

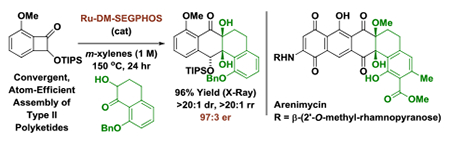
Abstract
The first enantioselective intermolecular metal catalyzed cycloadditions of benzocyclobutenones via C-C bond oxidative addition are described. In the presence of a ruthenium(0) complex modified by (R)-DM-SEGPHOS, tetralone-derived ketols and benzocyclobutenones combine to form cycloadducts with complete regio- and diastereoselectivity and high enantioselectivity. Using this method, the “bay region” substructure of the angucycline natural products arenimycin was prepared.
Graphical Abstract
Following pioneering work by Liebeskind,1,2 Jun3 and Murakami,4 the insertion of π-bonds into metallacycles obtained through C-C bond oxidative addition has matured into an active area of research.5 Despite substantial progress in this field, several key challenges persist. The use of directing groups is often required to overcome the kinetic and thermodynamic barriers posed by the activation of relatively strong and stable C-C bonds. Many C-C bond activation initiated C-C couplings are restricted to intramolecular processes. Finally, beyond reactions of donor-acceptor cyclopropanes,6,7 enantioselective intermolecular metal-catalyzed C-C bond activation initiated C-C couplings are exceptionally uncommon.8
In connection with studies of transfer hydrogenative C-C bond formation,9 we recently developed a novel class of ruthenium(0)-catalyzed cycloadditions of vicinal diols, ketols or diones.9d,10 These processes operate through a common mechanistic motif wherein oxidative formation of ruthenium(II) metallacycles, which is accompanied or triggered by dione addition, is followed by diol- or ketol-mediated transfer hydrogenolysis of the ruthenacycle to release product and regenerate the reactive dione (Figure 1).9d Recently, we found that ruthenacycles obtained via C-C bond oxidative addition to benzocyclobutenones are catalytically competent in transformations of this type, providing a kinetic pathway for the insertion of adjacent carbon atoms of vicinal diols, ketols or diones into C-C σ-bonds.10i Based on this finding, we now report the first examples of enantioselective intermolecular metal catalyzed cycloadditions of benzocyclobutenones.11–14
Figure 1.

General catalytic mechanism for ruthenium(0) catalyzed cycloaddition of diols, ketols or diones.
In an initial set of experiments (Table 1), a series of benzocyclobutenone derivatives la-ld were exposed to equimolar quantities of the tetralone-derived ketols 2a-2c (or diols) in the presence of the mononuclear ruthenium(0) catalyst15 derived from Ru3(CO)12 and (R)-SEGPHOS in m-xylene (1 M) at 150 °C. The reaction of the unsubstituted benzocyclobutenone la with the unsubstituted diol 2a led to the formation of cycloadduct 3a with complete regio- and diastereoselectivity, but in nearly racemic form (Table 1, entry 1). However, using the corresponding ortho-methoxy-substituted benzocyclobutenone lb, cycloadduct 3b was formed with significant levels of enantiomeric enrichment (Table 1, entry 2). Based on this result, an effort was made to define substituents that would both enforce optimal levels of enantiomeric enrichment and facilitate entry to naturally occurring type II polyketides of the angucycline class (vide infra). Toward this end, incorporation of a triisopropylsilyl (TIPS) ether at C2 of the benzocyclobutenone, as in lc, and, for the ketol partner, a benzyloxy substituent at C8, as in 2c, conspired to improve the degree of asymmetric induction. Thus, the reaction of benzocyclobutenone lc and ketol 2c delivered cycloadduct 3h with complete regio- and diastereoselectivity as a 94:6 ratio of enantiomers (Table 1, entry 8). Finally, using (R)-DM-SEGPHOS as ligand and a slight excess of benzocyclobutenone lc, cycloadduct 3h was generated in 96% yield as a single regio- and diastereomer as a 97:3 ratio of enantiomers (Table 1, entry 9).
Table 1.
