

Abstract
Objective
Oxidative stress is a central event linked with endothelial dysfunction and inflammation in several vascular pathologies, marked by over-production of ROS and concomitant decreases in antioxidants e.g. GSH. Here we distinguish endothelial oxidative stress regulation and associated functional disparities in the two main vascular conduits, (arteries and veins) following decreases in GSH.
Methods
MAECs and VCECs were used as models of arterial and venular endothelium, respectively and BSO (0–100μM) was used to indirectly increase cellular oxidative stress. Inflammatory responses were measured using immune cell attachment and immunoblotting for endothelial cell adhesion molecule (ICAM-1, VCAM-1) expression, altered cell proliferation and wound healing.
Results
MAECs and VCECs exhibited differential responses to oxidative stress produced by GSH depletion with VCECs exhibiting greater sensitivity to oxidative stress. Compared to MAECs, VCECs showed a significantly increased inflammatory profile and a decreased proliferative phenotype in response to decreases in GSH levels.
Conclusions
Arterial and venous endothelial cells exhibit differential responses to oxidant stress and decreases in GSH:GSSG are more exacerbated in venous endothelial cells. Specific pathogenesis in these vascular conduits, with respect to oxidant stress handling warrants further study, especially considering surgical interventions such as Coronary Artery Bypass Grafting that use both interchangeably.
Introduction
While the greater arteries and veins of the circulatory system are both conduits responsible for blood transport, these two divisions show distinct differences in their development, patterning, structure and function [1–3]. Several unique pathologies are also associated with each vessel type; arteries exhibit more atherosclerotic plaque formation in conduit vessels and endothelial dysfunction in resistance arteries (arterioles). By comparison, veins and venules classically mediate leukocyte extravasation during inflammation [4, 5].
With respect to clinical applications of veins vs. arteries for vascular grafting, important differences are also seen in the patency rates of grafts used for coronary artery bypass grafting (CABG). In particular, CABG procedures which use saphenous vein grafts are statistically more likely to fail compared to their arterial counterparts because of more frequent development of thrombosis and intimal thickening in vein to artery substitutions [6, 7]. Although the disparities in pathogenesis between arteries and veins have been largely attributed to a failure of veins to adapt to pressure and flow conditions in arteries, the redox status of these two vessel types has not been studied as much. Therefore, a clearer understanding of the molecular differences between these two endothelial cell types that may be involved in aberrant vascular remodeling may lead to improved interventional strategies focusing on improved vascular integration in grafting as well as treatments for several vascular pathologies.
As the inner lining of all blood vessels (tunica intima) endothelial cells are known to play critical roles in mediating several vascular pathologies. One of the earliest events in such pathologies is ‘endothelial dysfunction.’ This dysfunction is often characterized by loss of endothelial dependent vasodilation and an increase in inflammation mediated cellular adhesion to the endothelial luminal surface [8–11].
Oxidative stress is an important factor contributing to endothelial dysfunction often observed in cardiovascular pathologies and is linked with increased reactive oxygen species (ROS) production and decreased availability of antioxidants like glutathione (GSH) [12–15]. GSH is a major antioxidant whose main function is to attenuate ROS stress by reducing ROS (e.g. H2O2), which cause cellular and/or tissue injury. During oxidant catabolism, GSH is oxidized to form GSSG, (glutathione disulfide) which can be enzymatically reduced back to GSH. This recycling of GSSG to GSH maintains a homeostatic physiological balance of the GSH:GSSG ratio, whereas during several pathological states this ratio is not maintained [16, 17].
GSH:GSSG ratios have been demonstrated to dictate cell fates such as cell division, proliferation, and apoptosis [18, 19]. Along with decreased GSH, increases in GSSG often have cell-regulatory effects on signaling reflecting posttranslational modifications such as protein glutathionylation [20]. Although the protective role of GSH and its dysregulation in disease have been well-studied [17, 21], the role of the GSH:GSSG ratio itself and how this ratio differs between the arterial and venous vascular beds has been largely marginalized. The GSH:GSSG ratios in arterial and venous endothelia may underlie differences in redox regulation and its role in influencing endothelial health vs. pathology. Very few studies have compared how arteries and veins differentially respond to an imposed oxidative stress and fewer still have focused on glutathione dysregulation.
Here, our current study describes differences between arterial and venous endothelial cells including both basal differences in redox based on anatomic origin, as well as changes in protein expression and cell behavior produced by deliberately perturbing the GSH:GSSG ratios using BSO. BSO is an irreversible inhibitor of glutamate cysteine ligase (GCL), the rate-limiting enzyme required during the first step of GSH synthesis. Use of BSO results in decreased cellular GSH levels [22]. Our study shows that under basal conditions arterial and venous endothelial redox state differs significantly and that under redox stress the GSH:GSSG ratio/oxidative state may underlie differential immunological and functional outcomes between these two cell populations.
