

Abstract
The ubiquitin system regulates diverse biological processes, many involved in cancer pathogenesis, by altering the ubiquitination state of protein substrates. This is accomplished by ubiquitin ligases and deubiquitinases (DUBs), which respectively add or remove ubiquitin from substrates to alter their stability, activity, localization, and interactions. While lack of catalytic activity makes therapeutic targeting of ubiquitin ligases difficult, DUB inhibitors represent an active area of research and the identification of cancer-associated DUBs may lead to the development of novel therapeutics. A growing body of literature demonstrates that the DUB Otubain 1 (OTUB1) regulates many cancer-associated signaling pathways including MAPK, ERa, EMT, RHOa, mTORC1, FOXM1 and P53 to promote tumor cell survival, proliferation, invasiveness, and therapeutic resistance. In addition, clinical studies have associated elevated OTUB1 expression with high grade, invasiveness, and metastasis in several tumor types including lung, breast, ovarian, glioma, colon, and gastric. Interestingly, in addition to catalytic DUB activity, OTUB1 displays a catalytic-independent, non-canonical activity where it inhibits the transfer of ubiquitin onto protein substrates by sequestration of E2 ubiquitin conjugating enzymes. The aim of this review is to describe the canonical and non-canonical activities of OTUB1, summarize roles for OTUB1 in cancer-associated pathways, and discuss its potential therapeutic targeting.
Keywords: OTUB1, E2 enzyme, ubiquitin ligase, DUB inhibitor, metastasis
Introduction
Ubiquitination, the post-translational addition of the ubiquitin protein to target proteins, controls various cellular processes by altering protein stability, localization, activity, or interactions. This process is accomplished by ubiquitin ligases, E2 ubiquitin conjugating enzymes (E2s), and E1 activating enzymes (E1s). E1s activate and transfer ubiquitin to E2s, forming an E2-ubiquitin conjugate. Ubiquitin ligases then bind the E2-ubiquitin conjugate and, in the case of RING and U-box type ubiquitin ligases, transfer the ubiquitin directly onto the substrate. For HECT and RBR type ubiquitin ligases, ubiquitin is initially transferred onto the ubiquitin ligase and subsequently transferred onto the substrate. In all instances, the ubiquitin carboxy terminus is ligated to the epsilon-amino group of a substrate lysine residue, forming an isopeptide bond. This process can occur once, resulting in monoubiquitination, or repeatedly, resulting in polyubiquitin chain formation. Polyubiquitin chain function is determined by which ubiquitin lysine is used to link adjacent ubiquitins. For example, lysine 48 (K48) chains typically signal for proteasomal degradation, while K63 chains often regulate lysosomal targeting and protein interactions (Komander & Rape 2012). In opposition to ubiquitin ligases, deubiquitinases (DUBs) cleave the isopeptide bond, partially or completely removing the polyubiquitin chain, with some DUBs displaying preference for certain linkages. Thus, DUBs modify or reverse the effects of ubiquitin ligases (Clague et al. 2013).
Several pathways critical for cancer development and progression such as apoptosis, cell-cycle progression, receptor downregulation, migration, and invasion are maintained in homeostasis by the balance between ubiquitination and deubiquitination (HuangFu & Fuchs 2010, Gallo et al. 2017). Indeed, many ubiquitin ligases and DUBs are critical regulators of tumor promoting or suppressive pathways. One of the most well studied examples is the regulation of the tumor suppressor P53 by the ubiquitin ligase MDM2 and the deubiquitinase USP7. MDM2 ubiquitinates P53, causing its proteasomal degradation and the overexpression of MDM2 in NIH3T3 cells renders them tumorigenic (Fakharzadeh et al. 1991). In contrast, USP7 deubiquitinates and stabilizes P53, leading to cellular growth suppression and apoptosis (Li 2002). Although ubiquitin ligases remain challenging drug targets due to their lack of catalytic activity, DUB inhibitors have entered the clinical setting for the treatment of cancer (D’Arcy et al. 2015, Wang et al. 2016). Thus, the identification of cancer-associated DUBs may reveal novel therapeutic targets.
Accumulating evidence suggests that the recently identified DUB OTUB1 has a critical role in cancer initiation and progression. Several studies demonstrate that OTUB1 regulates apoptosis, therapeutic resistance, proliferation, migration, and invasion in several cancer types. Interestingly, in addition to canonical DUB activity, OTUB1 possesses a non-canonical, catalytic-independent activity where it inhibits the transfer of ubiquitin onto protein substrates by binding to the ubiquitin-charged E2. This review will describe the canonical and non-canonical activities of OTUB1, summarize OTUB1’s regulation of cancer-associated pathways, and discuss OTUB1’s potential therapeutic targeting.
