

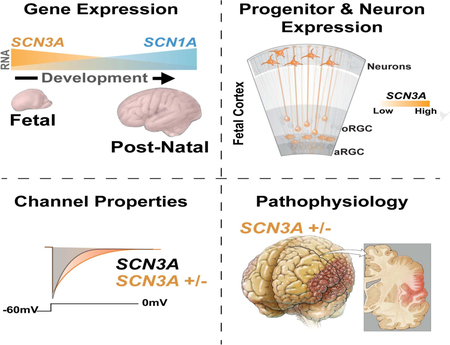
SUMMARY
Channelopathies are disorders caused by abnormal ion channel function in differentiated excitable tissues. We discovered a unique neurodevelopmental channelopathy resulting from pathogenic variants in SCN3A, a gene encoding the voltage-gated sodium channel NaV1.3. Pathogenic NaV1.3 channels showed altered biophysical properties including increased persistent current. Remarkably, affected individuals showed disrupted folding (polymicrogyria) of the perisylvian cortex of the brain but did not typically exhibit epilepsy; they presented with prominent speech and oral motor dysfunction, implicating SCN3A in prenatal development of human cortical language areas. The development of this disorder parallels SCN3A expression, which we observed to be highest early in fetal cortical development in progenitor cells of the outer subventricular zone and cortical plate neurons and decreased postnatally, when SCN1A (NaV1.1) expression increased. Disrupted cerebral cortical folding and neuronal migration were recapitulated in ferrets expressing the mutant channel, underscoring the unexpected role of SCN3A in progenitor cells and migrating neurons.
Keywords: Cortical Development, Polymicrogyria, Oromotor, Speech, SCN3A, NaV1.3, SCN1A, NaV1.1, Voltage-Gated Sodium Channel (VGSC), Outer Radial Glia
Graphical Abstract
INTRODUCTION
Human CNS channelopathies cause a range of brain disorders (Spillane et al., 2016). However, we lack a clear understanding of ion channel functions at early stages of cerebral cortical development, particularly in progenitor cells and migrating neurons (Spillane et al., 2016). Here we describe a disease of abnormal brain development, polymicrogyria (PMG), associated with pathogenic variants in SCN3A, which encodes brain-enriched voltage-gated sodium channel (VGSC) NaV1.3.
PMG is characterized by multiple abnormally small gyri in the cerebral cortex (Kuzniecky et al., 1993). PMG in language areas can affect speech (Stutterd and Leventer, 2014); its extent and location correlate with the severity of the resulting developmental language disorder (Guerreiro et al., 2002; Jansen et al., 2005). Although case reports suggest that SCN3A variants cause early infantile epilepsy, sometimes resolving in childhood (Holland et al., 2008; Lamar et al., 2017; Vanoye et al., 2014; Zaman et al., 2018), the pathogenic SCN3A alleles we discovered manifest as a fixed perisylvian anatomical defect with prominent orofacial weakness and/or speech delay, yet epilepsy was uncommon. Our findings reveal prenatal roles for SCN3A in cortical organization and neuronal migration, especially in speech and language areas, mirroring the enriched fetal expression of SCN3A. Prenatal cortical progenitor cells and neurons do not show detectable action potentials, supporting an unexpected role for sodium channel (SCN3A) in their proliferation and migration in the developing brain.
RESULTS AND DISCUSSION
Individuals with pathogenic SCN3A variants display aberrant cerebral cortical development and speech deficits
Magnetic resonance imaging (MRI) and neurological evaluations of six unrelated families with oral motor and/or speech deficits demonstrate bilateral perisylvian PMG (Families A & B; Figure 1A, 1B, Table S1), unilateral perisylvian PMG with abnormal sulcation (Families C & F), and bilateral frontoparietal PMG with microcephaly (Families D & E; Figure 1B). Families A and B are multigenerational with prominent dysarthric speech, and variable oral motor deficits and ID, clinically described previously (Guerreiro et al., 2000), compatible with autosomal dominant inheritance with incomplete penetrance (Figure 1C). Linkage and haplotype mapping, exome sequencing, and Sanger sequencing in Family A detected only one rare, predicted deleterious variant that segregated perfectly with the condition: it is heterozygous in affected individuals and obligate carriers, and absent in unaffected individuals (Figures 1C, S1A-C). The variant occurs in SCN3A, which encodes NaV1.3 (NM_006922.3), a VGSC alpha subunit (NaV). This channel’s pore-forming structure contains four homologous domains (DIDIV), each consisting of six membrane-spanning segments (S1-S6; Figure 2A). The SCN3A variant in Family A, c.5276T>A, p.Phe1759Tyr (F1759Y) affects a phenylalanine in the S6 segment of the fourth transmembrane domain (DIV-S6; Figures 2A, S1A), an important porefacing subunit regulating voltage-dependent inactivation (McPhee et al., 1995; Savio-Galimberti et al., 2012). A screen of 258 individuals with PMG and variable oral motor or speech deficits for SCN3A variants revealed five additional families. In Family B, the variant at c.4030A>C, p.Ile1344Leu (I1344L) affects an isoleucine in the S5 segment of the third domain (DIII-S5; Figures 1C, 2A, S1E), and was inherited by two affected siblings from their mother who had a normal MRI, suggesting she is a non-manifesting carrier, a feature of channelopathies (Cooper et al., 2013). The siblings’ maternal uncle, now deceased, reportedly never developed speech and had seizures but did not have brain imaging. The de novo SCN3A variant in Family C, c.2549T>C, p.Leu850Pro (L850P), present in one affected child and absent from both parents, affects a leucine in the S4 voltage-sensing segment of the second domain (DII-S4; Figures 1C, 2A, S1F). Unrelated Families D and E each have one affected child with the same de novo variant absent from their parents, SCN3A c.2624T>C, p.Ile875Thr (I875T), which is in the intracellular loop connecting the S4-S5 subunits in DII, close to the NaV voltage sensor (Savio-Galimberti et al., 2012) (Figures 1C, 2A, S1H). The SCN3A variant in Family F, 1862G>A, p.Arg621His (R621H), present in one affected child and absent in maternal DNA, affects an arginine in the S6-S1 cytoplasmic linker between DI and DII (Figures 1C,S1G).
Figure 1. Pathogenic variants in SCN3A disrupt cerebral cortical formation and oral motor function.

(A) MRI reconstruction of control (upper and middle panels) and age-matched affected individual B03 (lower panel). Red box outlines the PMG of perisylvian and surrounding areas. (B) Representative MRIs of affected individuals (Families A-F) reveal cortical malformations, PMG, abnormal gyral folding patterns, and shallow sulci. White dotted circles denote affected brain regions. MRI of asymptomatic individual A09 did not reveal visible PMG. Control, unaffected 11-year-old. Scale bar = 2cm. (C) Family A pedigree with a dominantly inherited point mutation causing amino acid change Phe1759Tyr in SCN3A. Family B pedigree with a dominantly inherited point mutation causing amino acid change Ile1344Leu in SCN3A. Single affected individual in Family C with a de novo point mutation causing amino acid change Leu850Pro in SCN3A. Single affected individuals in Families D and E with a de novo point mutation causing amino acid change Ile875Thr in SCN3A. Single affected individual in Family F with a point mutation causing amino acid change Arg621His in SCN3A; paternal sample unavailable (NA). Square, male; circle, female; black quadrant shading, affected individual (see phenotype legend). See also Figure S1 and Table S1.
Figure 2. Pathogenic SCN3A variants alter channel physiology.

