICT) process.40 We used a special functional group that would mask the fluorophore in the “off” state either by interrupting the conjugated π-electron system46 or holding the fluorophores in a nonionizable form.47 Fluorescence would be turned on by removing or changing the functional group in a tetrazine bioorthogonal reaction. Inspired by previous “click-to-release” strategies,48 we hypothesized that the masking functional group could be a dienophile such as a vinyl ether,49 which would unmask an alcohol after cycloaddition and thus act as a trigger for fluorogenic probe formation.
The phenoxide group and its resonance structure exist in various fluorophore skeletons, such as coumarins, fluoresceins, and some novel quinone cyanine dyes50 (Figure 4A). Our objective was to use the novel dienophile phenyl vinyl ether as a cage to mask the phenol group and thus quench fluorescence. After a tetrazine ligation followed by cascade elimination, the cage is released, resulting in free phenol and recovery of different fluorogenic structures (Figure 4B). To verify our design, we prepared the model vinyl ether cyanine probe 26 from a commercially available compound in a straightforward fashion. The reaction between 26 and dipyridyl tetrazine 27 was highly efficient (Figure 4C), restoring the original cyan color of 28 in buffer. The observed fluorescence was 70-fold greater than that of the caged light-yellow precursor (Figure 4D). The shift in absorption spectrum from that of 26 to that of 28 correlated with ICT efficiency. Caging the probe with vinyl ether caused a hypsochromic shift of the absorption peak from 620 to 550 nm (Figure 4E), indicating the effectiveness of our fluorogenic design. Using this strategy, we prepared a highly fluorogenic coumarin probe 29 (turn-on ratio = 162) and a less effectively quenched fluorescein probe 30 (turn-on ratio = 11).
Figure 4.

(A) Phenoxide anion and its resonance structure within various fluorophore scaffolds. (B) Schematic of fluorogenic probe design based on a decaging reaction. (C) Fluorogenic decaging reaction of probe 26 with tetrazine 27. (D) Fluorescence spectra of 26 and its corresponding uncaged cyanine compound 28. (E) Absorption spectra of 26 and 28. (F) Caged fluorogenic probes with different cassettes.
In order to apply our vinyl ether probes at the low concentrations typically used for cellular imaging, we needed to improve the sluggish turn-on kinetics. We employed nucleic-acid-templated chemistry to increase the effective molarity51 of the dienophile and tetrazine. Modifying oligonucleotides with our probes and then adding the appropriate antisense template dramatically accelerated the turn-on half-life to t1/2 = 54.9 min. Such reaction kinetics are suitable for live-cell imaging. We then engineered a plasmid expressing EGFP mRNA genetically incorporating our target binding sequence within the 3′ untranslated region. We transfected cells with this plasmid, delivered our fluorogenic RNA hybrid probes, and imaged the live cells without any washing step. We clearly observed mRNA labeling (Figure 5A), which was specific to our probe system and sense-antisense binding, as verified in several control experiments. This established the applicability of our system for synthesizing a series of tetrazine fluorogenic probes that span the visible and near-IR spectrum (Figure 5B).
Figure 5.

(A) Schematic of mRNA imaging using probe-decorated antisense oligonucleotides. (B) Fluorogenic probes covering the visible and near-IR light spectrum based on tetrazine bioorthogonal chemistry.