Otubain 1: a non-canonical deubiquitinase
Identified by Borodovsky and colleagues, OTUB1 is the founding member of the ovarian tumor (OTU) domain family of DUBs (Borodovsky et al. 2002). The OTU domain characterizes a larger superfamily of predicted cysteine proteases that possess a catalytic triad typically comprised of aspartate, cysteine, and histidine residues (Makarova et al. 2000). A subsequent study purified OTUB1 from HeLa cells using the DUB probe ubiquitin aldehyde (Ubal) and demonstrated that OTUB1 is an active DUB. This study also coined the name Otubain for OTU domain containing Ubal binding protein, identified the OTU domain, and defined the catalytic residues of OTUB1 as D88, C91, and H265 (Balakirev et al. 2003) (Figure1). Biochemical analysis determined OTUB1 is specific for cleaving K48-linked polyubiquitin chains, but has no activity for K63-linked chains. OTUB1 also demonstrated weak activity for NEDD8, an ubiquitin-like modifier (Edelmann et al. 2009).
Figure 1).
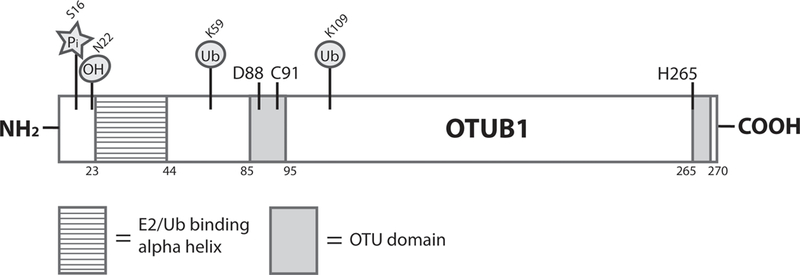
Schematic diagram of the OTUB1 protein. Experimentally confirmed post-translational modifications are shown above the structure with their corresponding residue designations. The E2/Ub binding alpha helix (residues 23–44) [lined shading], ovarian tumor (OTU) domain (residues 85–95 and 265–270) [gray shading], and catalytic residues (D88, C91, H265) are also depicted. Pi (phosphorylation), OH (hydroxylation), and Ub (ubiquitination/ubiquitin).
Located at locus 11q13.1, OTUB1 is expressed in a variety of human tissues, with high expression observed in the brain (Fagerberg et al. 2014). In the NIH3T3 mouse fibroblast cell line, OTUB1 is one of the most highly expressed DUBs (Schwanhausser et al. 2011). In addition, full knockout of OTUB1 results in embryonic lethality in mice, while heterozygous knockout leads to a reduction in grip strength and a decrease in lean body mass (mousephenotype.org [5–29-18]). As mentioned above, in addition to canonical deubiquitinase activity, OTUB1 displays a catalytic-independent, non-canonical activity where it inhibits ubiquitin transfer onto protein substrates by binding to the ubiquitin-charged E2 (Nakada et al. 2010) (Figure2). Thus, OTUB1 can decrease a protein’s ubiquitination state by removing ubiquitin (canonical-activity) or preventing ubiquitin conjugation (non-canonical activity).
Figure 2).
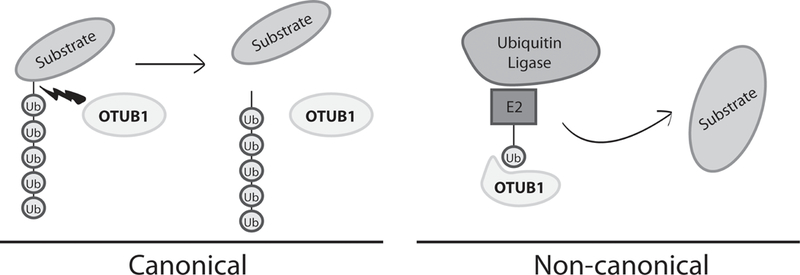
The canonical and non-canonical activities of OTUB1. On the left, the canonical, deubiquitinase activity of OTUB1 is depicted, with OTUB1 using its catalytic activity to cleave a polyubiquitin chain from a protein substrate. On the right, the non-canonical activity of OTUB1 is depicted, with OTUB1 inhibiting ubiquitin transfer onto a protein substrate by binding to the ubiquitin charged E2. Ub (ubiquitin).