Selected optimization experiments in the enantioselective intermolecular ruthenium catalyzed cycloaddition of benzocyclobutenones.a
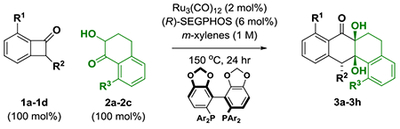
| entry | R1 | R2 | 1a-1d | R3 | 2a-2c | 3a-3h | yield (%), dr, rr | er |
|---|---|---|---|---|---|---|---|---|
| 1b | H | H | 1a | H | 2a | 3a | 87, >20:1, >20:1 | 53:47 |
| 2b | OMe | H | 1b | H | 2a | 3b | 83, >20:1, >20:1 | 81:19 |
| 3 | OMe | H | 1b | OMe | 2b | 3c | 77, >20:1, >20:1 | 58:42 |
| 4 | OMe | H | 1b | OBn | 2c | 3d | 79, >20:1, >20:1 | 76:24 |
| 5b | OMe | OTIPS | 1c | H | 2a | 3e | 75, >20:1, >20:1 | 87:13 |
| 6 | OMe | OTIPS | 1c | OMe | 2b | 3f | 78, >20:1, >20:1 | 89:11 |
| 7 | OMe | OTBS | 1d | OMe | 2b | 3g | 74, >20:1, >20:1 | 80:20 |
| 8 | OMe | OTIPS | 1c | OBn | 2c | 3h | 79, >20:1, >20:1 | 94:6 |
| ➡ 9c | OMe | OTIPS | 1c | OBn | 2c | 3h | 96, >20:1, >20:1 | 97:3 |
Yields are of material isolated by silica gel chromatography. Enantioselectivity values were determined by HPLC analysis on a chiral stationary phase. (R)-SEGPHOS (Ar = phenyl).
Reaction was conducted from the diol oxidation level, 1a-1c (200 mol%).
1c (130 mol%), (R)-DM-SEGPEIOS (Ar = 3,5-xylyl). See the Supporting Information for further experimental details.
The scope of the enantioselective ruthenium catalyzed benzocyclobutenone-ketol cycloaddition was evaluated (Table 2). Retaining key structural features required to enforce high levels of asymmetric induction, an otherwise diverse set of benzocyclobutenones 1c, 1e-1l, were reacted with ketol 2c to furnish cycloadducts 3h-3p. In each case, complete regio- and diastereoselectivity were accompanied by excellent levels of enantiomeric enrichment. The formation of 3o and 3p illustrates tolerance of activated aryl chlorides. Variation of the ketol partner also was explored. As demonstrated by the formation of cycloadducts 3q-3s, electron releasing substituents that reside in para-relationship with respect to the ketone improve enantioselectivity, precluding the need for an ortho-benzyloxy-group. For example, in the formation of cycloadduct 3r, a 99:1 enantiomeric ratio is observed. The formation of 3r also demonstrates compatibility with amine functional groups.
Table 2.