(A) Top, Schematic of SCN3A alpha subunit where colored circles indicate approximate locations of mutations identified in Families A-F. Bottom, WT and pathogenic sodium channels are similarly expressed in transfected HEK293T cells. (B) Representative voltage clamp recordings from SCN3A transfected HEK293T cells evoked by the voltage step activation protocol (5mV increments; -100 to +50 mV). Scale bar = 1nA/3ms. (C) Left: Average voltage activation curve fit with Boltzmann equation (see STAR Methods), plotted as normalized conductance against step voltage demonstrate F1759Y (red) and I875T (blue) variants produce depolarizing and hyperpolarizing shifts in voltage dependent activation, respectively. Right upper: Bar graph shows conductance of largest evoked SCN3A current (normalized to cell capacitance) as decreased in F1759Y (red) and I875T (blue) variants; lower: Bar graph shows a positive and negative shift in V½ voltage of activation for F1759Y and I875T variants compared to WT-SCN3A. (D) Upper: Representative sodium currents evoked by inactivation protocol. Lower: Voltage dependence of NaV current inactivation demonstrate F1759Y and I875T variants having shifted inactivation compared to WT-SCN3A. Boltzmann fit plotted as normalized current (channel availability) at 0 mV against the conditioning pre-pulse potential (-100 to 0 mV, See Table S2). (E) Left: Peak normalized sodium transient currents. Right: Voltage step plotted vs. persistent current (I-NaP), I-NaP measurement collected as the mean of the last 30 ms of the voltage step and presented as percent maximal peak inward current, demonstrate increase persistent current in F1759Y and I875T variants, compared to WT. (F) Left: Representative voltage clamp recordings, current density measurements indicated from area regions in yellow (15 ms). Right: Na+ currents plotted as current density against the stepping potential demonstrate F1759Y and I875T variants differentially affect current flow compared to WT. See also Table S2. (G) Primary culture of dissociated human fetal cortical neurons generated from 19 weeks gestation (WKSG) cortical plate. (H) Representative voltage clamp recordings from neurons in (G) evoked by the voltage step activation protocol (20mV increments; -80 to +40 mV) reveal sodium influx (black arrow). Scale bar = 100pA/1ms. (I) Representative current clamp recordings from same neurons (resting Vm = -57mV) evoked by current stepping protocol (10pA increments, 500ms) demonstrate fetal neurons lack action potentials, but have small voltage-activated depolarizing potentials that likely represent immature voltage gated Na+ influx (black arrow). Scale bar = 10mV/200ms. Measurements presented as mean ± S.E.M. (t-test; *P < 0.05, **P < 0.01).
R621, L850, I875, I1344, and F1759, are highly conserved among human VGSC subtype genes (SCN1A-SCN11A) and invariant in other vertebrate SCN3A orthologs (Figures S1 D-H). L850P, I1344L, and F1759Y are completely absent in large human genetics databases, such as the Genome Aggregation Database (gnomAD.broadinstitute.org), which contains 4877782 exome or genome sequences for > 138,000 individuals, including ~13,000 Finns. Four other I875T alleles have been reported, two individuals in large ID and developmental disability cohorts reported without further phenotypic detail (Deciphering Developmental Disorders Study, 2017; Lelieveld et al., 2016), and two individuals with PMG and severe epilepsy (one the same as reported herein) (Zaman et al., 2018), suggesting I875T is a recurrent, severely damaging allele. In silico pathogenicity predictions using SIFT, MutationTaster, and PolyPhen2 suggested that R621H, L850P, I875T, I1344L, and F1759Y are all damaging and likely disease-causing (Supplemental text). The mutations suggest a range of severity, from inheritance of variably penetrant perisylvian PMG and ID without epilepsy (F1759Y, I1344L) to the severely damaging recurrent I875T allele, which is de novo in the 5 families identified to date with widespread PMG, microcephaly, and severe seizures.
Pathogenic SCN3A (NaV1.3) variants alter channel physiology
Sodium current recordings from HEK293T cells that expressed mutated SCN3A harboring the F1759Y or I875T alleles displayed abnormal channel properties. Using whole-cell patch recordings, we evoked transient sodium currents over a range of voltages and measured the conductance-voltage curve in transfected cells (Figure 2B). Both mutant channels exhibited abnormal peak conductance (Ipeak) and half-maximal voltage of activation (V½; Figure 2C). Compared to wild type (WT), the F1759Y mutation produced a depolarizing shift (+7.8 ± 1.6 mV; t-test, p<0.01, n = 28), while the I875T mutation produced a hyperpolarizing shift (-5.4 ± 1.7 mV; t-test, p<0.01, n = 17; Figure 2C; see Table S2 for complete sodium current values). NaV channel availability calculated by measuring steady-state (fast) inactivation with a stepwise conditioning protocol (100 ms pre-pulse from −100 to 0 mV; Figure 2D) showed a trend towards depolarizing shifts in half-maximal voltage of inactivation (F1759Y, 5.2 ± 1.1 mV, ttest, p = 0.24, n = 28; and I875T, 5.5 ± 0.6 mV; t-test, p = 0.27 n = 17; Figure 2D).
Whereas SCN3A channels are already distinct from SCN1A channels by their larger, non-inactivating persistent current (I-NaP) (Lampert et al., 2006; Sun et al., 2007), F1759Y and I875T increased this difference further. The I-NaP measured at depolarized potentials for I875T (+10 to +50 mV; t-test, n = 17, p<0.05), and at all potentials tested for F1759Y (-10 to +50mV; t-test, n = 28, p<0.01), was greater than WT NaV1.3 controls (Figure 2E, Table S2).
Because I-NaP depolarizes cells (Stafstrom, 2007) and mutated SCN3A can enhance excitability (Estacion et al., 2010), we next evaluated total charge (current density) conducted by channels during a voltage step depolarization (Figure 2F). The F1759Y pathogenic variant conducted more sodium current through depolarizing potentials (-10mV to +25 mV; t-test, n=28, p<0.05), while the I875T pathogenic variant conducted less sodium current than controls (t-test, n=17, p<0.01; Figure 2F, Table S2). Because I-NaP is a characteristic of SCN3A channels and is similarly increased by both variants, it might underlie PMG associated with these variants.
Patch clamping human primary fetal cortical neurons from early-middle gestation showed small reproducible sodium currents (110pA ± 45pA, n = 12; Figure 2H), but no regenerative action potentials under the tested conditions (Figure 2I), consistent with prior reports (Moore et al., 2009). The absence of action potentials prenatally suggests that during human fetal development, SCN3A sodium currents may have functions beyond the classical role of action potential electrogenesis.
SCN3A expression is developmentally regulated in human brain
Unlike SCN1A, SCN3A is robustly expressed across human cortical regions during fetal periods but downregulated after birth (Figures 3A, S3C). RNA in situ analyses of fetal (20 weeks gestation) human brain revealed the highest SCN3A expression in the cortical plate (CP), which contains immature neurons, with significant expression in the outer subventricular zone (oSVZ) and intermediate zone (IZ), but low expression in the ventricular zone (VZ; Figures 3C, 3E, S3B). Consistent with this pattern, single-cell RNA sequencing (scRNA-seq) data at 16–18 weeks gestation (Pollen et al., 2015) showed SCN3A enrichment in intermediate progenitors and neurons (Figures 3B, S3A), and in situ analysis for outer radial glia (oRG; Figures S3B), a progenitor cell type present in developing gyrencephalic brains (non-human primates and humans), but rare or absent in mice (Florio and Huttner, 2014). oRG cells, but not ventricular RG cells, display small sodium currents (Hansen et al., 2010), suggesting SCN3A underlies sodium conductance in these cortical progenitors, which are key to gyrification. Adult human cortex (38 years old) showed very low SCN3A expression across all cortical layers (Figures 3D, 3F, S3C).
Figure 3. SCN3A expression is developmentally regulated in human brain.

(A) SCN3A transcripts are enriched during fetal gestational weeks (WKSG) and decrease postnatally. SCN1A (NaV1.1) expression starts lower and increases postnatally. Raw data analyzed from Allen Brain Atlas, presented as log2 RPKM (reads per kilobase per million) values. See also Figure S3C. (B) Single cell RNAseq dataset generated from 16–18 WKSG fetal cortex (Pollen et al., 2015), show higher SCN3A vs. SCN1A expression in radial glial cells (RGCs), intermediate progenitor cells (IPCs) and neurons (RGCs: p < 0.005; IPCs: p < 0.001; neurons: p < 0.001). Graph depicts two-part Wilcoxon test; **p < 0.01, ***p < 0.001 with Bonferroni adjusted alpha level of 0.017 (0.05/3). See also Figure S3A. (C) SCN3A mRNA in situ hybridization at 20 WKSG shows higher expression in the cortical plate (CP), and lower expression in the subventricular zone (SVZ). (D) SCN3A mRNA in situ hybridization in adult cortex. NeuN (RBFOX3) mRNA in situ hybridization reveals architecture and neurons in tissue section (38 y.o.; scale bar = 1cm). (E) Chromogenic mRNA in situ hybridization (i) and corresponding fluorescence imaging (ii) of 20 WKSG brain. Cell type specific markers for intermediate progenitors (TBR2) and neural progenitors (Vimentin, VIM) show SCN3A transcripts in SVZ/VZ. Scale bar left = 500μm; right = 25μm. See also Figure S3A. (F) mRNA in situ for SCN3A (i) and NeuN (ii) with fluorescence imaging (iii) of adult cortex (38 y.o., Brodmann area 27). Scale bar = 500μm. VZ, ventricular zone; IMZ, intermediate zone; SP, subplate; WM, white matter; Roman numerals (I - VI) correspond to approximate cortical layers. See also Figure S3.