The non-canonical activity of OTUB1 was discovered by the Durocher laboratory in 2010, when they identified OTUB1 as a DUB that suppresses K63 chromatin ubiquitination in response to DNA damage (Nakada et al. 2010). This observed K63-directed activity conflicted with previous reports that OTUB1 DUB activity is specific for K48 linkages. To resolve the discrepancy, the investigators demonstrated, with a series of OTUB1 mutants, that the reduction in ubiquitination was independent of OTUB1’s DUB activity. Further biochemical analysis revealed that OTUB1 binds to the E2 UBC13 and prevents the ubiquitin ligase RNF168 from ubiquitinating chromatin at sites of DNA damage. In addition, OTUB1 displayed an increased affinity for the ubiquitin-charged form of UBC13. Other studies have confirmed this interaction and identified other OTUB1-interacting E2s (Markson et al. 2009, Sowa et al. 2009, Zulkifle 2013) (Figure3).
Figure 3).
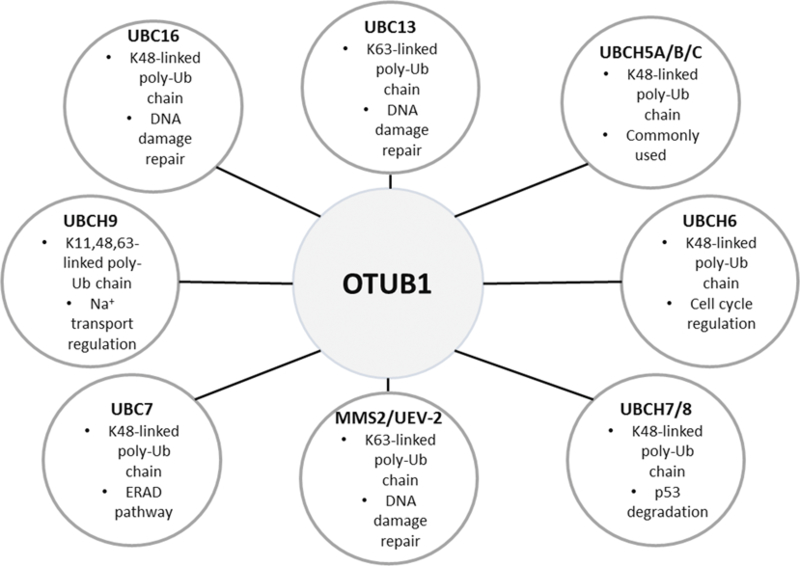
E2 ubiquitin conjugating enzymes that interact with OTUB1. Depicted are the E2 ubiquitin conjugating enzymes that have been experimentally determined to interact with OTUB1. Also given are brief descriptions of the polyubiquitin linkage type(s) synthesized and biological functions for each. (ERAD) Endoplasmic reticulum associated protein degradation, K (lysine), Ub (ubiquitin).
Regulation of OTUB1 activity
Since the discovery of OTUB1’s non-canonical activity, multiple studies have demonstrated that it non-canonically inhibits the ubiquitination of many proteins, suggesting that OTUB1 regulates several biological pathways in this manner. In addition, targets of OTUB1’s DUB activity have been identified. Importantly, critical interaction surfaces, key residues, and modes of regulation for the non-canonical and canonical activities have been elucidated. These will be discussed below.
Crystallographic analysis by Juang and colleagues of OTUB1 in complex with the ubiquitin-charged E2 UBCH5B identified three interaction surfaces important for OTUB1’s non-canonical activity. These include interaction of the E2 with an N-terminal alpha helix and the C-terminus of OTUB1, interaction of the donor ubiquitin (ubiquitin conjugated to the E2) with the N-terminal alpha helix and OTU core of OTUB1 and, unexpectedly, interaction of a free ubiquitin with the C-terminus of OTUB1. Strikingly, the structure revealed that the donor and free ubiquitins are positioned in a manner that mimic the cleavage product of a K48 DUB reaction, with the epsilon-amino group of K48 from the donor ubiquitin facing the C-terminus of the free ubiquitin, both positioned near the catalytic cysteine of OTUB1 (Juang et al. 2012) (Figure4). Termed “product inhibition mimicry” by the authors, this OTUB1-E2 inhibition complex is likely conserved across E2s non-canonically targeted by OTUB1, as they share high sequence homology (Nakada et al. 2010). Indeed, a different study modeling the E2 UBC13 interaction with OTUB1 found almost identical OTUB1 residues are critical for mediating an inhibitory complex (Sato et al. 2012).
Figure 4).