Enantioselective intermolecular ruthenium(0) catalyzed cycloaddition of benzocyclobutenones 1c, 1e-1l with ketols 2c-2f to form cycloadducts 3h-3s.a
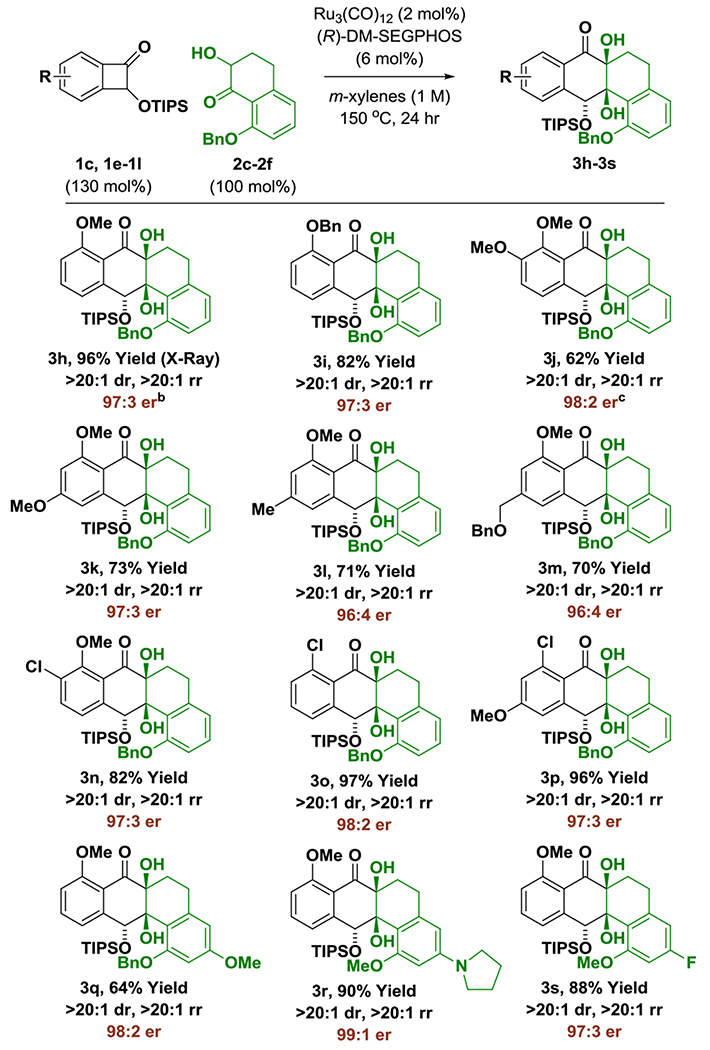 |
All reactions were performed on a 0.20 mmol scale. Yield of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
X-ray structure obtained after removal of silyl ether (3h′).
36 h.
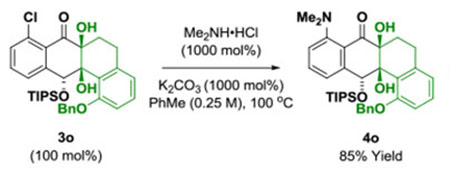
Underscoring the utility of this method vis-à-vis construction of type II polyketides, in particular, the angucycline antibiotics arenimycins A-D, collinone and SF22446A1-A3/SF22446B1-B3,16–19 benzocyclobutenone lc was exposed to ketol 2g under standard conditions for ruthenium-catalyzed cycloaddition (Scheme 1). The cycloadduct 3t was formed in good yield and incorporates the highly congested “bay region” motif found in arenimycin. To further demonstrate relevance to type II polyketide construction, cycloadduct 3h was subjected to silyl-deprotection followed by oxidation of the resulting alcohol to form the dione 4h (eq 1). Additionally, cycloadduct 3o was subjected to an SNAr reaction to form compound 4o, which incorporates a dimethylamino motif. Dimethylamino groups frequently occur in type II polyketide antibiotics(e.g. the tetracyclines) and, as recently demonstrated by Hergenrother, introduction of unhindered amines endows certain rigid “Gram-(+) only” antibacterials with Gram-(−) activity by conferring membrane permeability.20
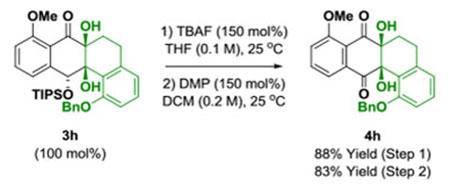 |
(eq. 1) |
 |
(eq. 2) |
Scheme 1.

Arenimycins A-D and collinone, antibacterial angucyclines that incorporate a bridgehead diol motif, and reaction of benzocyclobutenone 1c with ketol 2g to form cycloadduct 3ta.
aFor structurally related angucycline natural products, SF22446A1-A3 and SF22446B1-B3, see Ref. 19. Reaction was performed on 0.20 mmol scale. Yield of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase E1PEC analysis. See Supporting Information for further experimental details.