DISCOVERY OF “MINI-TAG” DIENOPHILES
In addition to the development of fluorogenic probes, we wanted to expand the scope of available dienophiles that could undergo effective bioorthogonal coupling with tetrazines. Some biomedical applications such as metabolic labeling and target identification of bioactive compounds impose steric constraints that mandate the use of small tags. In order to discover novel biologically stable dienophiles that could act as “mini-tags”, we were drawn to explore cyclopropenes, the smallest highly strained cyclic alkenes. From the work of Sauer et al., cyclopropenes were known to react rapidly with tetrazine,52 but unsubstituted cyclopropenes are unstable and prone to polymerize. Substitution of cyclopropenes can enhance stability but also dramatically lowers the reactivity to cycloaddition. Thus, our goal was to prepare biologically stable cyclopropenes as “mini-tags” while maintaining adequate reactivity.26
As mentioned, the number of ring substituents can strongly affect cyclopropene stability but can also perturb reactivity.52 We therefore began our investigation by varying the degree of substitution on cyclopropene. As expected, cycloprop-2-enecarboxylic acid 31 reacted rapidly with tetrazines but did not remain stable overnight at −20 °C. A clue to a possible solution came from work exploring a cyclopropene toxic trigger in a Japanese poisonous mushroom.53 In addition to studying the natural product, the researchers demonstrated that a single methyl substitution greatly improved stability to nucleophilic attack, which was a key for activating toxicity. Inspired by this study, we synthesized methylcyclopropene 33, which we obtained from commercial starting material in a straightforward manner. 33 showed dramatically higher stability and reacted with a second-order rate constant of 0.0047 ± 0.0004 M−1 s−1 at 37 °C.26 Since reaction kinetics are inversely proportional to the energy difference between cyclization reactants, we speculated that increasing electron density in the cyclopropene would raise the HOMO energy, narrowing the energy gap between cyclization reactants (LUMOdiene–HOMOdienophile) and speeding up the reaction.
We therefore synthesized a series of cyclopropenes with different functional handles to optimize reactivity (Figure 6A).26,29 By reducing the ester 32, we obtained the key synthon 34 in 51% yield. Starting from 34, we prepared 35 via carbamate formation in three steps. A process involving Dess–Martin oxidation followed by Horner–Wadsworth–Emmons reaction as key steps smoothly generated three cyclopropenes 36–38 with unsaturated functional handles. Amide or secondary amine derivatives 39–41 were converted from alcohol 34 via azide formation and reduction. Then we examined the stability of this panel of cyclopropenes and their kinetics of reaction with tetrazine. By transforming the electron withdrawing carbonyl group, the kinetics of reaction with tetrazine was dramatically improved by up to 2 orders of magnitude (Figure 6B). The cyclopropenes remained stable in solution overnight, except for α,β-unsaturated derivatives 36 and 37. Cyclopropenes with an amide linkage 41 or carbamate linkage 35 not only showed rapid kinetics but also remained stable overnight in the presence of cysteine. The amide 41 remained stable over an extended period involving multiple freeze–thaw cycles, while degradation of carbamate 35 was detected after 1 week. Based on these results, cyclopropene 35 is easy to prepare and can be appropriate for many applications, but cyclopropene 41 may be more appropriate for applications requiring lengthy incubations.
Figure 6.

(A) Synthetic of cyclopropene derivatives. (B) Kinetic of cyclopropene–tetrazine reactions.
We also investigated the rate constant of the reaction involving cyclopropene 41 or TCO with different tetrazines. Tetrazines with electron-withdrawing substituents reacted faster, and TCO led to faster reaction than cyclopropene 41 in most cases. To our surprise, sterically hindered tert-butyl substituted tetrazines reacted faster with cyclopropene than with TCO (Figure 7A). This is interesting, since tert-butyl substituted tetrazines are extraordinarily stable reactive groups. We hypothesized that this result reflected the steric difficulty of TCO in approaching the tetrazine. Density functional theory calculations performed by the Houk group suggested that the mini-tag cyclopropene 46 would react with the more sterically hindered tetrazine at rate similar to methyltetrazine (Figure 7B). In contrast, changing the methyl group to a tert-butyl group on the tetrazine would reduce the reactivity with TCO by more than 600-fold. These results suggest that small, stable methylcyclopropenes may be superior bioorthogonal partners for biological applications that require the use of bulky tetrazines or that occur in a sterically hindered environment.
Figure 7.

(A) Rate constants of the reaction involving cyclopropene or TCO with selected tetrazines. (B) M06–2X/6–31G(d)-optimized transition-state structures for the tetrazine ligation and M06–2X/6–311+G(d,p)//6–31G(d)-computed activation free energies in water and relative rate constants. (C) Chemical structure of cyclopropene tags.