SCN3A expression differs sharply from that of other VGSCs, including SCN1A, which was lower during gestation and upregulated postnatally (Figures 3A, S3C). Low SCN1A expression during gestation was confirmed across all fetal cortical cell types by scRNA-seq (two-part Wilcoxon test, p<0.005; Figure 3B). Notably, postnatal upregulation of SCN1A correlates with the age of onset of epilepsy associated with SCN1A variants (Spillane et al., 2016). Clinical reports have associated other SCN3A variants with infantile epilepsy that resolves with age (Vanoye et al., 2014), perhaps because fetal SCN3A expression later decreases. Mice also display post-natal Scn3a downregulation (Beckh et al., 1989; Gordon et al., 1987), and mice with loss-of-function (LOF) Scn3a mutations do not display spontaneous seizures (Lamar et al., 2017). Thus, different expression patterns between SCN1A and SCN3A may relate to the ages of onset and severity of VGSC channelopathies, possibly explaining the limited adult epilepsy observed in individuals with SCN3A pathogenic variants in this study (Families A, B, C, E), and the absence of brain malformations seen with SCN1A mutations (Spillane et al., 2016).
Overexpression of SCN3A in human cortical neurons stimulates neurite branching, but is impaired by I875T and F1759Y mutants
Expression of SCN3A in cultured human fetal neurons produced a modest increase in neurite branching without an overall increase in neurite length, and this branching effect was attenuated by both I875T and F1759Y mutants (Figure S2). Fetal neurons overexpressing the I875T mutant displayed modestly shorter neurites overall, similar to neuropathy-associated NaV1.7 mutations which disrupts neurites (Estacion et al., 2015), whereas SCN3A-F1759Y showed no differences compared to controls (Figure S2C). Over expression of the β-NaV subunit; which affects persistent current (Aman et al., 2009), increased neurite length (Figure S2C), consistent with previous reports in rodent cortical neurons (Brackenbury et al., 2008). These results further support the possibility that SCN3A regulates neuronal development through action potential-independent mechanisms, and that the disease-associated mutations impact these processes.
Altered SCN3A expression disrupts ferret cerebral cortical development
SCN3A-F1759Y mutation disrupted cortical gyral formation in vivo in the developing ferret cerebral cortex (Figure 4). Unlike rodents, ferret brains possess a stereotyped pattern of gyri and sulci, a human-like molecular diversity of progenitor cells, and a developmental timeline that permits transient gene overexpression in embryos via in utero electroporation (IUE) (Johnson et al., 2015; Kawasaki et al., 2012). Following IUE delivery of F1759Y or WT SCN3A at embryonic day 33 (E33), we analyzed ferret brains at: (1) birth (postnatal day 0, P0), coincident with mid-corticogenesis (cortical layers are still forming and cortex largely smooth), and (2) P33, when gyri and sulci are more fully formed. At P0, F1759Y overexpression severely disrupted neuronal migration: Tuj1-positive neurons were displaced near the VZ and SVZ instead of arriving at their age-appropriate cortical laminae (Figure 4C). At P33 (Figure 4E), F1759Y-transfected embryos developed atypical gyrification patterns, including the formation of additional sulci and gyri (5 of 5 ferrets examined; Figures 4E, S4C). At both time points (P0 & P33), the overexpression of SCN3A-WT had modest effects: neuronal migration was slightly altered at P0 (Figure 4B), perhaps resulting from enhanced sodium entry, and the gyral phenotype was modestly affected at P33 (2 of 6 ferrets examined; Figures 4H, S4C). Similar injections of a control vector did not alter the gyrification pattern (0 of 5 ferrets examined; Figures 4J, S4C).
Figure 4. Expression of mutant SCN3A disrupts neuronal migration and ferret cerebral cortical gyrification.

(A) Schematic of human and ferret development and study design. (B, C) Cerebral cortex of kits (P0) following an IUE at E33 with mCherry together with SCN3A-WT or -F1759Y; Tiled confocal images; scale bar = 25μm. Colocalization of mCherry-positive cells in the VZ/SVZ with cell type specific markers: progenitor (Ki67, TBR2), glial-astrocyte (GFAP), and neuronal (TUJ1) show mCherry expression in neurons. (D) Distribution pattern across cortex (bins 1–5) of mCherry positive cells in (A) reveals SCN3A alters neuronal migration (t-test; *p < 0.05, **p < 0.01). See also Figure S4F. (E) Upper: schematics of study design and stereotyped ferret gyri/sulci at P33. Lower: P30 ferret brain expressing electroporated SCN3A-F1759Y resulting in atypical gyrification pattern. (F) MRI images of P33 brain expressing SCN3A-F1759Y; scale bar = 5mm. Right, arrow denotes clusters of gray matter heterotopia. (G) Upper: mCherry positive (red) and Nissl (green) positive cells in brain sections from (F); scale bar = 2mm. Inset, higher magnification image suggesting a non-cell autonomous effect of SCN3A activation (right panel scale bar = 200μm). Lower: mCherry positive cells analyzed as in (C). Examples of brains expressing WT SCN3A (H, I) and mCherry vector (J, K), show stereotyped brain development. Scale bar = 5mm. See also Figure S4.
Finally, reconstruction of MRI images of adult ferret cortices revealed formation of cortical gray matter heterotopia in SCN3A-F1759Y-expressing brains (Figure 4F), but not in SCN3A-WT or empty vector controls (Figures 4I, 4K). Heterotopia consisted of displaced F1759Y mCherry-positive cells (Figure 4G), intermixed with mCherry-negative and SATB2positive cells, an upper layer cortical maker (Figure S4E), suggesting that some of the migratory defect involved WT cells disrupted non-cell-autonomously, perhaps by loss or damage to radial glia that do not normally express SCN3A. Evaluation of SATB2-positive cells at P15 also confirmed robust displacement of these cells in electroporated regions (Figure S4D). MRI analyses of SCN3A-WT and empty vector-expressing controls did not show displaced cells or heterotopia (Figures 4I, 4K). Together, these findings further support the model that SCN3A gene dosage, in addition to acquired pathological mutations, alters early cerebral cortical development, with sodium channel dysregulation leading to both cell-autonomous effects in neurons and potential effects on other cell types as well.
Prenatal SCN3A may not always involve classic action potential upstroke generation. Human oRG and astrocytes show functional sodium channels, with small and prolonged conductances, but no action potentials (Black et al., 1995; Hansen et al., 2010; Sontheimer and Waxman, 1993). Here, immature cortical neurons isolated when the sylvian fissure is developing did not show regenerative action potentials in vitro, but SCN3A may contribute Na+ conductances that modulate other voltage-dependent processes and downstream pathways, e.g., calcium signaling. In glia and neurons, changes to sodium flux can reverse Na+/Ca++ exchangers, triggering calcium waves and neurite degeneration, as well as activating Rac1 and ERK1/2 (Estacion et al., 2015; Pappalardo et al., 2014; Persson et al., 2014). Additional noncanonical roles of VGSCs in nonexcitable cells that might be affected include the release of bioactive molecules, regulation of Na+/K+-ATPase activity and cell motility, or a role in intracellular membranes to modulate phagocytosis (Black and Waxman, 2013). Fetal SCN3A mutations may activate different mechanisms than mutations in other VGSCs, like SCN1A, that are expressed postnatally, when both GOF and LOF are likely to modify excitability, altering circuit function (Blanchard et al., 2015; Escayg and Goldin, 2010), and SCN2A, where LOF (but not GOF) is reported in individuals with autism spectrum disorders without epilepsy (Ben-Shalom et al., 2017). As channels that affect membrane potential are important for cortical development (Ackman and Crair, 2014; LoTurco et al., 1995; Rash et al., 2016), and modifications to membrane potential in progenitor cells can disrupt cortical lamination in mice (Hurni et al., 2017), regulation of ion flux may represent a conserved mechanism governing development of several cell types.