Finally, the ability to achieve high levels of enantioselectivity in cycloadditions of chiral racemic benzocyclobutenones 1c, 1e-1l has implications regarding the nature of the C-C bond oxidative addition event. Fligh levels of enantiomeric enrichment persist at equimolar loadings of benzocyclobutenone and ketol (Table 1, entry 8) and do not depend on stoichiometry, which suggests kinetic resolution of benzocyclobutenones lc, le-11 is not operative. Rather, these data corroborate a catalytic mechanism wherein stereoablative cycloreversion to form a discrete α-oxo-ortho-quinodimethane or “ortho-quinoid ketene methide”21 is followed by enantiodetermining C-C bond oxidative addition to form a ruthenaindanone complex (Figure 2).22 The indicated stereochemical model for ruthenaindanone formation accounts for structural features of the benzocyclobutenone that are required to enforce high levels of enantioselectivity, specifically, non-bonded interactions between the methoxy and triisopropylsiloxy groups of the α-oxo-ortho-quinodimethane with the xylyl moieties of DM-SEGPHOS.
Figure 2.

Stereoablative cycloreversion precedes oxidative addition and stereochemical model for ruthenaindanone formation.
In summary, we report the first enantioselective intermolecular metal catalyzed cycloadditions of benzocyclobutenones via C-C bond oxidative addition. Using a mononuclear15 ruthenium(0) complex derived from Ru3(CO)12 and (R)-DM-SEGPHOS, tetralone-derived ketols and benzocyclobutenones react to form cycloadducts with complete regio- and diastereoselectivity and high enantioselectivity. Whereas the majority of methods for convergent type II polyketide construction require use of stoichiometric carbanions under cryogenic conditions (e.g. the Hauser-Kraus annulation,23,24 aryne-mediated cycloadditions25), the present method assembles type II polyketide motifs in the absence of stoichiometric byproducts under non-cryogenic conditions. In an initial synthetic application, this method was used to prepare the congested “bay region” substructure characteristic of the angucycline antibiotics arenimycin, collinone and SF22446A1-A3/SF22446B1-B3.16–19 Future studies are aimed at defining ketol partners for the synthesis of linear tetracycline antibiotics.26
Supplementary Material
Acknowledgments.
The Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM093905). Dr. Ping-Xin Zhou is acknowledged for technical assistance. Swiss National Science Foundation (SNSF) is acknowledged for early postdoctoral mobility fellowship (SRS, P2BEP2_172231).
Footnotes
Supporting Information Available: Experimental procedures and spectral data. HPLC traces of racemic and enantiomerically enriched products. Crystallographic data for 3h′. Tins material is available free of charge via the internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Huffman MA; Liebeskind LS J. Am. Chem. Soc 1991, 113, 2771. [Google Scholar]
- (2).The nickel catalyzed reaction cited above is conducted at 0 °C and is applicable to unactivated alkynes. There is an earlier report of a thermal reaction that requires elevated temperatures (80-150 °C) and heteroatom substituted alkynes:; Danheiser RL; Gee SK J. Org. Chem 1984. 49, 1672. [Google Scholar]