We further studied the applicability of methylcyclopropene as a mini-tag for live-cell imaging. We synthesized cyclopropene phospholipid derivative 47 (Figure 7C) which distributes in cell membranes and can be imaged using tetrazine fluorogenic probes.26 We also used cyclopropene-decorated mannosamine 48a (Figure 7C) as a metabolic probe to image glycans in living human cancer cells.31 We simultaneously labeled glycans using 48a and 48b, then visualized the probes orthogonally using appropriate bioorthogonal reactions.
DIENOPHILES FOR NUCLEIC ACID-TEMPLATED SIGNAL AMPLIFICATION
In order to specifically detect low abundant nucleic acid targets, we sought appropriate tetrazines and dienophiles that would undergo a nucleic acid templated fluorogenic reaction.51,54 In this strategy, a nucleic acid template (the detection target) binds with a pair of fluorogenic antisense probes. The hybridization brings two reactive groups into proximity, inducing the fluorogenic reaction in low concentration and producing a detectable readout.51 To minimize background, the reaction should be sluggish in the absence of target template; as a result, fast-reacting TCOs cannot be used.
We began our investigation using methylcyclopropene as dienophile.30 We used classical N-hydroxysuccinimide labeling to decorate variable-length oligonucleotides with cyclopropene. We used the same approach to generate tetrazine oligonucleo-tide probes with a quenched fluorophore. In the presence of 1 μM 27-mer template strand, the pair of 13-mer oligonucleotide probes reacted rapidly leading to a 9.3-fold increase in fluorescence (Figure 8A). The reaction half-life was 36 s, much shorter than the half-life of ~5 days predicted in the absence of template (assuming a previously reported second-order rate constant).26 LC/MS showed the reaction to be efficient, generating ligation product in ~92% yield. Next we examined how probe structure can affect reaction rate. The most rapid kinetics were observed when a single-nucleotide gap separated the two oligonucleotide probes. Shortening oligonu cleotide length from 13 to 5 bases weakened template binding and prolonged reaction half-life by 300-fold (Figure 8C). Minimal background signal was detected in the absence of template or the presence of mismatched template.
Figure 8.

(A). Schematic of templated fluorogenic tetrazine ligations. (B) Sequences of the template strand and corresponding DNA probes. (C) Rates of the templated reaction for different probes.
Then we applied this oligonucleotide-based templated tetrazine chemistry to the detection of low-abundant nucleic acid targets.27 In our effort to develop highly fluorogenic probes to achieve subnanomolar detection limits, we drew on the previously reported strategy of templated fluorogenic reactions with turnover-driven signal amplification.55 In this strategy, probes bind to the template and dissociate from it in dynamic equilibrium; the reaction produces a pair of nonligating products with similar affinity for the template. Reacted probes can be sequentially displaced by unreacted probes, allowing turnover of multiple reactions on a single template. In this way, the low-abundance targets catalyze the reaction and thereby amplify the signal (Figure 9A). However, the ligation of tetrazine and cyclopropene generates a product with higher affinity for the template, inhibiting turnover. We hypothesized that we could react norbornadiene derivatives as novel strained dienophiles with tetrazines via a tetrazine ligation/retro-Diels–Alder cascade. This tetrazine-mediated transfer (TMT) reaction would produce a pair of nonligating products that would allow displacement of reacted probes, facilitating reaction turnover and signal amplification (Figure 9B). As hypothesized, 7-azabenzonorbornadiene derivative 49 and quenched tetrazine-BODIPY 50 reacted to produce nonligating product pyridazine 51 in 95% isolated yield (Figure 9C). We next decorated the oligonucleotide with tetrazine and dienophile using standard NHS coupling. In the presence of template, two probes reacted smoothly with a half-life of 12.7 min and a turn-on ratio of 108. Next we fine-tuned these probes to optimize template-driven turnover. We shortened the probes to 10 bases such that their melting temperatures were 38.8 and 43.8 °C, facilitating dynamic strand exchange at physiological temperature. We incubated d21′-Tz and d21′-ABN with decreasing amounts of DNA template for 7 h. Substantial fluorescence amplification was observed at substoichiometric template concentrations: signal could be distinguished from background at template concentrations as low as 500 fM (Figure 9D).