To date, genetic evidence connecting cortical malformations with oral motor and developmental language disorders is limited. Guerreiro et al., (2002) and Jansen et al., (2005) demonstrated the presence of a developmental language delay in individuals with bilateral perisylvian PMG, and the high prevalence of PMG in persons with developmental language delay. Both studies correlated the location and extent of the cortical malformation with the type and severity of developmental language delay. The perisylvian region contains key language areas which can be affected by PMG (Stutterd and Leventer, 2014). Notably, infantile epilepsy cases with SCN3A variants not showing PMG may display variable abnormalities in oral motor and speech development (Lamar et al., 2017; Vanoye et al., 2014). SCN3A deficits also overlap with features of syndromes caused by mutations in the N-methyl-D-aspartate receptor subunit GRIN2A and the transcription factor FOXP2 (Adam et al., 1993; Turner et al., 2015) as well as the inherited “Worster-Drought” syndrome (Clark et al., 2000) which resembles several of our SCN3A families. Understanding the early function of these genes will illuminate development of the Sylvian fissure and its adjacent perisylvian language and oral motor areas.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Maria K Lehtinen, (maria.lehtinen@childrens.harvard.edu )
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects and samples
Research performed on samples of human origin was conducted according to protocols approved by the institutional review boards of Boston Children’s Hospital, Beth Israel Deaconess Medical Center, Montreal Neurological Hospital and Institute, Erasmus MC UMC Rotterdam, and by The Children’s Hospital of University of Helsinki. Subjects were identified and evaluated in a clinical setting, and biological samples collected after obtaining written informed consent. Fetal brain tissue was received after release from clinical pathology, with a maximum post-mortem interval of 4 h. Cases with known anomalies were excluded. Male and female brain tissue was collected from 17–20 gestation weeks. Tissue was transported in HBSS medium on ice to the laboratory for research processing.
Phenotypic assessment
All affected individuals and clinical data were examined by neurologists and radiologists, and polymicrogyria was diagnosed using criteria described previously (Guerrini et al., 2000). Detailed clinical information on affected individuals in Family A was collected over a 20-year period, summarized in Table S1. The affected individuals of Families B - F were evaluated by neurologists over many years and detailed clinical and radiographic evaluations are described in supplemental results text.
Clinical description of families
Family A, a multigenerational Finnish family, is the largest family with bilateral perisylvian polymicrogyria (PMG) reported to date (Guerreiro et al., 2000), having four pairs of affected siblings with variable penetrance and expression of PMG and oral motor dysfunction (dysarthric speech and deficient tongue movement) (Figures 1B & C; Table S1). With the exception of a single rolandic seizure in one individual (A12), neurological evaluation of the affected individuals performed throughout life confirmed the absence of epilepsy in Family A. Two affected individuals (A11 and A12) showed slightly subnormal language comprehension and intellectual ability, and these features ranged from borderline normal to subnormal in others (Figure 1C; Table S1).
Family B is a multigenerational French Canadian family with bilateral perisylvian PMG and oral motor dyspraxia (Figures 1B & C). As previously described, the two affected siblings (B02 and B03) had delayed speech development and a history of choking in infancy, and have dysarthria, drooling and significant restriction of tongue movements. Additionally, they were both noted to have difficulty whistling and blowing, abnormal brisk jaw jerk, increased reflexes, absent gag reflex and automatic involuntary dissociation of facial movements (Guerreiro et al., 2000). Upon reevaluation at 43 years of age, B03 had macrocephaly with a head circumference of 57 cm (99th percentile, 2.4 standard deviations), her height was 163 cm (48th percentile) and her weight was 126 pounds (49th percentile). She reported a history of having difficulty manipulating objects in childhood, and was observed to have normal muscle tone and gross motor movements, but had a mild tremor at the end of intentional movements and mild difficulty with rapid alternating movements. Prior clinical testing for B03 included a normal karyotype (46, XX) and normal IQ. B02 was evaluated on the same occasion, was 50 years of age, and presented with a very similar clinical picture as his sister, B03. B01, the daughter of B02 and previously described to have mild dysarthria and a normal brain MRI (Guerreiro et al., 2000), was reported at 16 years of age to have intellectual disability but no speech disorder. A sister of B02 and B03 died at 16 months of age of unknown cause, and a maternal uncle never developed speech, had epilepsy and died in adulthood. Brain imaging was not available for either the deceased sister or uncle. B04, sister of B02 and B03, was unaffected. B05, the mother of B02 and B03, was unaffected and had a normal brain MRI (Guerrini et al., 2000) and normal fluorescence in situ hybridization study of the DiGeorge syndrome region [ish 22q11.2 (TUPLE1×2)].
Family C is of Dutch descent and has a single male affected with unilateral perisylvian PMG and contralateral abnormal perisylvian sulcation with developmental motor and speech delay (Figure 1B & C). The proband has two healthy older sisters and healthy parents. He was born by induced delivery at 41 weeks gestation, following an uncomplicated pregnancy, and had a birth weight of 3.5 kg (46th percentile). In infancy, he was unable to breastfeed and learning to chew was slow and required therapy. He presented at 1 year of age with delay of motor development and hypotonia. A brain MRI was obtained at 2 years of age, noting right-sided perisylvian PMG, involving the posterior-frontal, anterior-parietal and postero-superior temporal lobes. The posterior body and isthmus of the corpus callosum was thin, the right cerebral ventricle was mildly enlarged and white matter volume was slightly diminished in the right hemisphere. At 3 years of age, his height is 93 cm (-1.5 standard deviations) and head circumference is 52.0 cm (0.8 standard deviations). He is noted to have a prominent forehead and downward-slanting eyelids. Neurologic examination revealed axial hypotonia and very mild left pyramidal signs but no differences in strength. Vision and hearing are normal; he sleeps well and has not had any seizures. At 3 years, he is able to stably walk and tries to jump and run, has good social skills but weak executive functions. Speech is delayed, as he uses two to three word sentences only, but oral motor abilities, including articulation, swallowing and chewing, are normal. He has a normal gag reflex and no mouth breathing.
Family D is of mixed European descent (Poland, Germany and Ireland) and has a single male affected with frontoparietal PMG, seizures, microcephaly, and neurodevelopmental delay (Figures 1B & C). The proband was born at 42 weeks gestation following an uncomplicated pregnancy and presented with seizures at 2 weeks of age. At 2 months of age, he had a normal head circumference and length (40th and 80th percentiles respectively by report), a small penis (<10th percentile reportedly), was non-dysmorphic, exhibited hypotonia, had feeding difficulties and apneic episodes, as well as developmental delay (not smiling or rolling). Brain MRI at 2 months of age revealed extensive coarse PMG in the frontal and parietal lobes with subcortical calcifications and abnormal frontal white matter. A thin but fully formed corpus callosum was noted and the white matter was markedly diminished, especially frontally. Upon evaluation at 1 year, he had a mixed seizure disorder with complex and simple partial seizures described as occurring 1 per day initially, worsening when he was ill, and better controlled more recently. There was significant generalized hypotonia with spasticity and upper extremity hypertonia, and he did not have complete head control. He had oral motor discoordination (unable to bottle feed but able to breastfeed), recurrent otitis media and genu valgum that did not require intervention. Developmentally, he was rolling over at 1 year but did not fully grab, hold or transfer toys from his left to right hand; he said “ma” and responded to his name but did not fix or follow consistently. At that time he received physical, occupational and vision therapies. Head circumference at 10 years 10 months was 49 cm (3.89 standard deviations below the mean). Ophthalmologic evaluation noted strabismus and significant cortical visual impairment with normal retina and optic nerves. At 11 years 8 months of age he has remained non-verbal, with dysphagia and mixed seizure disorder, hypotonia and severe global delay.