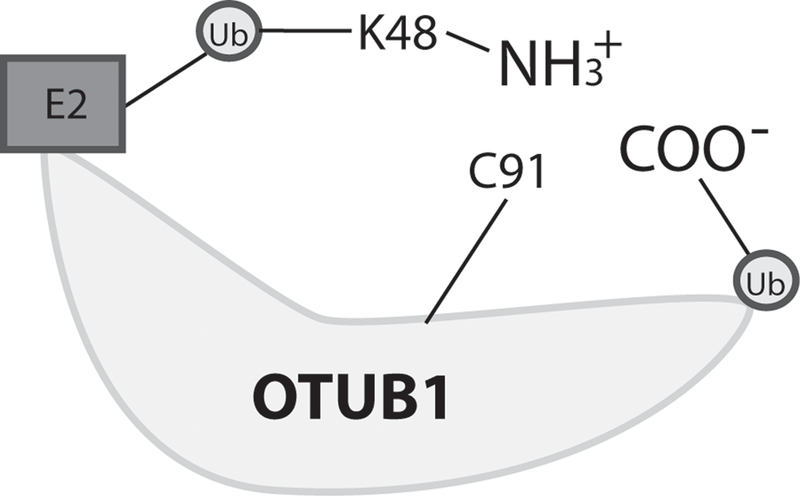
The non-canonical “product inhibition mimicry” complex. Depicted is a ternary complex of OTUB1, an ubiquitin-charged E2, and a free ubiquitin. This complex mimics the products of a K48 deubiquitinase reaction, with the epsilon-amino group (NH3+) of K48 from the donor ubiquitin facing the C-terminus (COO-) of the free ubiquitin, both positioned near the catalytic cysteine (C91) of OTUB1. K (lysine), Ub (ubiquitin).
Expanding on this study, Weiner and colleagues found that free ubiquitin binding to OTUB1’s C-terminus allosterically triggers the formation of the E2/ubiquitin binding alpha helix in OTUB1’s N-terminus and that this is essential for E2 inhibition. Consistent with this observation, deletion of the first 45 residues of OTUB1 abolishes the ability of OTUB1 to non-canonically inhibit E2s. In addition, in vitro work demonstrated that in the absence of free ubiquitin, OTUB1 does not bind to ubiquitin-charged E2s and that free ubiquitin enhances E2 binding in a concentration dependent manner (Wiener et al. 2012). Interestingly, OTUB1’s DUB activity is also regulated by E2 enzymes. In this study, under physiological concentrations of free ubiquitin, uncharged E2s stimulate DUB activity by stabilizing OTUB1’s N-terminal E2/ubiquitin binding alpha helix, thus increasing OTUB1’s affinity for K48 ubiquitin chains. In contrast, charged E2s inhibit DUB activity, which the authors attribute to the formation of the non-canonical inhibition complex described above. This complex would preclude the binding sites for polyubiquitin chains and inhibit catalysis. (Wiener et al. 2013).
Transcriptional and post-transcriptional regulation of OTUB1
Regulation of OTUB1
Transcriptional and translational controls of OTUB1 have been described. In a recent study, upregulation of OTUB1 is demonstrated to be downstream of the CXCL12/CXCR7/ERK signaling axis in MCF7 breast cancer cells. The authors suggest that the upregulated OTUB1 mediates endocrine therapy resistance by deubiquitinating ERa and enhancing ERa-mediated transcription (Hao et al. 2018). As the CXCL12/CXCR7 signaling axis contributes to the development and progression of other endocrine-related cancers, such as prostate (Sun et al. 2010) and ovarian (Yu et al. 2014), this pathway may be activated by a variety of tumors to stimulate OTUB1’s tumorigenic effects.
Translational regulation of OTUB1 has also been described. MiR-542–3p, a tumor suppressor miRNA, was demonstrated to directly downregulate OTUB1 in gastric adenocarcinoma (Yaun et al. 2017)(see below). Importantly, the tumor suppressive effects of miR-542–3p were shown to be partially dependent on OTUB1 downregulation. As miR-542–3p also functions as a tumor suppressor in breast (Wu et al. 2017) and lung (Liu et al. 2017) carcinomas, use of this miRNA to suppress OTUB1 could offer clinical benefit.
Monoubiquitination of OTUB1 regulates its non-canonical activity
While investigating OTUB1’s ability to inhibit P53 ubiquitination (see below), the Dai laboratory identified a slightly larger form of OTUB1 that was consistent with a monoubiquitination event. Further study confirmed that OTUB1 is monoubiquitinated primarily at lysine 59 or 109 by UBCH5 and that this is required for non-canonical regulation of P53. Indeed, mutation of all lysines on OTUB1, generating a form of OTUB1 unable to be ubiquitinated, failed to inhibit P53 ubiquitination and bind to UBCH5 (Li et al. 2014). This suggests UBCH5-mediated monoubiquitination may regulate the non-canonical activity of OTUB1 for a broad range of targets and could be a site for therapeutic intervention.