Materials and Methods
Cell culture and BSO treatment:
Endothelial cell lines (mouse aortic endothelial cells; MAECs and mouse vena cava endothelial cells; VCECs) were cultured in Dulbecco’s modified Eagle Media-DMEM (Caisson labs, UT) with 10% Fetal bovine serum-FBS (Caisson labs, UT), added L-glutamine (Genesee Scientific, CA), penicillin and streptomycin (Genesee Scientific, CA) at 37oC in 5% CO2. Cells were grown to confluence followed by BSO (Sigma, MO) treatments of increasing concentrations (0, 5, 25, 50, 75, and 100 μM) for 19hrs in starvation media (with 0.5% FBS).
GSH and GSSG measurement:
High performance liquid chromatography (HPLC) was used to measure GSH and GSSG as described previously [23]. Confluent cell monolayers of MAECs and VCECs with and without treatment were washed in PBS, lysed, and protein precipitated using trichloroacetic acid (TCA). The acid precipitate was then centrifuged and the supernatant used for GSH and GSSG measurements.
Proliferation assay:
Confluent cell monolayers with and without treatment were collected using trypsin (Lonza, MD), stained with Trypan blue (Lonza, MD) and counted manually using a hemocytometer.
Wound healing assay:
Confluent cell monolayers were subjected to a wound made with a p200 pipette tip as shown in Fig. 5 and subjected to BSO treatment for 16hrs under observation in a custom designed microscope slide holder (Bioscience Tools, San Diego, CA) at 37oC in 5% CO2 using a Nikon Eclipse Ti-E microscope (Nikon Instruments Inc., Melville, NY) for image acquisition. Cells were treated with Hydroxyurea during BSO treatment to attenuate proliferation and exclusively study migratory functions of the endothelial cells.
Figure 5:
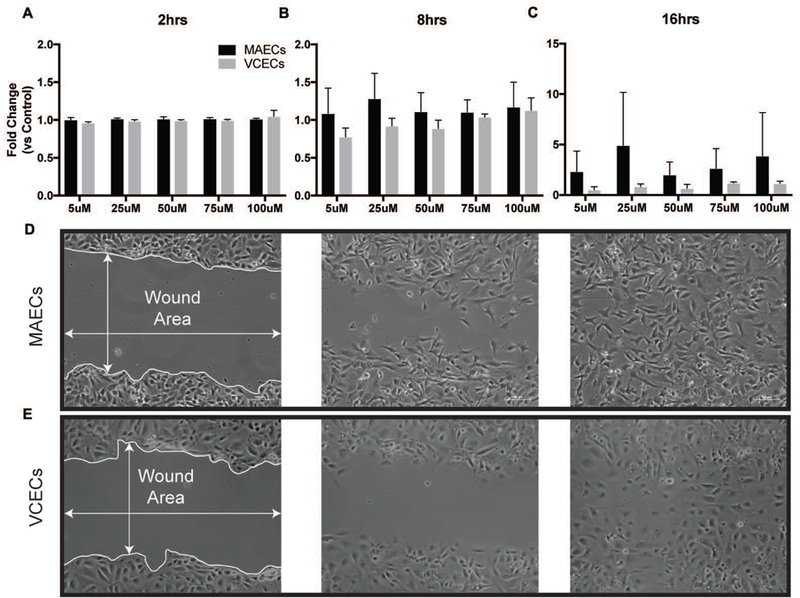
Wound closure rates of MAECs and VCECs following administration of increasing µM levels of BSO (0,5,25,50,75,100) for 16hrs. Bars represent fold change compared to initial wound area and normalized to untreated control for 2hrs (A), 8hrs (B) and 16hrs (C). D. Wound healing assay showing representative bright field images for MAECs (D) and VCECs (E) with closing wound areas at 2hrs, 8hrs and 16hrs.
Western blot analysis of adhesion molecules:
Following treatment with BSO for 19hrs, cells were collected in Laemmli buffer (Biorad, CA) and stored at −80oC until probing for ICAM-1 and VCAM-1 (Novus biologicals, CO) using Western Blot.
Macrophage staining and adhesion:
Murine bone marrow-derived macrophages (BMDMs) were isolated from C57BL/6 mice femurs, flushed and cultured in DMEM with L929 fibroblast conditioned medium at 37°C and 5% CO2 as described previously [24]. Macrophages used for experiments were 98% pure as assayed by flow cytometry using macrophage markers CD68APC and CD11bPE. Confluent macrophages were collected and stained with Vybrant® CFDA SE Cell Tracer Kit (Invitrogen, CA) as per manufacturer protocols and spiked onto BSO treated MAECs and VCECs. 15 minutes later, cells were triple washed with PBS (VWR, PA) and replaced with 10% FBS DMEM for imaging. Images were collected using a Nikon Eclipse Ti-E microscope (Nikon Instruments Inc., Melville, NY) and adherent macrophages were counted using the cell counting software feature on NIS Elements Advanced Research (Nikon Instruments Inc., Melville, NY).