Family E is a non-consanguineous family from the United Arab Emirates and has a single female affected with bilateral frontoparietal PMG, microcephaly and severe global developmental delay (Figures 1B & C). The proband was born at full term following an unremarkable pregnancy and delivery, and her birth weight was 3 kg (21st percentile; -0.8 standard deviations). Hypotonia and developmental delay prompted a brain MRI at 5 months of age, which revealed bilateral PMG sparing the posterior temporal and occipital lobes; the cerebral ventricles were mildly enlarged and white matter volume slightly reduced. At 10 months of age, she had microcephaly; her head circumference was 40.7 cm (-3.2 standard deviations). At that time she was able to roll over but not sit or crawl, she had a palmar grasp only and had stereotyped movements. Neurologic evaluation revealed right esotropia, no response to visual or auditory stimuli, generalized hypotonia with head lag and normal deep tendon reflexes. At 13 months, she had not made further motor developmental progress but responded well to visual and auditory stimuli, and was social. She did not have seizures, though an EEG was reported to have shown bilateral synchronous epileptiform discharges but no medication was prescribed. Karyotype and serum lactic acid and ammonia levels were reported as normal. Another brain MRI was performed at 9 years of age that reported a thickened cortex bilaterally, shallow Sylvian fissures, slightly prominent cerebral ventricles, sparse white matter of normal signal intensity and minimally increased extra-axial cerebral spinal fluid. Follow-up evaluation at 13 years of age revealed spastic quadriplegia and global developmental delay. She was wheelchair bound, could roll over but not sit up or stand. She was nonverbal, had excessive drooling and feeding difficulties, taking soft and pureed foods orally with slow swallowing. Decreased range of motion, flexion deformity of the left wrist, equinus deformity of the left foot, and significant scoliosis with right curvature were noted. She was observed to be very attentive and smiling, and to both fix and follow appropriately. She was reported to menstruate, have urinary incontinence and have sleep disturbances.
Family F is of French Canadian, Russian and Irish descent and has a single female affected with unilateral right-sided PMG and seizures (Figures 1B & C). The proband has three siblings and a father who are healthy, and her mother has a history of thyroid adenoma and left frontal meningioma. She was born at term, had a birth weight of 4.9 kg (>99th percentile, 3.2 standard deviations), and had an uncomplicated neonatal course. Seizures were first noted at 2 years of age and she had a history of motor developmental delay but normal speech and oral motor development. Her ability to reason and to comprehend others’ language was reportedly reduced.
Brain MRI at 21 years of age revealed right hemispheric PMG with the right hemisphere being smaller than the left, fully formed and thick corpus callosum, small right basal ganglia and thalamus, significant enlargement of the right lateral ventricle and severe diminution of the right hemispheric white matter. On evaluation at 28 years of age, she had macrocephaly with a head circumference of 60.5 cm (5.1 standard deviations), her height was 168 cm (77th percentile, 0.74 standard deviations), and her weight was 188 lbs (95th percentile, 1.7 standard deviations). Notably, her mother was also macrocephalic with a head circumference of 61 cm (6 standard deviations). Treated with Tegretol, a seizure frequency of once or twice a year was reported. She had good vision with normal visual field and extraocular movements.
METHODS DETAILS
Human genetics
Leukocyte-derived DNA was utilized for studies of all families. Genome-wide genotyping was performed on individuals 1–12 in Family A using the Illumina Infinium HumanHap550-Duo BeadChip. These data were used to perform linkage and haplotype analysis. Genotypes were called using GenomeStudio Software (Illumina). Low performing SNPs (those not called in all individuals in this study or not called in >5% of individuals run on the same chips), low variance SNPs (those with minor allele frequencies (MAF) of 0 in all individuals in this study; or that have an MAF < 10% or that deviate from HWE with P-value < 0.00000001 among unrelated individuals run on the same chips), and SNPs with Mendelian and non-Mendelian errors (calculated with Merlin) were removed from the analysis using PLINK. PLINK was used to thin SNPs to reduce linkage disequilibrium. Linkage analysis was performed to generate a LOD score from the remaining SNPs using Merlin under a dominant mode of inheritance assuming a disease allele prevalence of 0.0001 and a penetrance of 0.6. Haplotype analysis was performed on the SNPs around the maximum LOD score using Merlin. Whole exome sequencing (WES) was performed at the Broad Institute using Agilent Sure-Select Human All Exon v2.0 capture array and sequenced on Illumina HiSeq2000 sequencers.
Identification and segregation of alleles
Linkage and haplotype analysis in Family A identified a candidate locus at Chromosome 2 (Figures S1B & C) with the highest LOD score of 2.48 at rs10930488-rs1489635; chr2:164,028,745–172,490,226 (hg19). Targeted sequencing was performed and the mutation identified in Family A is Chr2(GRCh37):g.165947387A>T, NM_006922.3:c.5276T>A, p.Phe1759Tyr (Figure S1A). Algorithms suggest that the mutation is of a highly conserved nucleotide (phyloP: 5.29 [-14.1;6.4]), and predict the amino acid change to be deleterious [SIFT: deleterious (score: 0, median: 4.32), MutationTaster: disease-causing (p-value: 1), PolyPhen-2: probably damaging (score: 1.0)]. The mutation identified in Family B is Chr2(GRCh37):g.165953971T>G, NM_006922.3:c.4030A>C, p.Ile1344Leu. Algorithms suggest that the mutation is of a highly conserved nucleotide (phyloP: 5.21 [-14.1;6.4]), and predict the amino acid change to be deleterious [SIFT: deleterious (score: 0, median: 4.32), MutationTaster: disease-causing (p-value: 1), PolyPhen-2: possibly damaging (score: 0.88)].
The de novo mutation identified in Family C is Chr2(GRCh37):g.165987770A>G, NM_006922.3:c.2549T>C, p.Leu850Pro. Algorithms suggest that the mutation is of a highly conserved nucleotide (phyloP: 5.13 [-14.1;6.4]), and predict the amino acid change to be deleterious [SIFT: Deleterious (score: 0, median: 4.32), MutationTaster: disease-causing (pvalue: 1), PolyPhen-2: probably damaging (score: 0.999). The de novo mutation in Families D and E is Chr2(GRCh37):g165986748A>G, NM_006922.3:c.2624T>C, p.Ile875Thr. Algorithms suggest that the mutation is of a highly conserved nucleotide (phyloP: 4.89 [-14.1;6.4]), and predict the amino acid change to be deleterious [SIFT: Deleterious (score: 0.01, median: 4.32), MutationTaster: disease causing (p-value: 1), PolyPhen2: probably damaging (score: 1.0)]. I875 was previously reported in two large intellectual and developmental disability cohorts, ClinVar ID 373960 (Deciphering Developmental Disorders Study, 2017; Lelieveld et al., 2016). The mutation in Family F is Chr2(GRCh37):g.165997318C>T, NM_006922.3:c.1862G>A, p.Arg621His. Algorithms suggest that the mutation is of a highly conserved nucleotide (phyloP: 4.48 [-14.1;6.4]), and predict the amino acid change to be deleterious [SIFT: Deleterious (score: 0, median: 4.32), MutationTaster: disease causing (prob: 1), PolyPhen2: probably damaging (score: 0.99)].
Sanger sequencing confirmed familial segregation of the inherited mutations in Family A and B, and the de novo occurrence of the mutations in Family C, D, and E, being absent from both parents (Figures S1F & H). In Family F, the mutation was confirmed by Sanger sequencing in the proband and was absent from her mother but DNA was not available from the father to verify its de novo occurrence (Figure S1G). Further, amino acids upstream and downstream of R621, L850, I875, I1344 and F1759 in SCN3A are highly conserved in orthologs as well, 16 of 20 amino acids, 19 of 20 amino acids, 19 of 20 amino acids, 20 of 20 amino acids, and 20 of 20 amino acids respectively (Figures S1 D-H). Taken together, these data demonstrate that the SCN3A gene and the families’ mutation sites and surrounding alleles are highly conserved.