Hydroxylation of Asparagine 22 restricts the OTUB1 interactome
Expanding on their previous finding that OTUB1 is hydroxylated (Scholz et al. 2013), Scholz and colleagues determined the factor inhibiting HIF hydroxylates OTUB1 at N22 and that this restricts the OTUB1 interactome (Scholz et al. 2016). Interestingly, although the authors demonstrate that an absence of hydroxylation has no effect on OTUB1 DUB activity, their observations emphasize that N22 lies at the junction between the N-terminal E2/ubiquitin binding helix and the remaining, unstructured N-terminus (Figure1). Thus, this modification may regulate OTUB1’s non-canonical activity.
Phosphorylation of Serine 16 triggers nuclear localization
Several studies have determined that OTUB1 is phosphorylated on multiple residues (Edelmann et al. 2010, Herhaus et al. 2013). Subsequent investigation determined that CK2 phosphorylates OTUB1 at S16 and that this triggers OTUB1 nuclear localization (Herhaus et al. 2015). While the authors determined S16 phosphorylation does not affect the DUB or non-canonical activities of OTUB1, they did demonstrate that cytoplasmic localization was required for OTUB1 to inhibit chromatin ubiquitination in response to DNA damage. This suggests that CK2 may broadly control OTUB1 activity by restricting its access to substrates in the cytosol or nucleus.
OTUB1 regulates several cancer-associated pathways and correlates with aggressive clinical disease
A growing number of studies have identified OTUB1 as a regulator of pathways important in cancer development and progression. In addition, clinical data from multiple cancer types demonstrates that high OTUB1 expression is associated with aggressive clinical disease and poorer patient survival. The majority of these studies support an oncogenic role for OTUB1; especially in the context of tumor migration, invasion, and metastasis. However, some studies demonstrate OTUB1 potentiates growth suppressive pathways such as P53 and TGFb, suggesting that the contribution of OTUB1 to cancer development and progression may be context or tumor type-dependent. A list of cancers and cancer-associated pathways that OTUB1 targets is depicted in Table 1, while key individual studies are summarized below.
Table 1).
Cancers and cancer-associated pathways associated with OTUB1. From left to right; the cancer or cancer-associated pathway that OTUB1 associates with, the protein of the cancer/pathway directly regulated by OTUB1 activity (if known), the mode of regulation (canonical [C] or non-canonical [NC]) (if known), the ubiquitin ligase and E2 enzyme pair inhibited by OTUB1 (if known), and the biological result.
| Cancer/Pathway | Target | Mode | Ligase/E2 | Result |
|---|---|---|---|---|
| TGFb Signaling | SMAD2 | NC | NEDD4L/UBCH5A | Increased migration |
| DSB DNA Repair | Histones | NC | RNF168/UBC13 | Reduced histone ubiquitination |
| Lung | RAS | NC | RABEX5/UBCH5C | Increased growth |
| Apoptosis | P53 | C/NC | MDM2/UBCH5C | Increased apoptosis |
| Apoptosis | MDMX | NC | MDM2/UBCH5C | Increased apoptosis |
| Apoptosis | c-IAP | C | Decreased apoptosis | |
| mTORC1 | DEPTOR | NC | Autophagy | |
| Endometrial | ERa | C/NC | Decreased ERa transcription | |
| Breast | FOXM1 | C | Increased proliferation, epirubicin resistance | |
| Ovarian | FOXM1 | C | Increased proliferation, migration, invasion | |
| Esophageal | SNAIL | C | EMT, increased migration, invasion | |
| Liver* | Increased migration, invasion | |||
| Glioma | EMT, increased migration | |||
| Colon | EMT, increased migration, invasion | |||
| Gastric | EMT, increased migration |
The liver cancer study (Ni et al. 2017) was not summarized in this review. DSB (double strand break), EMT (epithelial-mesenchymal transition).