Statistical analysis:
All statistical analyses were performed using GraphPad Prism 7 (GraphPad, La Jolla, CA). Comparisons within each cohort of MAECs or VCECs between untreated controls and treatment groups were done using one-way ANOVA (with n=3, X±SE significant at P ≤ 0.05), while comparisons between the two cell types (MAECs and VCECs) across treatment groups were done using 2-way ANOVA (with n=3, X±SE significant at P ≤ 0.05) as mentioned specifically in the figure legends.
Results
3.1. Endothelial cells of arteries and veins show different basal redox profiles (GSH:GSSG ratio) and varying susceptibilities to oxidative stress.
Levels of the cellular antioxidant GSH and its oxidized form (GSSG) provide an insight into the redox state of a cell and any differences in these quantities between MAECs and VCECs could help identify, to some degree, the pathological disparities seen within these blood vessel types. HPLC measurements shown in Figure 1 compare the individual GSH and GSSG levels between MAECs (A) and VCECs (B) following increasing concentrations of BSO (0, 5, 25, 50, 75, and 100μM) for 19hrs. Results show a significant decrease in GSH following BSO treatment in both MAECs and VCECs across all concentrations. The sensitivity of MAECs to BSO treatment is significant at the least amount of BSO used (5µM), and remains constant with increasing concentrations of BSO. VCECs treated with BSO show decreases at 5µM with continued decreases with increasing concentrations of BSO (25µM - 100µM). Comparison of GSH:GSSG ratios as shown in Figure 1 show that veins have a lower GSH:GSSG ratio (20:1) and less GSH at basal physiological levels (32.2 µM/mg protein) than arterial endothelial cell GSH:GSSG ratio (30:1) and basal GSH levels (142.6 µM/mg protein). Interestingly, treatment with BSO tends to drastically decrease the GSH:GSSG ratio in VCECs compared to MAECs as shown in the Figure 1 table inset. These results suggest that veins are more sensitive to decreases in GSH and can possibly be overloaded with ROS under an oxidative stress insult. Veins may be less capable of handling oxidant stress. Comparatively arteries have a much higher expression of the antioxidant GSH, allowing for a smaller decrease in the total pool of GSH following exposure to BSO. A better ability to handle cellular redox fluctuations could help arteries tolerate the increased accumulation of ROS, neutralizing any likely pathological insults due to oxidative stress in this experimental system.
Figure 1:
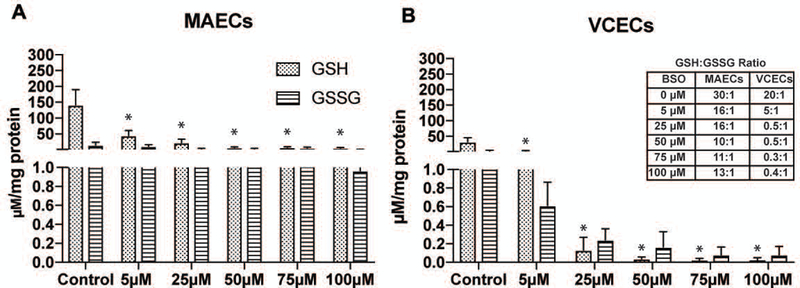
Altered GSH and GSSG levels in MAECs (A) and VCECs (B) measured with HPLC under untreated conditions (control) and treatment with increasing µM levels of BSO (0,5,25,50,75,100) for 19hrs. * Represents significance in GSH levels within either MAECs or VCECs compared to respective untreated control (n=3, X±SE significant at *p ≤ 0.05). Table inset shows changing GSH:GSSG ratio in MAECs and VCECs with increasing concentrations of BSO as shown in graphs (A and B). MAECs: Mouse Aortic Endothelial cells, VCECs: Vena Cava endothelial cells.
3.2. Assessment of macrophage recruitment and expression of adhesion molecules disclose differential immune regulation patterns between heterogeneous endothelial cells.
Interactions between endothelial and immune cells are known to be an important initial event in vascular pathologies [25, 26]. Many cardiovascular pathologies are initiated by endothelial phenotype changes causing them to produce chemoattractant factors and adhesion molecules that stimulate monocyte recruitment to the endothelial cells and support infiltration into the surrounding parenchyma [27]. Results from macrophage recruitment by endothelial cells in this study is shown in Figure 2 and depicts a distinct disparity between arterial and venous endothelial cells in their capacity to recruit these cells. Macrophages more readily adhere to VCECs under conditions of decreased GSH following exposure to increasing concentrations of BSO. Macrophages show significantly increased adherence to VCECs compared to MAECs with BSO concentrations of 50 µM and 100 µM. VCECs also show a pattern of increase in recruitment of macrophages albeit not significant with increasing concentrations of BSO (5 µM to 100 µM) compared to untreated control. Despite enhanced recruitment of macrophages indicating biological function, Figure 3 demonstrates no striking differences between two of the most commonly associated adhesion molecules, VCAM and ICAM, expression in arteries and veins regardless of BSO concentration. The data demonstrating increased recruitment of macrophages suggest that venous endothelial cells are more susceptible to an inflammatory response following stress due to redox imbalance.