Figure 9.

(A) Schematic of templated fluorogenic reactions with turnover-driven signal amplification. (B) Proposed mechanism of the TMT reaction. (C) Reaction of tetrazine-BODIPY with azabenzonorbornadiene. (D) Fluorescence from 100 nM d21′-Tz and 200 nM d21′-ABN with variable d21 concentrations. (E) Distinguish the target mir21 from two single-mismatch variants. (F) Sequences of probes and templates. (G) Normalized fluorescence from different cell lysates. The SKBR3 (–) contained probe lacking reactive groups (10-fold excess over mir21′-Tz and mir21′ABN) as a competitive inhibitor. (H) Detection of miRNAs in living human cancer cells.
We next turned our attention to detecting endogenous microRNAs (miRNAs), which are ~22-mer single-stranded noncoding RNAs that regulate many biological processes. Altered miRNA levels are associated with many human diseases.56 We decorated a 2-O-methyl RNA backbone with probes; this RNA derivative was chosen as backbone because it is more stable, hybridizes faster, and allows better mismatch detection than the natural backbone. Excellent signal amplification with a detection limit of ~5 pM was observed after 7 h incubation. At 1% template concentration, our probes discriminated the target mir21 from the single-base mismatch variant mir21A with a signal-to-background ratio of 9.9 and from the single-base mismatch variant mir21B with a signal-to-background ratio of 24.4 (Figure 9E). We detected strong fluorescence signal after transfecting our probes into the cell lines SKBR3 and MCF-7, both of which express high levels of the target mir21. We detected weak signal in HeLa cells, which express low levels of mir21 (Figure 9H). We confirmed that the observed signal specifically reflects mir21 expression in control experiments in which unmodified probe was added to cell lysates as a competitive inhibitor (Figure 9G).
ELECTROCHEMICAL CONTROLLED TETRAZINE CHEMISTRY FOR MICROELECTRODES FUNCTIONALIZATION
While much of our research focuses on expanding the bioorthogonal toolbox for live-cell imaging and target detection, our lab is also interested exploring other implementations of tetrazine chemistry for in vitro applications. Microelectrode arrays are a sensitive, controllable, noninvasive method for recording signals in many contexts, including high-throughput screening and biosensor development.57 We have begun to employ our improved tetrazine synthetic methods to generate tetrazine probes for the design of novel redox-active micro-electrodes.32 Tetrazines have the ability to be reduced to dihydrotetrazines, which would be inert to cycloaddition. We therefore sought to explore whether electrochemical control over the tetrazine ligation would be feasible.
We synthesized thiolated tetrazines 51 and 52 using our Lewis-acid-promoted approach, diluted them with 1-nonanethiol, and allowed them to form self-assembled monolayers (SAM) on gold surfaces. We observed constant redox behavior in cyclic voltammetry experiments, reflecting the interconversion between tetrazine and dihydrotetrazine58 [first scan for 51: reduction at Epc = −0.47 V, oxidation at Epc = 0.28 V vs Ag/ AgCl/ (3 M) KCl]. In the presence of TCO, the signal peak of the monolayer decreased rapidly with a measured second-order rate constant of k = 4800 M−1 s−1 (Figure 10C). Maintaining the electrode potential below the reduction potential eliminated the monolayer’s bioorthogonal reactivity, which was restored by opening the circuit. Using this potential-dependent reactivity, we functionalized, in site-selective fashion, ferrocene-modified TCO 53 into an interdigitated array with 10 μm spacing (Figure 10D). We also used potential-dependent reactivity to control the attachment of horseradish peroxidase to the monolayer via a tetrazine bioorthogonal reaction, while maintaining enzyme activity. The ability to rapidly functionalize microelectrodes using tetrazine biorthogonal chemistry without any diffusible catalyst will help support advances in high-throughput biomolecular screening.
Figure 10.

(A) Chemical structures of thiolated tetrazine probes. (B) Tetrazine redox behavior. (C) Sequential cyclic voltammograms of a mixed SAM during reacting with 1 μM TCO-PEG3-amine. (D) Electrochemically controlled tetrazine ligation at a SAM.