Activation of RAS in lung cancer
The RAS GTPases are the most frequently mutated oncoproteins in cancer and activate many downstream signaling cascades such as MAPK, PLC, and RAC to promote tumor cell growth, survival, and migration (McCormick 2015). While several mechanisms exist to regulate their activity, ubiquitination has been shown to suppress RAS activity and relocalize RAS from the plasma membrane to endosomes (Xu et al. 2010). In an effort to identify factors that regulate RAS ubiquitination, Baietti et al. demonstrated that OTUB1 non-canonically inhibits the monoubiquitination of RAS in lung carcinoma cells and stimulates the accumulation of RAS at the plasma membrane (Baietti et al. 2016). Additionally, overexpression of OTUB1 potentiated serum-induced phosphorylation of ERK1/2 in cells expressing wild type (WT) RAS but had no effect in the presence of dominant negative (DN) or constitutively active RAS, indicating that the effects of OTUB1 on ERK1/2 are RAS dependent. Further, overexpression of OTUB1 enhanced soft agar colony formation and xenograft tumor growth of lung carcinoma cell lines harboring WT RAS but not in those expressing hyperactive, mutant RAS. Interestingly, silencing of OTUB1 in the mutant RAS lung carcinoma cell line A549 suppressed ERK1/2 signaling, soft agar colony formation, and xenograft tumor growth. The authors suggest that while endogenous expression levels of OTUB1 are required to maintain mutant RAS activity, overexpression of OTUB1 does not further potentiate mutant RAS activity.
To further validate this data, the investigators analyzed human lung tumors and patient data sets for gene expression and copy number aberrations. They found that OTUB1 copy number gain occurs in human lung tumors and that it is mutually exclusive with KRAS mutational status. In addition, OTUB1 copy number is constant throughout tumor stage, indicating that OTUB1 gain is an early event; in contrast, KRAS mutational status increases with stage. Immunohistochemical analysis of human tumors demonstrated that in WT KRAS tumors, high OTUB1 levels are significantly correlated with increased ERK1/2 phosphorylation. Survival analysis of individuals with WT RAS tumors demonstrated that patients expressing moderate levels of OTUB1 have significantly poorer survival compared to tumors with low OTUB1 expression. No survival difference with respect to OTUB1 expression was observed in mutant RAS tumors, supporting a role for OTUB1 overexpression in the WT RAS setting. As RAS is considered “undruggable” (McCormick 2015), therapeutic inhibition of OTUB1 could offer a successful avenue for the in-direct inhibition of RAS in lung and other RAS-dysregulated cancers.
Suppression of the estrogen receptor
Activation of the estrogen receptor alpha (ERa) by estrogen affects the development of certain breast and endometrial carcinomas (Ali et al. 2004, Yue et al. 2010). In 2009, Stanisic and colleagues identified OTUB1 as an ERa interacting protein that deubiquitinates ERa in a manner dependent on its canonical DUB activity, as the C91S catalytically-inactive OTUB1 mutant restored ERa ubiquitination. Surprisingly, when the effect of OTUB1 on ERa-mediated transcription was analyzed, both OTUB1 WT and C91S inhibited transcriptional activity, whereas an OTUB1 mutant incapable of binding ubiquitin, potentiated ERa-mediated transcription. Strengthening these findings, silencing of OTUB1 lead to an increase in several ERa-regulated transcripts when endometrial cancer cells were treated with estrogen (Stanisic et al. 2009). Taken together, these observations suggests that OTUB1 regulates ERa canonically, by directly interacting with and deubiquitinating ERa and non-canonically, by possibly suppressing the activity of an unidentified ubiquitin ligase important for ERa transcriptional activity. Future studies will need to investigate whether OTUB1 suppresses ERa-mediated tumor growth, a question not answered by this study.
Deubiquitination of FOXM1 in breast and ovarian carcinomas
The FOXM1 transcription factor promotes cell cycle progression and Epithelial-Mesenchymal Transition (EMT) in various tumor types by promoting the expression of several genes including CCND1, CCNB1, AURKB, BIRC5, SNAI2, and VEGFA (Saba et al. 2016). In addition, several studies demonstrate that FOXM1 mediates therapeutic resistance across multiple tumor types (Alvarez-Fernandez & Medema 2013). Together, these studies highlight the critical role for FOXM1 in tumor development, progression, and recurrence. Recently, two studies revealed that OTUB1 deubiquitinates FOXM1 in breast and ovarian cancer cells.
In breast cancer, OTUB1 is proposed to mediate epirubicin resistance by deubiquitinating and stabilizing FOXM1. In support of this, OTUB1 was observed to diminish the cytotoxic effects of epirubicin and potentiate cell proliferation in a manner dependent on FOXM1 and OTUB1 DUB activity. Furthermore, analysis of 116 patient samples demonstrated that OTUB1 and FOXM1 expressions significantly correlate, however no association of OTUB1 expression with histological type, lymph node status, or stage was observed. Although no association with patient survival was initially seen, multivariate cox regression analysis controlling for clinicopathological parameters demonstrated that OTUB1 did significantly associate with poorer overall and disease-specific survival. In addition, survival analysis of the chemotherapy treated portion of this group (n=60) demonstrated that high OTUB1 expression significantly associates with poorer overall and disease-specific survival (Karunarathna et al. 2016).