- (3).(a) Jun C-H; Lee H J. Am. Chem. Soc 1999, 121, 880. [Google Scholar]; (b) Jun C-H; Lee H Lim S-G J. Am. Chem. Soc 2001, 123, 751. [DOI] [PubMed] [Google Scholar]
- (4).(a) Murakami M; Itahashi T; Ito Y J. Am. Chem. Soc 2002, 124, 13976. [DOI] [PubMed] [Google Scholar]; (b) Matsuda T; Fujimoto A; Ishibashi M; Murakami M Chem. Lett 2004, 55, 876. [Google Scholar]
- (5).For selected reviews on metal catalyzed C-C bond activation see:; (a) Bishop KC III., Chem. Rev 1976, 76, 461. [Google Scholar]; (b) Mitsudo T.-a.; Kondo T Synlett 2001, 309. [Google Scholar]; (c) Kondo T; Mitsudo T.-a. Chem. Lett 2005, 34, 1462. [Google Scholar]; (d) Murakami M; Matsuda T Chem. Comm 2011. 47, 1100. [DOI] [PubMed] [Google Scholar]; (e) Kondo T Bull. Chem. Soc. Jpn 2011. 84, 441. [Google Scholar]; (f) Nakao Y Top. Curr. Chem 2014, 346, 33. [DOI] [PubMed] [Google Scholar]; (g) Dreis AM; Douglas CJ Top. Curr. Chem 2014, 346, 85. [DOI] [PubMed] [Google Scholar]; (h) Souillart L; Cramer N Chem. Rev 2015, 115, 9410. [DOI] [PubMed] [Google Scholar]; (i) Shaw MH; Bower JF Chem. Comm 2016, 52, 10817. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Kondo T Eur. J. Org. Chem 2016, 1232. [Google Scholar]; (k) Chen P.-h.; Dong G Chem. Eur. J 2016, 22, 18290. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Murakami M; Ishida N J. Am. Chem. Soc 2016, 138, 13759. [DOI] [PubMed] [Google Scholar]; (m) Chen P.-h.; Billet BA; Tsukamoto T; Dong G ACS Catal 2017, 7, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Fumagalli G, Stanton S, Bower JF, Chem. Rev 2017107, 9404. [DOI] [PubMed] [Google Scholar]
- (6).For a recent review on donor-acceptor cyclopropanes, see:; Schneider TF; Kaschel J; Werz DB Angew. Chem. Int. Ed. 2014, 53, 5504. [DOI] [PubMed] [Google Scholar]
- (7).For selected examples of donor-acceptor cyclopropanes in enantiose-lective intermolecular metal catalyzed C-C bond activations, see:; (a) Parsons AT; Johnson JS J. Am. Chem. Soc 2009, 131, 3122. [DOI] [PubMed] [Google Scholar]; (b) Moran J; Smith AG; Carris RM; Johnson JS; Krische MJ J. Am. Chem. Soc 2011, 133, 18618. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trost BM; Morris PJ Angew. Chem., Int. Ed 2011, 50, 6167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Trost BM; Morris PJ; Sprague SJ J. Am. Chem. Soc 2012, 134, 17823. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu Q-S; Wang D-Y; Yang Z-J; Luan Y-X; Yang J-F; Pu Y-G; Ye M J. Am. Chem. Soc 2017, 139, 18150. [DOI] [PubMed] [Google Scholar]
- (8).(a) Shibata T; Nishizawa G; Endo K Synlett 2008, 765. [Google Scholar]; (b) Yada A; Fujita S; Murakami M J. Am. Chem. Soc 2014, 136, 7217. [DOI] [PubMed] [Google Scholar]; (c) Guo R; Zheng X; Zhang D; Zhang G Chem. Sci 2017, 8, 3002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zheng X; Guo R; Zhang G; Zhang D Chem. Sci 2018, 9, 1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).For recent reviews on transfer hydrogenative C-C bond formation, see:; (a) Ketcham JM; Shin I; Montgomery TP; Krische MJ Angew. Chem. Int. Ed. 2014, 53, 9142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ Science 2016, 354, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim SW; Zhang W; Krische MJ Ace. Chem. Res 2017, 50, 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sato H; Turnbull BWH; Fukaya K; Krische MJ Angew. Chem. Int. Ed 2018, 57, 3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Geary LM; Glasspoole BW; Kim MM; Krische MJ J. Am. Chem. Soc. 2013, 135, 3796. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McInturff EL; Mowat J; Waldeck AR; Krische MJ J. Am. Chem. Soc 2013, 135, 17230. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kasun ZA; Geary LM; Krische MJ Chem. Comm 2014, 7545. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Geary LM; Chen T-Y; Montgomery TP; Krische MJ J. Am. Chem. Soc 2014, 136, 5920. [DOI] [PubMed] [Google Scholar]; (e) Saxena A; Perez F; Krische MJ J. Am. Chem. Soc 2015, 137, 5883. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Saxena A; Perez F; Krische MJ Angew. Chem. Int. Ed 2016, 55, 1493. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Sato H; Fukaya K; Poudel BS; Krische MJ Angew. Chem. Int. Ed 2017, 56, 14667. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Sato H; Bender M; Chen W; Krische MJ J. Am. Chem. Soc 2016, 138, 16244. [DOI] [PubMed] [Google Scholar]; (i) Bender M; Turnbull BWH; Ambler BR; Krische MJ Science 2017, 357, 779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For intermolecular nickel catalyzed cycloadditions of cyclobutenone derivatives with π-unsaturated partners, see reference 1 and the following reports:; (a) Murakami M; Ashida S; Matsuda T J. Am. Chem. Soc 2005, 127, 6932. [DOI] [PubMed] [Google Scholar]; (b) Auvinet A-L; Harrity JPA Angew. Chem. Int. Ed 2011, 50, 2769. [DOI] [PubMed] [Google Scholar]; (c) Ho KYT; Aïssa C Chem. Eur. J 2012, 18, 3486. [DOI] [PubMed] [Google Scholar]; (d) Ishida N; Yuhki T; Murakami M Org. Lett 2012, 14, 3898. [DOI] [PubMed] [Google Scholar]; (e) Kumar P; Louie J Org. Lett 2012, 14, 2026. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Thakur A; Evangelista JL; Kumar P; Louie J J. Org. Chem 2015, 80, 9951. [DOI] [PubMed] [Google Scholar]
- (12).For intermolecular rhodium catalyzed cycloadditions of cyclobutenone derivatives with π-unsaturated partners, see:; (a) Kondo T; Taguchi Y; Kaneko Y; Niimi M; Mitsudo TA Angew. Chem. Int. Ed. 2004, 43, 5369. [DOI] [PubMed] [Google Scholar]; (b) Kondo T; Niimi M; Nomura M; Wada K; Mitsudo T Tetrahedron Lett. 2007, 48, 2837. [Google Scholar]
- (13).For intermolecular ruthenium catalyzed cycloadditions of cyclobutenone derivatives with π-unsaturated partners, see reference 12a and the following report:; Kondo T; Nakamura A; Okada T; Suzuki N; Wada K; Mitsudo T.-a. J. Am. Chem. Soc. 2000, 122, 6319. [Google Scholar]
- (14).For enantioselective organocatalyzed cycloadditions of non-benzannulated cyclobutenones, see:; (a) Li B-S; Wang Y; Jin Z; Zheng P; Ganguly R; Chi YR Nat. Commun 2015, 6, 6207. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li Y; Su X; Zhou W; Li W; Zhang J Chem. Eur. J 2015, 21, 4224. [DOI] [PubMed] [Google Scholar]
- (15).The reaction of Ru3(CO)12 with dppe in benzene solvent provides Ru(CO)3(dppe):; Sanchez-Delgado RA; Bradley JS; Wilkinson G J. Chem. Soc. Dalton Trans. 1976, 399. [Google Scholar]; As shown in reference 10b, the mononuclear ruthenium(II) adamantanecarboxylate complex, Ru(CO)(dppp)(C10H15CO2)2, is obtained in a similar manner.