CONCLUSIONS
Tetrazine bioorthogonal chemistry is an efficient strategy to illuminate biological processes at the molecular level. We have focused on improving tetrazine synthetic methodology and dienophile expansion. We have developed two practical tetrazine synthetic methods that generate tetrazine derivatives with unique characteristics useful for fluorogenic labeling and microelectrode probes. In parallel, we have developed a new generation of dienophiles for versatile applications, including methylcyclopropenes as mini-tags for metabolic labeling, norbornadiene for templated turnover chemistry to detect miRNA in low-abundance, and vinyl ether for the design of decaging probes. The efforts from our group and others to expand the tetrazine bioorthogonal repertoire have facilitated applications in chemical biology, imaging, and drug discovery, as reflected in the rapidly growing number of examples reported in the literature. These reactions are likely to continue to be important for a wide range of disciplines in the future.
Now tetrazine bioorthogonal chemistry faces the difficult transition from “proof-of-concept” to preclinical study. Efforts to optimize and expand the toolbox face several challenges, and further improvements in synthetic methodology will continue to broaden biomedical applications. Probes with large Stokes shifts will help to minimize in vivo autofluorescence, and novel dienophiles enabling cascade bioorthogonal reactions will promote the integration of diagnostics and therapy. Two-photon imaging based on tetrazine bioorthogonal chemistry still needs to be developed for deeper tissue penetration, while probes for multimodality molecular imaging require optimization. However, through creative synthesis of new probes geared to unique applications, we believe the future looks bright.
ACKNOWLEDGMENTS
H.W. acknowledge financial support from National Natural Science Foundation of China (Grant No. 21602146). N.K.D. acknowledges financial support from the University of California, San Diego, the National Institutes of Health (DP2DK111801, R01GM123285, K01EB010078), and the Department of Defense (W911NF-13–1-0383).
Biographies
Haoxing Wu received his Ph.D. in Medicinal Chemistry from Sichuan University in 2011. He was a postdoctoral fellow in the Devaraj lab (2013–2016). He is currently a professor at Sichuan University. His research focuses mainly on molecular imaging probe design.
Neal K. Devaraj received a dual B.S. in Chemistry and Biology from the Massachusetts Institute of Technology in 2002 and his Ph.D. in Chemistry from Stanford University. After a postdoctoral position in molecular imaging at the Harvard Medical School, he joined the faculty of the University of California, San Diego in 2011. His research focuses on the design of bioconjugation reactions for addressing problems in bottom-up synthetic biology and molecular imaging. He is the recipient of the 2017 ACS Award in Pure Chemistry and the recipient of 2016 National Fresenius Award.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Patterson AM Words about Words. Chem. Eng. News 1953, 31, 462. [Google Scholar]
- (2).Miyawaki A Proteins on the move: insights gained from fluorescent protein technologies. Nat. Rev. Mol. Cell Biol 2011, 12, 656. [DOI] [PubMed] [Google Scholar]
- (3).Das J Aliphatic Diazirines as Photoaffinity Probes for Proteins: Recent Developments. Chem. Rev 2011, 111, 4405–4417. [DOI] [PubMed] [Google Scholar]