Figure 2:
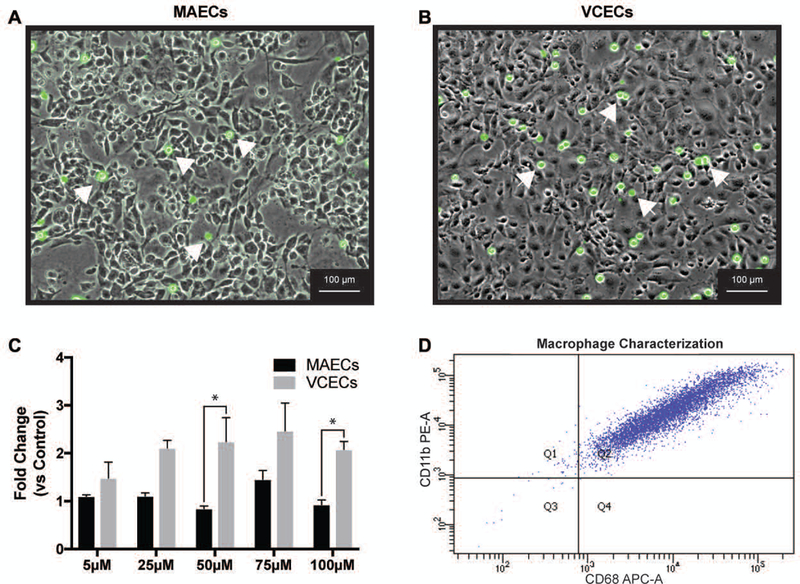
Macrophage recruitment in MAECs and VCECs treated with increasing μM levels of BSO (0,5,25,50,75,100) for 19hrs (A and B) are representative images of MAECs and VCECs (respectively) with attached macrophages (identified with red arrows). (C) Graphical representation of macrophages adhesion assays with statistical significance between the groups represented with *. (D) FACS data demonstrating purity of macrophages. * Represents significance between MAECs and VCECs normalized to respective untreated control shown as fold change (n=3, X±SE significant at *p ≤ 0.05).
Figure 3:
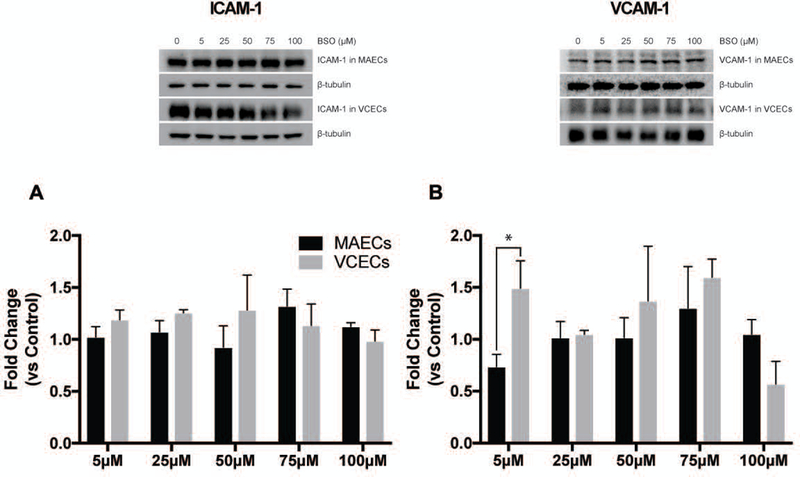
Western blot representative images and quantification of ICAM-1 (A) and VCAM-1 (B) in MAECs and VCECs treated with increasing μM levels of BSO (0,5,25,50,75,100) for 19hrs. * Represents significance between MAECs and VCECs normalized to respective untreated control shown as fold change (n=3, X±SE significant at *p ≤ 0.05).
3.3. Varying patterns in proliferation and migration are observed between the aortic and vena cava endothelial cells resultant from decreasing levels of GSH.