In ovarian cancer, deubiquitination and stabilization of FOXM1 by OTUB1 was also observed. Here, OTUB1 overexpression potentiated proliferation, migration, and invasion of SKOV3 ovarian cancer cells in vitro, while xenografts demonstrated a significant increase in tumor growth rate and size. Importantly, FOXM1 dependence was demonstrated in all studies, as knockdown of FOXM1 attenuated the effects of OTUB1 overexpression. Analysis of 200 patient samples demonstrated that OTUB1 expression significantly correlates with stage, tumor size, spread to local tissues, and recurrence. Survival analysis revealed that patients with high OTUB1 and FOXM1 expression have a significantly shorter disease-free and disease-specific survival (Wang et al. 2016).
As FOXM1 is upregulated in many tumor types (Alvarez-Fernandez & Medema 2013), these studies underscore the broad role OTUB1 may play in promoting therapeutic resistance and tumor progression across multiple tumor types, highlighting the OTUB1-FOXM1 axis as a promising therapeutic target.
OTUB1 promotes invasion in glioma, colon, gastric, and esophageal cancer via EMT
Glioblastoma, a high grade glioma, is the most deadly form of brain cancer, with a 5 year survival rate of 5% (Goodenberger & Jenkins 2012). One factor contributing to this poor prognosis is its diffuse and infiltrative growth pattern which primarily arises through cell migration and invasion (Noroxe et al. 2016). In a recent study (Xu et al. 2017), high OTUB1 expression in human glioma samples (n=89) was significantly associated with tumor grade and poor survival, but not tumor size. In addition, a significant inverse relationship was observed between OTUB1 and E-cadherin protein expression, suggesting a role for OTUB1 in EMT activation. To investigate this, the U87MG glioblastoma cell line, which demonstrated high expression of OTUB1 and EMT-associated genes, was used in OTUB1 knockdown in vitro experiments. OTUB1 silencing decreased the expression of the EMT-associated genes MMP2, MMP9, SNAI2, and VIM. In addition, a significant reduction in wound closure and transwell migration was observed with OTUB1 knockdown. As OTUB1 stabilizes Snail to promote EMT in esophageal cancer (see below), it’s possible that OTUB1 initiates a Snail-induced EMT in glioma and other tumor types to promote tumor progression.
Two independent studies also observed OTUB1 overexpression in colorectal cancer (CRC). In the first (Liu et al. 2014), analysis of 27 CRC patient samples revealed that OTUB1 transcript is significantly elevated in tumors relative to adjacent, normal mucosa. In addition, an examination of clinicopathological variables found that OTUB1 expression significantly correlates with tumor size, poor tumor differentiation, and lymph node metastasis in 108 patient samples. In the second study (Zhou et al. 2014), survival analysis (n=260) revealed that OTUB1 expression was negatively correlated with overall and progression-free survival and that OTUB1 serves as an independent prognostic factor for both. Correlation of OTUB1 with clinicopathological features revealed that OTUB1 expression was significantly associated lymph node and distant metastasis. Consistent with these clinical data, in vitro migration and invasion assays using CRC cell lines revealed that OTUB1 overexpression promoted, while knockdown inhibited, migration and invasion. OTUB1 did not affect proliferation. To identify an underlying mechanism, the authors assessed whether OTUB1 affected EMT-associated markers. ZEB1, B-catenin, E-cadherin, N-cadherin, and Vimentin protein expression were all modulated by OTUB1 overexpression and knockdown. In addition, immunohistochemical analysis revealed that OTUB1 expression was negatively correlated with E-cadherin protein expression in CRC tissue samples. A role for OTUB1 in promoting CRC liver metastasis in vivo was evaluated by spleen and tail vein injections using OTUB1-overexpressing CRC cells. In these mouse models, OTUB1 significantly increased the number of metastatic liver nodules. In vitro analysis of several EMT-promoting pathways demonstrated that OTUB1 stimulates the nuclear translocation of NF-kB, a known promoter of EMT in CRC (Bates et al. 2004). Therefore, NF-kB induced EMT may be a pathway OTUB1 engages to potentiate CRC metastasis. Interestingly, NF-kB can also stimulate the unfolded protein response (UPR) pathway by driving the transcription of stress-response genes (Tam et al. 2012). OTUB1 has also been demonstrated to interact with c-IAP1, a constitutive partner of the UPR initiating TRAF2 protein (Goncharov et al. 2013). As several reports indicate that the UPR promotes EMT (Zhong et al. 2011, Shen et al. 2015), it is possible that OTUB1 activates the UPR pathway to enhance EMT and/or that OTUB1 activates the UPR to mediate distinct tumorigenic effects.