- (4).Weissleder R; Nahrendorf M Advancing biomedical imaging. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 14424–14428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Saxon E; Bertozzi CR Cell Surface Engineering by a Modified Staudinger Reaction. Science 2000, 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- (6).Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Patterson DM; Nazarova LA; Prescher JA Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol 2014, 9, 592–605. [DOI] [PubMed] [Google Scholar]
- (8).Blackman ML; Royzen M; Fox JM Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J. Am. Chem. Soc 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Devaraj NK; Weissleder R; Hilderbrand SA Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjugate Chem 2008, 19, 2297–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Devaraj NK; Weissleder R Biomedical Applications of Tetrazine Cycloadditions. Acc. Chem. Res 2011, 44, 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Oliveira BL; Guo Z; Bernardes GJL Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev 2017, 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- (12).Patterson DM; Nazarova LA; Xie B; Kamber DN; Prescher JA Functionalized cyclopropenes as bioorthogonal chemical reporters. J. Am. Chem. Soc 2012, 134, 18638–18643. [DOI] [PubMed] [Google Scholar]
- (13).Rieder U; Luedtke NW Alkene-tetrazine ligation for imaging cellular DNA. Angew. Chem 2014, 126, 9322–9326. [DOI] [PubMed] [Google Scholar]
- (14).Erdmann RS; Takakura H; Thompson AD; Rivera-Molina F; Allgeyer ES; Bewersdorf J; Toomre D; Schepartz A Super-resolution imaging of the Golgi in live cells with a bioorthogonal ceramide probe. Angew. Chem., Int. Ed 2014, 53, 10242–10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Agarwal P; Beahm BJ; Shieh P; Bertozzi CR Systemic Fluorescence Imaging of Zebrafish Glycans with Bioorthogonal Chemistry. Angew. Chem., Int. Ed 2015, 54, 11504–11510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lang K; Chin JW Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev 2014, 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- (17).Li J; Chen PR Development and application of bond cleavage reactions in bioorthogonal chemistry. Nat. Chem. Biol 2016, 12, 129–137. [DOI] [PubMed] [Google Scholar]
- (18).Ramil CP; Dong M; An P; Lewandowski TM; Yu Z; Miller LJ; Lin Q Spirohexene-Tetrazine Ligation Enables Bioorthogonal Labeling of Class B G Protein-Coupled Receptors in Live Cells. J. Am. Chem. Soc 2017, 139, 13376–13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Blizzard RJ; Backus DR; Brown W; Bazewicz CG; Li Y; Mehl RA Ideal Bioorthogonal Reactions Using A Site-Specifically Encoded Tetrazine Amino Acid. J. Am. Chem. Soc 2015, 137, 10044–10047. [DOI] [PubMed] [Google Scholar]
- (20).Selvaraj R; Fox JM trans-Cyclooctene—a stable, voracious dienophile for bioorthogonal labeling. Curr. Opin. Chem. Biol 2013, 17, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Haun JB; Devaraj NK; Hilderbrand SA; Lee H; Weissleder R Bioorthogonal chemistry amplifies nanoparticle binding and enhances the sensitivity of cell detection. Nat. Nanotechnol 2010, 5, 660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Liu S; Zhang H; Remy RA; Deng F; Mackay ME; Fox JM; Jia X Meter-long multiblock copolymer microfibers via interfacial bioorthogonal polymerization. Adv. Mater 2015, 27, 2783–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liu F; Liang Y; Houk KN Theoretical elucidation of the origins of substituent and strain effects on the rates of Diels-Alder reactions of 1,2,4,5-tetrazines. J. Am. Chem. Soc 2014, 136, 11483–11493. [DOI] [PubMed] [Google Scholar]