GSH and ROS levels have been shown to regulate cell growth, proliferation and cell cycle under normal physiological conditions [19, 28, 29]. For our experiments we wanted to study the effects of decreases in GSH on proliferation and migration of MAECs and VCECs. We performed these experiments by decreasing levels of GSH using increasing concentrations of BSO. Cell proliferation in VCECs is significantly lower compared to MAECs following exposure to increasing levels of BSO (25µM – 75µM) as measured by cell counting; data is represented as fold change compared to untreated control (Figure 4). In contrast MAECs exposed to BSO proliferate similar to untreated control, showing no significant effects following decrease in GSH. Results showing VCECs have decreased proliferation highlights the important role of GSH in proliferation, with these cells having significantly decreased levels of GSH compared to MAECs. On the other hand MAECs do not show a decrease in proliferation following decreases in GSH. This may be attributed to their significantly higher GSH reserves that are not depleted to the same degree as VCECs and therefore may not influence proliferation. Cell monolayer recovery assessed by a scratch assay measures cellular migration as another functional outcome of decreasing GSH levels. We measured monolayer recovery at 2hrs, 8hrs and 16hrs post scratch (Figure 5). Both MAECs and VCECs have similar rates of cell monolayer healing for the 2hrs and 8hrs time points, regardless of concentration of BSO exposure. However, by 16hrs, there is a trend for MAECs migrating faster. Although this is not a significant difference it does support our evidence that the arterial cells are better able to handle the stress of decreased GSH:GSSG ratios. Since migration and proliferation are events regulated by different signaling pathways the effect of decreased GSH:GSSG may cause different signaling changes as well.
Figure 4:
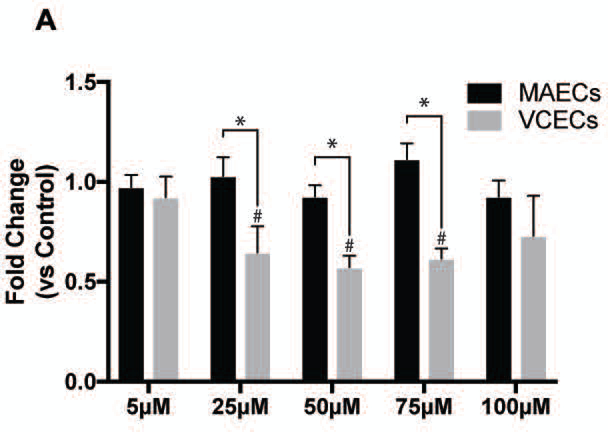
Proliferation of MAECs and VCECs post treatment with increasing µM levels of BSO (0,5,25,50,75,100) for 19hrs. * Represents significance between MAECs and VCECs normalized to respective untreated control shown as fold change. # Represents significance within VCECs compared to untreated control (n=3, X±SE significant at */# p ≤ 0.05).
Discussion
Vein and arterial endothelial cells basally contain different levels of GSH and GSSG and most importantly, have very different GSH:GSSG ratios. Different basal GSH:GSSG ratios and departures from ‘normal’ GSH:GSSG have been linked to changes in many cellular processes. More severe GSH:GSSG disturbances can be extremely harmful to cells and reduce survival. The importance of GSH:GSSG ratios and regulation have also been highlighted previously by our group where we demonstrated that variations in GSH levels can have opposing effects in response to different pathological insults [23]. Ratios of GSH:GSSG that are 13–1 and lower have been associated with non-enzymatically driven protein glutathionylation [30], this contributes to a relatively tight balance between GSH:GSSG and functional outcomes, both beneficial and detrimental. Antioxidant therapies to treat cardiovascular diseases have largely failed, if antioxidant concentrations are too low there is oxidative stress, however if antioxidants are too high then there is a state of reductive stress where all physiological signaling is abolished. These considerations make supplementation experiments to rescue antioxidant depletion problematic. We need a better understanding of the contributions of each component of the redox system to better tackle therapy development.
To study the effect of varying tissue levels of GSH on angiogenesis following ischemia, Gclm−/− KO mice in which (GSH decreased by >80%), Gclm+/− HET mice (GSH decreased by >20%) and Gclm+/+ WT mice (highest levels of GSH) were subjected to hind limb ischemia. At days 3 and 7 post-ligation, GSH levels were significantly decreased in all groups [23]. Interestingly, our results showed that blood flow and cellular proliferation, (both necessary for revascularization) were most robust in HET mice compared to their KO and wild type counterparts. These results demonstrate that relatively small decreases in GSH synthesis can actually be extremely beneficial in healing, whereas large decreases in GSH synthesis are detrimental to revascularization. Therefore, GSH regulation can improve or intensify the severity of pathological outcomes through the control of GSH:GSSG redox ratios; these ratios may also play major roles influencing initiation and progression of several vascular disease states [31, 32].