Electrophysiology
Neurons were visualized using an inverted Olympus IX73 microscope with epifluorescence capabilities. Whole-cell voltage-clamp currents (series resistance compensated 85%) were recorded with an Axopatch 200B amplifier and pClamp 9.2 (Molecular Devices, Sunnyvale, CA). Glass pipettes were formed from borosilicate glass (1.5–3 Mohm) on a Sutter Puller (P-97 Sutter Instrument) and filled with intracellular solution for voltage clamp (in mM: 80 CsMES, 25 NaCl, 10 HEPES, 10 Cs4-BAPTA, 2 MgCl2, free Ca 90uM, OsM 295 mOs. pH 7.3 with CsOH) or for current clamp (in mM: 120 K-gluconate, 10 Na-gluconate, 4 NaCl, 10 HEPES-K, 10 Na phosphocreatine, 2 Na-ATP, 4 Mg-ATP, and 0.3 GTP, adjusted to pH 7.3 with KOH). Control extracellular solution contained the following (in mM): 150 NaCl, 10 HEPES, 1.8 CaCl2, pH to 7.4 with NaOH, 300mOsM.
HEK293T cells (from ATCC, CRL-3216) were grown under standard culture conditions (5% CO2, 37°C) in Dulbeccos's Modified Eagle's Medium with 10% FBS and antibiotics. The sex of the cell line was undetermined. Site directed mutagenesis of SCN3A-F1759Y and SCN3A-I875T was performed using a bridge PCR method followed by Sanger sequencing of the entire plasmid to control for off-target mutations. Cells were transfected (Lipofectamine 3000, ThermoFisher) with one of the SCN3A alpha subunits: SCN3A-WT, SCN3A-F1759Y, or SCN3A-I875T, together with the human auxiliary NaV-Beta subunits (β1-IRES-CD4 and β2-IRES-GFP; using a plasmid ratio of 1:6 for β:SCN3A) to support membrane localization and physiological current kinetics. Data were analyzed with IGOR-Pro (Wavemetrics) and reported as mean ± S.E.M. Conductancevoltage plots (from measured reversal potentials) were fit with a Boltzmann function of the following form (IImax/{1exp[(VVhalf)/ks]}), where Imax is the maximal current, V is the prepulse potential, Vhalf is the half activation potential of the current, and ks is the slope factor.
Statistical significance was assessed with Student’s two-tailed t-test. For measuring the noninactivating persistent current (I-NaP), the mean current from the last 30 ms of the voltage step was measured and this value was divided by the maximal peak inward current. These I-NaP data are presented as a percentage of the maximal peak inward current.
Human Tissue Brain Preparation and mRNA in situ hybridization
Following fixation (4% PFA) and cryoprotection (30% sucrose), brains (17 – 20 gestational weeks, both male and female) were frozen using Isopentane on dry ice. Samples were sectioned at 20 – 30 μm thickness (Leica Cryostat), mounted immediately onto warm charged SuperFrost Plus slides (Fisher), then dried at -20°C for 20 minutes before storage at –80°C. After applying a hydrophobic barrier around the tissue (ImmEdge Pen, Vector Labs), sections were baked for 20 minutes at 60°C before beginning with the manufacturer’s standard protocol for multiplex fluorescent in situ hybridization (Multiplex Version 2 kit, Advanced Cell Diagnostics). In situ probe ACD catalog numbers are as follows: Vimentin: 479411, SCN3A: 460121, GFAP: 311801, RBFOX: 415591, Eomes: 429691. Whole tissue mRNA in situ imaging was performed on a Zeiss Axio Observer with image tiling at 20X magnification. Bright-field images were background corrected by Zen Blue Software for center intensity illumination and stitched together (Figure 3C, D).
Ferret in utero electroporation
All animal experimentation was carried out under protocols approved by the IACUC of Boston Children’s Hospital. Ferrets (Mustela putorius furo) were obtained from Marshall Bioresources and animals were same-sex housed within a dedicated Large Animal Facility of Boston Childrens Hospital on a 12-hr light/dark cycle at 18°C–23°C, with rotating sensory enrichment. Food and water were available ad libitum. In utero electroporations were performed according to standard procedures (Kawasaki et al., 2012). Briefly, pregnant Jills (embryonic day 32–33) were anesthetized with 5% isoflurane and maintained at 3% utilizing a nose cone. Following laparotomy, 1.5–3μl (2 μg/μl) of the SCN3A and mCherry constructs were introduced into the lateral ventricle using a pulled glass micropipette and Hamilton syringe (Hamilton Company). 150V square electric pulses were passed 5 times at 1s intervals (ECM830, BTX). Tissues were harvested on postnatal days P0 and ~P33, and fixed in 4% PFA. Successful plasmid expression in embryos was evaluated based on mCherry fluorescence (Figure S4B). Both male and female embryos were targeted for injection.
Ferret Tissue Preparation and Immunohistochemistry
Following perfusion with ice cold 4% PFA in PBS, tissues were drop-fixed for 8 hours in 4% PFA at 4°C. For cryosections, brains were cryoprotected in 30% sucrose overnight, embedded in OCT, frozen in isopentane, sectioned at 15 – 35 um thickness (Leica Cryostat), and mounted onto charged SuperFrost Plus slides (Fisher). After applying a hydrophobic barrier around the tissue (ImmEdge Pen, Vector Labs), slides were washed in cold 0.1M PB followed by antigen retrieval and endogenous fluorescence quenching (Retrievagen A pH6.0, BD Biosciences) at 80–90°C in a hybridization oven for 45 minutes. Sections were then cooled to room temperature in Retrievagen A, washed in cold 0.1M PB, and blocked for 1 hour at room temperature (5% Normal Donkey Serum, 1% w/v BSA, 0.2% w/v each glycine/lysine, in PBS). Slides were incubated with primary antibodies for two nights on a rotary shaker at 4°C in blocking buffer plus 0.3% Triton X-100, washed in PBS, and incubated for two hours at room temperature with secondary antibodies diluted in blocking buffer (1:500; Jackson Immunoresearch). Finally, slides were washed in PBS, and coverslipped with Fluoromount-G containing Dapi (Southern Biotech). Images were obtained with a Zeiss LSM700 confocal microscope and Leica MZ16 F fluorescence stereomicroscope. Primary antibodies include mouse anti-phosphorylated Vimentin (4A4; 1:500; MBL), rabbit anti-Ki67 (1:250; Abcam ab15580), chicken anti-GFAP (1:1000; Abcam ab4674), mouse anti-TUJ1 (beta-III tubulin; 1:500; Abcam ab7751), rabbit anti-RFP (1:250; Rockland 600–901-379S), rabbit anti-SATB2 (1:400; Bethyl A301–864A). Secondary antibodies included AlexaFluor (1:500; 488nm, 563nm, 650nm) donkey anti-mouse and donkey anti-rabbit (Invitrogen).
Membrane protein extraction and immunoblotting
HEK293T cells were harvested and membranes were isolated according to manufacturer’s instructions (Mem-PER Plus Membrane Protein Extraction Kit for Mammalian Cells, Thermo Fisher Scientific). Membrane proteins (25ug) were resolved on 4–15% SDS-polyacrylamide gels (Bio-Rad), transferred to PVDF membrane (GE Healthcare Life Science), and probed with rabbit anti-Pan Na Channel α Subunit (clone D219C, 1:1000, Cell Signaling Technology), mouse antiATP1A1 (clone 464.6, 1:1000, Thermo Fisher Scientific), and chicken anti-GFP (1:1000, Abcam) antibodies, followed by anti-rabbit or anti-mouse horseradish peroxidase secondary antibodies (1:2000, Cell Signaling Technology). Immunoblots were developed with the Amersham ECL Western Blotting Detection Kit (GE Healthcare Life Science).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitative MRI analysis
The MRI images (B03 and control age-matched subject) were processed to extract the surfaces of the gray/white matter and gray matter/cerebrospinal fluid boundaries, and to define Brodmann areas on the cortical surface using FreeSurfer software.
Human Gene Expression Analysis
The Allen Human Brain Atlas (ABA) publishes a rich dataset of cortical genetic expression across cortical brain regions, from age 8 weeks post conception to adult ages (Jones et al., 2009). BrainSpan data analysis of SCN3A (ch2:165944030–166060577, GRCh37/hg19) and SCN1A (ch2: 166,845,670–166,930,180, GRCh37/hg19) was performed. RNA-seq expression measured in RPKM (reads per kilobase exon per million mapped reads) was obtained from the BrainSpan project data and summarized to Gencode v10 exons for all annotated neocortical tissues aged 8 weeks post conception to 38 years. Cortical areas for Figure 3A include: dorsolateral prefrontal cortex; ventrolateral prefrontal cortex; anterior (rostral) cingulate (medial prefrontal) cortex; orbital frontal cortex; primary motor-sensory cortex; parietal neocortex; posterior (caudal) superior temporal cortex (area 22c), inferolateral temporal cortex (area 20); occipital neocortex.