- (16).For reviews on angucycline natural products, see:; (a) Kharel MK; Pahari P; Shepherd MD; Tibrewal N; Nybo SE; Shaaban KA; Rohr J Nat. Prod. Rep 2012, 29, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Carreño MC; Urbano A Synlett 2005, 1. [Google Scholar]; (c) Krohn K; Rohr J Top. Curr. Chem 1997,188, 127. [Google Scholar]
- (17).For isolation and initial biological profiles of arenimycins A-D, see:; (a) Asolkar RN; Kirkland TN; Jensen PR; Fenical WJ Antibiot. 2010, 63, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kersten RD; Ziemert N; Gonzalez DJ; Duggan BM; Nizet V; Dorrestein PC; Moore BS Proc. Natl. Acad. Sci. U. S. A 2013, 110, E4407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kang H-S; Brady SF J. Am. Chem. Soc 2014,136, 18111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).For isolation and initial biological profiles of collinone, see:; Martin R; Sterner O; Alvarez MA; De Clercq E; Bailey JE; Minas WJ Antibiot. 2001, 54, 239. [DOI] [PubMed] [Google Scholar]
- (19).For isolation and initial biological profiles of SF2446, see:; (a) Takeda U; Okada T; Takagi M; Gomi S; Itoh J; Sezaki M; Ito M; Miyadoh S; Shomura T J. Antibiot. 1988, 41, 417. [DOI] [PubMed] [Google Scholar]; (b) Gomi S; Sasaki T; Itoh J; Sezaki M J. Antibiot 1988, 41, 425. [DOI] [PubMed] [Google Scholar]
- (20).Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ Nature 2017, 545, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wurm T; Turnbull BWH; Ambler BR; Krische MJ J. Org. Chem 2017, 82, 13751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Liebeskind LS; Baysdon SL; South MS; Blount JF J. Organomet. Chem. 1980, 202, C73. [Google Scholar]
- (23).For selected reviews on the Hauser-Kraus annulation, see:; (a) Mitchell AS; Russell RA Tetrahedron 1995, 51, 5207. [Google Scholar]; (b) Rathwell K; Brimble MA Synthesis 2007, 643. [Google Scholar]; (c) Mal D; Pahari P Chem. Rev 2007,107, 1892. [DOI] [PubMed] [Google Scholar]
- (24).For selected examples use of the Hauser-Kraus annulation in the synthesis of Type II polyketides, see:; (a) Hauser FM; Rhee RP J. Am. Chem. Soc. 1979, 101, 1628. [Google Scholar]; (b) Hauser FM; Mal D J. Am. Chem. Soc 1983,105, 5688. [Google Scholar]; (c) Hauser FM; Gauuan PJF Org. Lett 1999,1, 671. [DOI] [PubMed] [Google Scholar]; (d) Matsumoto T; Yamaguchi H; Tanabe M; Yasui Y; Suzuki K Tetrahedron Lett. 2000, 41, 8393. [Google Scholar]; (e) Nicolaou KC; Zhang H; Chen JS; Crawford JJ; Pasunoori L .Angew. Chem., Int. Ed 2007, 46, 4704. [DOI] [PubMed] [Google Scholar]; (f) Gibson JS; Andrey O; Brimble MA Synthesis 2007, 2611. [Google Scholar]; (g) Nicolaou KC; Becker J; Lim YH; Lemire A; Neubauer T; Montero A J. Am. Chem. Soc 2009,131, 14812. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Mal D; De SR Org. Lett 2009, 11, 4398. [DOI] [PubMed] [Google Scholar]; (i) Yang X; Fu B; Yu B J. Am. Chem. Soc 2011,133, 12433. [DOI] [PubMed] [Google Scholar]; (j) Liau BB; Milgram BC; Shair MD J. Am. Chem. Soc 2012,134, 16765. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Adachi S; Watanabe K; Iwata Y; Kameda S; Miyaoka Y; Onozuka M; Mitsui R; Saikawa Y; Nakata M Angew. Chem., Int. Ed 2013, 52, 2087. [DOI] [PubMed] [Google Scholar]
- (25).For selected examples of the use of aryne-mediated benzannulation in the synthesis of Type II polyketides, see:; (a) Hosoya T; Takashiro E; Matsumoto T; Suzuki K J. Am. Chem. Soc. 1994,116, 1004. [Google Scholar]; (b) Matsumoto T; Sohma T; Yamaguchi H; Kurata S; Suzuki K Synlett 1995, 263. [Google Scholar]; (c) Chen C-L; Sparks SM; Martin SF J. Am. Chem. Soc 2006,128, 13696. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) O’Keefe BM; Mans DM; Kaelin DE Jr.; Martin SF J. Am. Chem. Soc 2010,132, 15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).For selected reviews on tetracycline antibiotics, see:; (a) Nguyen F; Starosta AL; Arenz S; Sohmen D; Doenhoefer A; Wilson DN Biol. Chem 2014, 395, 559. [DOI] [PubMed] [Google Scholar]; (b) (b) Wright PM; Seiple IB; Myers AG Angew. Chem. Int. Ed 2014, 53, 8840 Liu F; Myers AG Curr. Opin. Chem. Biol 2016, 32, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.