ROS dependent endothelial cell proliferation and migration are well-documented phenomena [33–36]. It is important to recognize that cell redox state is extremely sensitive to altered GSH:GSSG ratios and even slight perturbations can powerfully influence cellular signaling mechanisms [16, 37]. We have demonstrated that venous endothelial cells have lower GSH:GSSG ratios and are more sensitive to an imposed oxidant stress than arteries. Consequently, venous endothelial cells may proliferate less compared to MAECs and are more prone to inflammatory activation, both of which are signs of cellular dysfunction. Arterial endothelial cells (MAECs) have higher basal levels of GSH (142.6 µM/mg protein) than VCECs (32.2 µM/mg protein) and appear to be more sensitive to changes in ROS. Administration of BSO causes a more severe phenotypic change in VCECs and results in enhanced macrophage adhesion, an important component of the inflammatory response. No significant changes can be observed in MAEC macrophage adhesion assays, however with VCECs we observed an increasing trend for macrophage adhesion between 25uM - 100uM, with significant changes seen at 50µM and 100 µM BSO. These results are quite interesting as protein glutathionylation may be playing a role here. Although not measured this posttranslational modification is known to occur enzymatically at low GSH:GSSG ratios [30]. Further study is warranted, especially measuring pools of GSH:GSSG in discreet compartments of the cell. We have measured the GSH:GSSG in the whole cell and this may not be sufficient to see smaller changes leading to large disturbances in the posttranslational modifications of proteins.
Interestingly, despite enhanced recruitment of inflammatory cells we observed no changes in VCAM-1 or ICAM-1 expression with increasing oxidant stress. These data suggest that changes in endothelial adhesion molecules may not be the main driving force underlying recruitment of macrophages in our hands (despite an increasing body of evidence demonstrating that oxidative stress enhances endothelial expression of adhesion molecules and altering their affinity/avidity [38–40]. Such studies induce oxidative stress indirectly through several environmental and cytokine stressors such as using endotoxin and inflammatory cytokine initiated monocyte recruitment to show differences between both of these vascular cells [41]. However in our current study, an induced redox imbalance itself was sufficient to recruit macrophages to endothelial monolayers. Once again this phenomena is potentially related to posttranslational modification of proteins through glutathionylation playing a role in macrophage attachment.
Our study demonstrates that arterial and venous endothelial cells respond differently to oxidant stressors. Both of these cells accomplish vastly different tasks in the vascular system; arteries are responsible for delivering oxygenated blood to the tissues, while veins return deoxygenated blood to the heart, where the lungs remove CO2 and replenish O2. We might speculate that arteries handle changes in oxidant stress better than veins (as shown here) as veins may be maladapted to handling large fluxes of oxygen and oxidant stress. Regardless of the ultimate causes for the difference in ‘stress handling’ between these two divergent anatomies, oxidant stress remains an important consideration when forming anastomoses, bypasses, etc. The difference between veins and arteries in recruiting inflammatory cells is only one potential consequence that may lead to graft failure in CABG interventional procedures.
Perspectives
Oxidative stress and lipid retention in saphenous and arterial vascular conduits show higher oxidative stress, LDL accumulation, and the presence of oxidized epitopes in vein grafts as compared to arterial grafts [42]. Similarly, human studies between CABG arteries and veins concluded that NADPH oxidase activity (a major ROS producer) was higher in veins than in arteries [43]. Differences in gene expression were also observed between human coronary artery (CAEC) and saphenous vein (SVEC) endothelial cells due to atherogenic stimuli [44]. It is clear from the data presented in this manuscript that veins and arteries have vastly different responses to lower GSH:GSSG ratios. These data were performed in mouse endothelial cells, however the translational aspect of this data is high as others have demonstrated that various redox components observed in rodents is conserved in humans, with interesting correlations found in a wide variety of tissues and processes (skin, brain, and fat handling) [45–47]. Overall the data presented in this current manuscript open up exciting possibilities to address disparities found when these vascular cells are used in therapeutic interventions; the development of new therapies might better transition these vascular cells when introducing them to starkly different environments (e.g. veins used for CABG).
Acknowledgements
This work was supported by an American Heart Association SDG and an NIH COBRE (15SDG25710038 and P20GM121307 to C.B.P.), an intramural Malcolm Feist Post-doctoral Fellowship (B.S.), the Department of Defense (W81XWH-11-1-0577 to J.S.A.), and the National Institutes of Health (R01 HL098435, HL133497 and P20GM121307 to A.W.O and R01 HL131844 to M.D.W.).