Analysis of Single Cell RNA-seq Data
Dataset was provided by Alex Pollen UCSF; see (Pollen et al., 2015) methods for additional information. Briefly, fetal cortex was dissociated and captured on C1 (Fluidigm). Classification of cells was completed using principal component analysis (PCA) and used expectation-maximization clustering to group cells based on their position in PC space. Gene expression values were normalized based on library size as counts per million reads (CPM) and log2(CPM+1) transformed. Libraries with fewer than 1,000 genes detected above 1 CPM were eliminated as outliers. Since these data contain many cells with zero CPM for the genes of interest, either due to lack of expression or to the limits of detection of single cell RNA-seq, a two-part Wilcoxon test was used to test differences in gene expression. A compound null hypothesis of no difference in the proportion of cells with zero CPM and of no difference in the mean CPM of the non-zero CPM cells for a given gene between two groups was tested. An R code, which applies a two-part Wilcoxon test with a continuity-corrected binomial test was implemented. These tests were conducted with a Bonferroni adjusted alpha level of 0.017 (0.05/3). Data are presented as box-plots.
Primary Neuronal Cultures and Neurite Analysis
Neuronal cultures were prepared according to established protocols with modifications. Briefly, the perisylvian cortex was dissected from 20 WKGS human samples, male and female cases, and dissociated according to the manufacturer’s protocol into a single cell suspension (MACs neural dissociation kit, Miltenyi Biotec). Cortical neurons were plated onto poly-ornithine and laminin treated coverslips at 5 × 104 cells/well in 24-well plates and maintained in DMEM/Neurobasal, N2, B27, glucose, NaHCO3, HEPES (4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid) with fibroblast growth factor 2 (FGF-2; 20 ng/ml; Invitrogen) and EGF (FGF-2; 20 ng/ml; Invitrogen) treatment for the first 24 h only. Neurons were transfected with mCherry vector together with each experimental condition at a 1:6 plasmid ratio as described above (Figure S2). Cultured medium was changed every other day. Neurons were fixed after 5 days in vitro (DIV 5) with 4% PFA in PBS for 15 min. at room temperature. Cells were blocked in 10% NGS in PBS with 0.3% Triton X-100 for 1 h at room temperature followed by immunostaining. Images were captured using a digital camera under a Zeiss Axio Observer fluorescent microscope. 30–50 neurons were analyzed from three separate coverslips for each experimental condition. Neurites that had lengths at least twice the diameter of the cell body were measured. Neurite lengths from the soma were traced and measured using ImageJ software in a blinded manner, and data were analyzed using Igor Pro Software. Statistical significance was assessed with Student’s two-tailed t-test.
Ferret Brain Magnetic Resonance Imaging
Three P30 ferret brains (expressing SCN3A-WT, SCN3A-F1759Y, and mCherry) were post-fixed in 4% paraformaldehyde in PBS and submerged into perfluorocarbon oil (Fomblin, Fisher Scientific) at 4°C for 3 days. The brains were scanned using a Bruker BioSpec 70/30 7T MRI scanner (Small Animal Imaging Laboratory) at BCH. This is a high-resolution (sub-millimeter) MRI scanner designed specifically for imaging small animals, and has a 30 cm bore and 450 mT/m gradient. Ferret MRI scans are isotropic 63μm voxels across the entire brain. Cortical gray matter and white matter were visualized using the FreeSurfer and OsiriX software.
DATA AND SOFTWARE AVAILABILITY
Links accession numbers generated in the manuscript GEOI (phs000492.v3.p1)
“Sodium channel SCN3A (NaV1.3) regulation of human cerebral cortical folding and oral-motor development,” (NEURON-D-18–00001) Highlights and eTOC for our study:
"Smith et al, define a role for sodium channel SCN3A (NaV1.3) in the developing human cerebral cortex, as well as a cortical malformation that can result from NaV1.3 dysfunction.”
Mutations in SCN3A (NaV1.3) associate with perisylvian polymicrogyria
Pathogenic SCN3A mutations increase persistent sodium current
SCN3A is expressed during human fetal development in progenitors and neurons
Mutated SCN3A alters cortical folding pattern in the ferret brain
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the families reported here. We thank F. Andermann, B. Bean, and members of the Clapham, Lehtinen, and Walsh labs for helpful discussions; B. Bean and D. Clapham for sharing electrophysiology equipment, M. Marcotrigiano, Y. Wu, and the Boston Children’s Hospital Small Animal Imaging Lab for MRI, E. Pollack and A. Nedder, and the Boston Children’s Hospital large animal facility, D. Gleason, J. Rodriguez, A. Malesz, R. Hu, for technical help, A. Pollen for sharing single cell sequencing data, J. Kearney for the WT SCN3A plasmid, D. Nguyen and L. Al-Gazali for assistance with clinical data. This work was supported by NIH 1F32NS100033801 (R.S.S.); the Erasmus MC Mrace project #104673 (G.M.S.M.); the Finnish Medical Society, Arvo and Lea Ylppö Foundation, and Finnish governmental subsidiaries TLK0278 (R.P.) and TRTR019 (O.S.); The Folkhälsan Research Foundation (AE.L.); Paul G. Allen Frontiers Program, and NIH R01NS032457 and R01NS035129 (C.A.W.), the New York Stem Cell Foundation (M.K.L.), and BCH IDDRC 1U54HD090255. C.A.W. is an Investigator of the Howard Hughes Medical Institute. M.K.L. is a New York Stem Cell Foundation – Robertson Investigator.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ackman JB, Crair MC, (2014). Role of emergent neural activity in visual map development. Current Opinion in Neurobiology 24, 166–175. doi: 10.1016/j.conb.2013.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mefford HC, Stephens K, Amemiya A, Ledbetter N, Morgan A, Fisher SE, Scheffer I, Hildebrand M, (1993). FOXP2-Related Speech and Language Disorders.