- (24).Yang J; Karver MR; Li W; Sahu S; Devaraj NK Metal-Catalyzed One-Pot Synthesis of Tetrazines Directly from Aliphatic Nitriles and Hydrazine. Angew. Chem., Int. Ed 2012, 51, 5222–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wu H; Yang J; Šečkutė J; Devaraj NK In Situ Synthesis of Alkenyl Tetrazines for Highly Fluorogenic Bioorthogonal Live-Cell Imaging Probes. Angew. Chem., Int. Ed 2014, 53, 5805–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yang J; Šečkutė J; Cole CM; Devaraj NK Live-Cell Imaging of Cyclopropene Tags with Fluorogenic Tetrazine Cyclo-additions. Angew. Chem., Int. Ed 2012, 51, 7476–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wu H; Cisneros BT; Cole CM; Devaraj NK Bioorthogonal Tetrazine-Mediated Transfer Reactions Facilitate Reaction Turnover in Nucleic Acid-Templated Detection of Micro-RNA. J. Am. Chem. Soc 2014, 136, 17942–17945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wu H; Alexander SC; Jin S; Devaraj NK A Bioorthogonal Near-Infrared Fluorogenic Probe for mRNA Detection. J. Am. Chem. Soc 2016, 138, 11429–11432. [DOI] [PubMed] [Google Scholar]
- (29).Yang J; Liang Y;Šečkutė J; Houk KN; Devaraj NK Synthesis and Reactivity Comparisons of 1-Methyl-3-Substituted Cyclopropene Mini-tags for Tetrazine Bioorthogonal Reactions. Chem. - Eur. J 2014, 20, 3365–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Seckute J; Yang J; Devaraj NK Rapid oligonucleotidetemplated fluorogenic tetrazine ligations. Nucleic Acids Res 2013, 41, e148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cole CM; Yang J;Šečkutė J; Devaraj NK Fluorescent Live-Cell Imaging of Metabolically Incorporated Unnatural Cyclopropene-Mannosamine Derivatives. ChemBioChem 2013, 14, 205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ehret F; Wu H; Alexander SC; Devaraj NK Electrochemical Control of Rapid Bioorthogonal Tetrazine Ligations for Selective Functionalization of Microelectrodes. J. Am. Chem. Soc 2015, 137, 8876–8879. [DOI] [PubMed] [Google Scholar]
- (33).Hofmann KA; Ehrhart O Einwirkung von Hydrazin auf Dicyandiamid. Ber. Dtsch. Chem. Ges 1912, 45, 2731–2740. [Google Scholar]
- (34).Clavier G; Audebert P s-Tetrazines as building blocks for new functional molecules and molecular materials. Chem. Rev 2010, 110, 3299–3314. [DOI] [PubMed] [Google Scholar]
- (35).Kukushkin VY; Pombeiro AJL Additions to Metal-Activated Organonitriles. Chem. Rev 2002, 102, 1771–1802. [DOI] [PubMed] [Google Scholar]
- (36).Bowie RA; Gardner MD; Neilson DG; Watson KM; Mahmood S; Ridd V Studies on some symmetrically and unsymmetrically 3,6-disubstituted 1,2-dihydro-1,2,4,5-tetrazines including their conversion into the corresponding tetrazines and 3,5-disubstituted 4-amino-1,2,4-triazoles. J. Chem. Soc., Perkin Trans. 1 1972, I, 2395–2399. [Google Scholar]
- (37).Meimetis LG; Carlson JCT; Giedt RJ; Kohler RH; Weissleder R Ultrafluorogenic Coumarin–Tetrazine Probes for Real-Time Biological Imaging. Angew. Chem., Int. Ed 2014, 53, 7531–7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Denk C; Svatunek D; Filip T; Wanek T; Lumpi D; Fröhlich J; Kuntner C; Mikula H Development of a (18) F-labeled tetrazine with favorable pharmacokinetics for bioorthogonal PET imaging. Angew. Chem., Int. Ed 2014, 53, 9655. [DOI] [PubMed] [Google Scholar]
- (39).Denk C; Svatunek D; Mairinger S; Stanek J; Filip T; Matscheko D; Kuntner C; Wanek T; Mikula H Design, Synthesis, and Evaluation of a Low-Molecular-Weight 11C-Labeled Tetrazine for Pretargeted PET Imaging Applying Bioorthogonal in Vivo Click Chemistry. Bioconjugate Chem 2016, 27, 1707–1712. [DOI] [PubMed] [Google Scholar]
- (40).de Silva AP; Gunaratne HQN; Gunnlaugsson T; Huxley AJM; McCoy CP; Rademacher JT; Rice TE Signaling Recognition Events with Fluorescent Sensors and Switches. Chem. Rev 1997, 97, 1515–1566. [DOI] [PubMed] [Google Scholar]