List of Abbreviations:
- ROS
reactive oxygen species
- GSH
glutathione
- GSSG
glutathione disulfide
- MAEC
mouse aortic endothelial cell
- VCEC
mouse vena cava endothelial cell
- BSO
buthionine sulfoximine
- CABG
coronary artery bypass grafting
- SVG
saphenous vein graft
- H2O2
hydrogen peroxide
- GCL
glutatmate cysteine ligase
- Gclm
glutamate cysteine ligase modifier subunit
- Gclm−/− KO
glutamate-cysteine ligase modifier subunit homozygous deletion
- Gclm+/− HET
glutamate-cysteine ligase modifier subunit heterozygous deletion
- Gclm+/+ WT
glutamate-cysteine ligase modifier subunit wild type control
- VCAM-1
vascular cell adhesion molecule 1
- ICAM-1
intercellular adhesion molecule 1
Literature Cited:
- 1.Aird WC, Endothelial cell heterogeneity. Crit Care Med, 2003. 31(4 Suppl): p. S221–30. [DOI] [PubMed] [Google Scholar]
- 2.Lawson ND and Weinstein BM, Arteries and veins: making a difference with zebrafish. Nat Rev Genet, 2002. 3(9): p. 674–82. [DOI] [PubMed] [Google Scholar]
- 3.Nolte A, et al. , Veins are no arteries: even moderate arterial pressure induces significant adhesion molecule expression of vein grafts in an ex vivo circulation model. J Cardiovasc Surg (Torino), 2011. 52(2): p. 251–9. [PubMed] [Google Scholar]
- 4.Corada M, Morini MF, and Dejana E, Signaling pathways in the specification of arteries and veins. Arterioscler Thromb Vasc Biol, 2014. 34(11): p. 2372–7. [DOI] [PubMed] [Google Scholar]
- 5.dela Paz NG and D’Amore PA, Arterial versus venous endothelial cells. Cell Tissue Res, 2009. 335(1): p. 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diodato M and Chedrawy EG, Coronary artery bypass graft surgery: the past, present, and future of myocardial revascularisation. Surg Res Pract, 2014. 2014: p. 726158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Favaloro RG, Critical analysis of coronary artery bypass graft surgery: a 30-year journey. J Am Coll Cardiol, 1998. 31(4 Suppl B): p. 1B–63B. [DOI] [PubMed] [Google Scholar]
- 8.Thorgeirsson G and Robertson AL Jr., The vascular endothelium-pathobiologic significance. Am J Pathol, 1978. 93(3): p. 803–48. [PMC free article] [PubMed] [Google Scholar]
- 9.Barthelmes J, et al. , Endothelial dysfunction in cardiovascular disease and Flammer syndrome-similarities and differences. EPMA J, 2017. 8(2): p. 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Scholten BJ, et al. , Markers of inflammation and endothelial dysfunction are associated with incident cardiovascular disease, all-cause mortality, and progression of coronary calcification in type 2 diabetic patients with microalbuminuria. J Diabetes Complications, 2016. 30(2): p. 248–55. [DOI] [PubMed] [Google Scholar]
- 11.Park KH and Park WJ, Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J Korean Med Sci, 2015. 30(9): p. 1213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papaharalambus CA and Griendling KK, Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc Med, 2007. 17(2): p. 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Libby P, Vascular biology of atherosclerosis: overview and state of the art. Am J Cardiol, 2003. 91(3A): p. 3A–6A. [DOI] [PubMed] [Google Scholar]
- 14.Mugge A, The role of reactive oxygen species in atherosclerosis. Z Kardiol, 1998. 87(11): p. 851–64. [DOI] [PubMed] [Google Scholar]
- 15.Kaneto H, et al. , Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm, 2010. 2010: p. 453892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schafer FQ and Buettner GR, Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med, 2001. 30(11): p. 1191–212. [DOI] [PubMed] [Google Scholar]
- 17.Smith CV, et al. , Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicol Appl Pharmacol, 1996. 140(1): p. 1–12. [DOI] [PubMed] [Google Scholar]
- 18.Nkabyo YS, et al. , Glutathione and thioredoxin redox during differentiation in human colon epithelial (Caco-2) cells. Am J Physiol Gastrointest Liver Physiol, 2002. 283(6): p. G1352–9. [DOI] [PubMed] [Google Scholar]
- 19.Pallardo FV, et al. , Role of nuclear glutathione as a key regulator of cell proliferation. Mol Aspects Med, 2009. 30(1–2): p. 77–85. [DOI] [PubMed] [Google Scholar]
- 20.Xiong Y, et al. , S-glutathionylation: from molecular mechanisms to health outcomes. Antioxid Redox Signal, 2011. 15(1): p. 233–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harding JJ, Blakytny R, and Ganea E, Glutathione in disease. Biochem Soc Trans, 1996. 24(3): p. 881–4. [DOI] [PubMed] [Google Scholar]
- 22.Thanislass J, Raveendran M, and Devaraj H, Buthionine sulfoximine-induced glutathione depletion. Its effect on antioxidants, lipid peroxidation and calcium homeostasis in the lung. Biochem Pharmacol, 1995. 50(2): p. 229–34. [DOI] [PubMed] [Google Scholar]