- Aman TK, Grieco-Calub TM, Chen C, Rusconi R, Slat EA, Isom LL, Raman IM, (2009). Regulation of Persistent Na Current by Interactions between Subunits of Voltage-Gated Na Channels. Journal of Neuroscience 29, 2027–2042. doi: 10.1523/JNEUROSCI.4531-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckh S, Noda M, Lübbert H, Numa S, (1989). Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J. 8, 3611–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shalom R, Keeshen CM, Berrios KN, An JY, Sanders SJ, Bender KJ, (2017). Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol. Psychiatry 0. doi: 10.1016/j.biopsych.2017.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JA, Waxman SG, (2013). Noncanonical roles of voltage-gated sodium channels. Neuron 80, 280–291. doi: 10.1016/j.neuron.2013.09.012 [DOI] [PubMed] [Google Scholar]
- Black JA, Westenbroek R, Minturn JE, Ransom BR, Catterall WA, Waxman SG, (1995). Isoform-specific expression of sodium channels in astrocytes in vitro: immunocytochemical observations. Glia 14, 133–144. doi: 10.1002/glia.440140208 [DOI] [PubMed] [Google Scholar]
- Blanchard MG et al. , (2015). De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. Journal of Medical Genetics 52, 330–337. doi: 10.1136/jmedgenet-2014-102813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackenbury WJ, Davis TH, Chen C, Slat EA, Detrow MJ, Dickendesher TL, Ranscht B, Isom LL, (2008). Voltage-Gated Na+ Channel 1 Subunit-Mediated Neurite Outgrowth Requires Fyn Kinase and Contributes to Postnatal CNS Development In Vivo. Journal of Neuroscience 28, 3246–3256. doi: 10.1523/JNEUROSCI.5446-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M, Carr L, Reilly S, Neville BG, (2000). Worster-Drought syndrome, a mild tetraplegic perisylvian cerebral palsy. Review of 47 cases. Brain 123, Pt 10, 2160–2170. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H, (2013). Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet 132, 1077–1130. doi: 10.1007/s00439-013-1331-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study, (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, Goldin AL, (2010). Sodium channel SCN1A and epilepsy: Mutations and mechanisms. Epilepsia 51, 1650–1658. doi: 10.1111/j.1528-1167.2010.02640.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estacion M, Gasser A, Dib-Hajj SD, Waxman SG, (2010). A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Experimental Neurology 224, 362–368. doi: 10.1016/j.expneurol.2010.04.012 [DOI] [PubMed] [Google Scholar]
- Estacion M, Vohra BPS, Liu S, Hoeijmakers J, Faber CG, Merkies ISJ, Lauria G, Black JA, Waxman SG, (2015). Ca2+ toxicity due to reverse Na+/Ca2+ exchange contributes to degeneration of neurites of DRG neurons induced by a neuropathy-associated Nav1.7 mutation. Journal of Neurophysiology 114, 1554–1564. doi: 10.1152/jn.00195.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Huttner WB, (2014). Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141, 2182–2194. doi: 10.1242/dev.090571 [DOI] [PubMed] [Google Scholar]
- Gordon D, Merrick D, Auld V, Dunn R, Goldin AL, Davidson N, Catterall WA, (1987). Tissue-specific expression of the RI and RII sodium channel subtypes. Proc Natl Acad Sci USA 84, 8682–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro MM et al. , (2000). Familial perisylvian polymicrogyria: a new familial syndrome of cortical maldevelopment. Annals of Neurology 48, 39–48. [PubMed] [Google Scholar]
- Guerreiro MM, Hage SRV, Guimarães CA, Abramides DV, Fernandes W, Pacheco PS, Piovesana AMSG, Montenegro MA, Cendes F, (2002). Developmental language disorder associated with polymicrogyria. Neurology 59, 245–250. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PRL, Kriegstein AR, (2010). Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464, 554–561. doi: 10.1038/nature08845 [DOI] [PubMed] [Google Scholar]
- Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, Glaaser IW, Kass RS, Meisler MH, (2008). Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neurosci. Lett 433, 65–70. doi: 10.1016/j.neulet.2007.12.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurni N, Kolodziejczak M, Tomasello U, Badia J, Jacobshagen M, Prados J, Dayer A, (2017). Transient Cell-intrinsic Activity Regulates the Migration and Laminar Positioning of Cortical Projection Neurons. Cereb. Cortex 27, 3052–3063. doi: 10.1093/cercor/bhx059 [DOI] [PubMed] [Google Scholar]
- Jansen AC, Leonard G, Bastos AC, Esposito-Festen JE, Tampieri D, Watkins K, Andermann F, Andermann E, (2005). Cognitive functioning in bilateral perisylvian polymicrogyria (BPP): clinical and radiological correlations. Epilepsy Behav 6, 393–404. doi: 10.1016/j.yebeh.2005.01.012 [DOI] [PubMed] [Google Scholar]
- Johnson MB, Wang PP, Atabay KD, Murphy EA, Doan RN, Hecht JL, Walsh CA, (2015). Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat Neurosci 18, 637–646. doi: 10.1038/nn.3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Iwai L, Tanno K, (2012). Rapid and efficient genetic manipulation of gyrencephalic carnivores using 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzniecky R, Andermann F, Guerrini R, (1993). Congenital bilateral perisylvian syndrome: study of 31 patients. The CBPS Multicenter Collaborative Study. Lancet 341, 608–612. [DOI] [PubMed] [Google Scholar]
- Lamar T, Vanoye CG, Calhoun J, Wong JC, Dutton SBB, Jorge BS, Velinov M, Escayg A, Kearney JA, (2017). SCN3A deficiency associated with increased seizure susceptibility. Neurobiology of Disease 102, 38–48. doi: 10.1016/j.nbd.2017.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampert A, Hains BC, Waxman SG, (2006). Upregulation of persistent and ramp sodium current in dorsal horn neurons after spinal cord injury. Exp Brain Res 174, 660–666. doi: 10.1007/s00221-006-0511-x [DOI] [PubMed] [Google Scholar]
- Lelieveld SH et al. , (2016). Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci 19, 1194–1196. doi: 10.1038/nn.4352 [DOI] [PubMed] [Google Scholar]
- LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR, (1995). GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15, 1287–1298. [DOI] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA, (1995). A Critical Role for Transmembrane Segment IVS6 of the Sodium Channel α Subunit in Fast Inactivation. J. Biol. Chem 270, 12025–12034. doi: 10.1074/jbc.270.20.12025 [DOI] [PubMed] [Google Scholar]
- Moore AR, Filipovic R, Mo Z, Rasband MN, Zecevic N, Antic SD, (2009). Electrical excitability of early neurons in the human cerebral cortex during the second trimester of gestation. Cereb. Cortex 19, 1795–1805. doi: 10.1093/cercor/bhn206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappalardo LW, Samad OA, Black JA, Waxman SG, (2014). Voltage-gated sodium channel Nav 1.5 contributes to astrogliosis in an in vitro model of glial injury via reverse Na+ /Ca2+ exchange. Glia 62, 1162–1175. doi: 10.1002/glia.22671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson A-K, Estacion M, Ahn H, Liu S, Stamboulian-Platel S, Waxman SG, Black JA, (2014). Contribution of sodium channels to lamellipodial protrusion and Rac1 and ERK1/2 activation in ATP-stimulated microglia. Glia 62, 2080–2095. doi: 10.1002/glia.22728 [DOI] [PubMed] [Google Scholar]
- Pollen AA et al. , (2015). Molecular Identity of Human Outer Radial Glia during Cortical Development. Cell 163, 55–67. doi: 10.1016/j.cell.2015.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rash BG, Ackman JB, Rakic P, (2016). Bidirectional radial Ca(2+) activity regulates neurogenesis and migration during early cortical column formation. Sci Adv 2, e1501733. doi: 10.1126/sciadv.1501733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio-Galimberti E, Gollob MH, Darbar D, (2012). Voltage-gated sodium channels: biophysics, pharmacology, and related channelopathies. Front. Pharmacol 3, 124. doi: 10.3389/fphar.2012.00124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer H, Waxman SG, (1993). Expression of voltage-activated ion channels by astrocytes and oligodendrocytes in the hippocampal slice. Journal of Neurophysiology 70, 1863–1873. doi: 10.1152/jn.1993.70.5.1863 [DOI] [PubMed] [Google Scholar]
- Spillane J, Kullmann DM, Hanna MG, (2016). Genetic neurological channelopathies: molecular genetics and clinical phenotypes. Journal of Neurology, Neurosurgery & Psychiatry 87, 37–48. doi: 10.1136/jnnp-2015-311233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE, (2007). Persistent sodium current and its role in epilepsy. Epilepsy Currents 7, 15–22. doi: 10.1111/j.1535-7511.2007.00156.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutterd CA, Leventer RJ, (2014). Polymicrogyria: a common and heterogeneous malformation of cortical development. Am J Med Genet C Semin Med Genet 166C, 227–239. doi: 10.1002/ajmg.c.31399 [DOI] [PubMed] [Google Scholar]
- Sun G-C, Werkman TR, Battefeld A, Clare JJ, Wadman WJ, (2007). Carbamazepine and topiramate modulation of transient and persistent sodium currents studied in HEK293 cells expressing the Na(v)1.3 alpha-subunit. Epilepsia 48, 774–782. doi: 10.1111/j.15281167.2007.01001.x [DOI] [PubMed] [Google Scholar]
- Turner SJ, Mayes AK, Verhoeven A, Mandelstam SA, Morgan AT, Scheffer IE, (2015). GRIN2A: an aptly named gene for speech dysfunction. Neurology 84, 586–593. doi: 10.1212/WNL.0000000000001228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoye CG, Gurnett CA, Holland KD, George AL, Kearney JA, (2014). Novel SCN3A variants associated with focal epilepsy in children. Neurobiology of Disease 62, 313–322. doi: 10.1016/j.nbd.2013.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman T, Helbig I, Božović IB, DeBrosse SD, Bergqvist AC, Wallis K, Medne L, Maver A, Peterlin B, Helbig KL, Zhang X, Goldberg EM, (2018). Mutations in SCN3A cause early infantile epileptic encephalopathy. Annals of Neurology 83, 703–717. doi: 10.1002/ana.25188 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.