- (41).Li X; Gao X; Shi W; Ma H Design Strategies for Water-Soluble Small Molecular Chromogenic and Fluorogenic Probes. Chem. Rev 2014, 114, 590–659. [DOI] [PubMed] [Google Scholar]
- (42).Devaraj NK; Hilderbrand S; Upadhyay R; Mazitschek R; Weissleder R Bioorthogonal Turn-On Probes for Imaging Small Molecules inside Living Cells. Angew. Chem 2010, 122, 2931–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kim TG; Castro JC; Loudet A; Jiao JGS; Hochstrasser RM; Burgess K; Topp MR Correlations of Structure and Rates of Energy Transfer for Through-Bond Energy-Transfer Cassettes. J. Phys. Chem. A 2006, 110, 20–27. [DOI] [PubMed] [Google Scholar]
- (44).Knorr G; Kozma E; Herner A; Lemke EA; Kele P New Red-Emitting Tetrazine-Phenoxazine Fluorogenic Labels for Live-Cell Intracellular Bioorthogonal Labeling Schemes. Chem. - Eur. J 2016, 22, 8972–8979. [DOI] [PubMed] [Google Scholar]
- (45).Wieczorek A; Werther P; Euchner J; Wombacher R Green-to far-red-emitting fluorogenic tetrazine probes - synthetic access and no-wash protein imaging inside living cells. Chem. Sci 2017, 8, 1506–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Vaughan JC; Dempsey GT; Sun E; Zhuang X Phosphine Quenching of Cyanine Dyes as a Versatile Tool for Fluorescence Microscopy. J. Am. Chem. Soc 2013, 135, 1197–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Cohen AS; Dubikovskaya EA; Rush JS; Bertozzi CR Real-Time Bioluminescence Imaging of Glycans on Live Cells. J. Am. Chem. Soc 2010, 132, 8563–8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Versteegen RM; Rossin R; ten Hoeve W; Janssen HM; Robillard MS Click to Release: Instantaneous Doxorubicin Elimination upon Tetrazine Ligation. Angew. Chem 2013, 125, 14362–14366. [DOI] [PubMed] [Google Scholar]
- (49).Boger DL; Schaum RP; Garbaccio RM Regioselective Inverse Electron Demand Diels–Alder Reactions of N-Acyl 6-Amino-3-(methylthio)-1,2,4,5-tetrazines. J. Org. Chem 1998, 63, 6329–6337. [DOI] [PubMed] [Google Scholar]
- (50).Gnaim S; Shabat D Quinone-Methide Species, A Gateway to Functional Molecular Systems: From Self-Immolative Dendrimers to Long-Wavelength Fluorescent Dyes. Acc. Chem. Res 2014, 47, 2970–2984. [DOI] [PubMed] [Google Scholar]
- (51).Silverman AP; Kool ET Detecting RNA and DNA with Templated Chemical Reactions. Chem. Rev 2006, 106, 3775–3789. [DOI] [PubMed] [Google Scholar]
- (52).Thalhammer F; Wallfahrer U; Sauer J Reaktivität einfacher offenkettiger und cyclischer dienophile bei Diels-Alder-reaktionen mit inversem elektronenbedarf. Tetrahedron Lett 1990, 31, 6851–6854. [Google Scholar]
- (53).Matsuura M; Saikawa Y; Inui K; Nakae K; Igarashi M; Hashimoto K; Nakata M Identification of the toxic trigger in mushroom poisoning. Nat. Chem. Biol 2009, 5, 465. [DOI] [PubMed] [Google Scholar]
- (54).Di Pisa M; Seitz O Nucleic Acid Templated Reactions for Chemical Biology. ChemMedChem 2017, 12, 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Shibata A; Uzawa T; Nakashima Y; Ito M; Nakano Y; Shuto S; Ito Y; Abe H Very Rapid DNA-Templated Reaction for Efficient Signal Amplification and Its Steady-State Kinetic Analysis of the Turnover Cycle. J. Am. Chem. Soc 2013, 135, 14172–14178. [DOI] [PubMed] [Google Scholar]
- (56).Li J; Tan S; Kooger R; Zhang C; Zhang Y MicroRNAs as novel biological targets for detection and regulation. Chem. Soc. Rev 2014, 43, 506–517. [DOI] [PubMed] [Google Scholar]
- (57).Haensch C; Hoeppener S; Schubert US Chemical modification of self-assembled silane based monolayers by surface reactions. Chem. Soc. Rev 2010, 39, 2323–2334. [DOI] [PubMed] [Google Scholar]
- (58).Kaim W The coordination chemistry of 1,2,4,5-tetrazines. Coord. Chem. Rev 2002, 230, 127–139. [Google Scholar]