- 23.Bir SC, et al. , Control of angiogenesis dictated by picomolar superoxide levels. Free Radic Biol Med, 2013. 63: p. 135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navratil AR, et al. , Francisella tularensis LVS induction of prostaglandin biosynthesis by infected macrophages requires specific host phospholipases and lipid phosphatases. Infect Immun, 2014. 82(8): p. 3299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antohe F, Endothelial cells and macrophages, partners in atherosclerotic plaque progression. Arch Physiol Biochem, 2006. 112(4–5): p. 245–53. [DOI] [PubMed] [Google Scholar]
- 26.Moore KJ, Sheedy FJ, and Fisher EA, Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol, 2013. 13(10): p. 709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Packard RR and Libby P, Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem, 2008. 54(1): p. 24–38. [DOI] [PubMed] [Google Scholar]
- 28.Diaz-Vivancos P, et al. , Glutathione—linking cell proliferation to oxidative stressq Free Radic Biol Med, 2015. 89: p. 1154–64. [DOI] [PubMed] [Google Scholar]
- 29.Kang YJ, Feng Y, and Hatcher EL, Glutathione stimulates A549 cell proliferation in glutamine-deficient culture: the effect of glutamate supplementation. J Cell Physiol, 1994. 161(3): p. 589–96. [DOI] [PubMed] [Google Scholar]
- 30.Mailloux RJ and Willmore WG, S-glutathionylation reactions in mitochondrial function and disease. Front Cell Dev Biol, 2014. 2: p. 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owen JB and Butterfield DA, Measurement of oxidized/reduced glutathione ratio. Methods Mol Biol, 2010. 648: p. 269–77. [DOI] [PubMed] [Google Scholar]
- 32.Zitka O, et al. , Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett, 2012. 4(6): p. 1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aminizadeh N, et al. , Stimulation of cell proliferation by glutathione monoethyl ester in aged bone marrow stromal cells is associated with the assistance of TERT gene expression and telomerase activity. In Vitro Cell Dev Biol Anim, 2016. 52(7): p. 772–81. [DOI] [PubMed] [Google Scholar]
- 34.Milovanova T, et al. , Endothelial cell proliferation associated with abrupt reduction in shear stress is dependent on reactive oxygen species. Antioxid Redox Signal, 2004. 6(2): p. 245–58. [DOI] [PubMed] [Google Scholar]
- 35.Luanpitpong S, et al. , Regulation of lung cancer cell migration and invasion by reactive oxygen species and caveolin-1. J Biol Chem, 2010. 285(50): p. 38832–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu SY, et al. , Correlation between cell migration and reactive oxygen species under electric field stimulation. Biomicrofluidics, 2015. 9(5): p. 054120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moriarty-Craige SE and Jones DP, Extracellular thiols and thiol/disulfide redox in metabolism. Annu Rev Nutr, 2004. 24: p. 481–509. [DOI] [PubMed] [Google Scholar]
- 38.Lee YW, et al. , IL-4-induced oxidative stress upregulates VCAM-1 gene expression in human endothelial cells. J Mol Cell Cardiol, 2001. 33(1): p. 83–94. [DOI] [PubMed] [Google Scholar]
- 39.Hemmingsen JG, et al. , Oxidative stress, genotoxicity, and vascular cell adhesion molecule expression in cells exposed to particulate matter from combustion of conventional diesel and methyl ester biodiesel blends. Environ Sci Technol, 2011. 45(19): p. 8545–51. [DOI] [PubMed] [Google Scholar]
- 40.Cooper D, et al. , Oxidative stress promotes blood cell-endothelial cell interactions in the microcirculation. Cardiovasc Toxicol, 2002. 2(3): p. 165–80. [DOI] [PubMed] [Google Scholar]
- 41.Kalogeris TJ, et al. , Differential monocyte adhesion and adhesion molecule expression in venous and arterial endothelial cells. Am J Physiol, 1999. 276(1 Pt 1): p. L9–L19. [DOI] [PubMed] [Google Scholar]
- 42.Shi Y, et al. , Oxidative stress and lipid retention in vascular grafts: comparison between venous and arterial conduits. Circulation, 2001. 103(19): p. 2408–13. [DOI] [PubMed] [Google Scholar]
- 43.Guzik TJ, et al. , Systemic regulation of vascular NAD(P)H oxidase activity and nox isoform expression in human arteries and veins. Arterioscler Thromb Vasc Biol, 2004. 24(9): p. 1614–20. [DOI] [PubMed] [Google Scholar]
- 44.Deng DX, et al. , Differences in vascular bed disease susceptibility reflect differences in gene expression response to atherogenic stimuli. Circ Res, 2006. 98(2): p. 200–8. [DOI] [PubMed] [Google Scholar]
- 45.Raza H, et al. , Glutathione S-transferases in human and rodent skin: multiple forms and species-specific expression. J Invest Dermatol, 1991. 96(4): p. 463–7. [DOI] [PubMed] [Google Scholar]
- 46.Hoos MD, et al. , The impact of human and mouse differences in NOS2 gene expression on the brain’s redox and immune environment. Mol Neurodegener, 2014. 9: p. 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang YC, et al. , Deficiency of NPGPx, an oxidative stress sensor, leads to obesity in mice and human. EMBO Mol Med, 2013. 5(8): p. 1165–79. [DOI] [PMC free article] [PubMed] [Google Scholar]