A mechanism for OTUB1 overexpression in CRC was recently elucidated, where OTUB1 was identified as a target of miR-542–3p, a tumor suppressor miRNA (Yaun et al. 2017). In this study, ectopic expression of miR-542–3p attenuated proliferation, migration, and invasion, while it promoted apoptosis. Bioinformatic analysis revealed a miR-542–3p consensus sequence in the 3’-UTR of OTUB1 and further analysis demonstrated that OTUB1 expression is directly downregulated by miR-542–3p. Importantly, expression of an OTUB1 construct resistant to miR-542–3p binding partially rescued the tumor suppressive effects of miR-542–3p by restoring the proliferative, migratory, and invasive potential of CRC cells. This suggests that the tumor suppressive effects of miR-542–3p are partially mediated by the downregulation of OTUB1. Supporting this, expression of a miR-542–3p resistant OTUB1 construct restored the expression of the EMT and anti-apoptotic markers BCL2, phospho-AKT, MMP9, MMP2, and N-cadherin, which were suppressed by miR-542–3p. As several studies demonstrate that OTUB1 engages EMT to promote tumor cell migration and invasion, therapeutic use of miR-542–3p to downregulate OTUB1 may suppress the development of an invasive phenotype in many tumor types.
Stimulation of migration and invasion by OTUB1 was also observed in gastric adenocarcinoma (Weng et al. 2016). In this study, OTUB1 significantly associated with gastric adenocarcinoma invasion in the clinical setting and promoted the expression of invasion-associated genes in vitro. Immunohistochemical analysis of OTUB1 expression in tumor, dysplastic, and normal tissue demonstrated that the percentage of samples with high OTUB1 expression significantly increased as the tissue became more malignant (normal 9%, dysplastic 33%, tumor 76%). In addition, high OTUB1 expression was significantly associated with aggressive clinical features including invasive depth, lymph node status, stage, and nerve invasion. No association with tumor size was found. Survival analysis demonstrated that patients with high OTUB1 expression exhibit a significantly shorter disease-free survival (DFS) and disease-specific survival (DSS). Importantly, OTUB1 was determined to be an independent risk factor for DSS but not DFS. Because of OTUB1’s association with invasive clinical properties, in vitro migration and invasion assays were performed. OTUB1 overexpression in AGS and HGC-27 gastric cancer cell lines significantly enhanced wound closure ability and transwell migration. Expression analysis of invasion-associated genes in these cell lines demonstrated that OTUB1 significantly increased the transcript and protein levels of N-cadherin, MMP2, and MMP9, while the opposite effect was observed for E-Cadherin. Similar to the CRC studies, this work suggests that OTUB1 promotes gastric adenocarcinoma invasion through EMT.
OTUB1 also promoted EMT in esophageal squamous cell carcinoma (ESSC) (Zhou et al. 2018). This study determined that WT OTUB1, but not a mutant harboring mutations at all catalytic triad residues, increased the protein expression of Snail, a master promoter of EMT (Wang et al. 2013). This effect was post-translational, as no effect on Snail transcript was observed but OTUB1 did extend Snail protein half-life. Next, the investigators assessed whether OTUB1 modulation of Snail altered the expression of EMT-associated genes and found that OTUB1 overexpression increased Snail, N-cadherin, and Vimentin expression while reducing E-cadherin expression. The opposite was observed with OTUB1 knockdown. Transwell assays revealed that OTUB1 overexpression increased, while knockdown decreased, migration and invasion of ESSC cells. Importantly, knockdown of Snail significantly attenuated the ability of OTUB1 to promote migration and invasion, demonstrating that Snail is required for these effects. In support of these in vitro data, in vivo studies demonstrated that OTUB1 knockdown significantly reduced the number of lung metastases in a tail-vein injection mouse model, suggesting that OTUB1 facilitates tumor cell survival in the circulation and/or the seeding of lung metastasis. In addition, analysis of patient samples revealed that OTUB1 is significantly upregulated in ESSC tissue compared to matched, normal tissue (n= 48). Importantly, OTUB1 is positively associated with Snail expression in these tissues. Survival analysis demonstrated that high OTUB1 expression is significantly associated with shorter overall survival (n=143). Taken together, the results of this study suggest that OTUB1 may potentiate the metastatic spread of ESSC by induction of a Snail-mediated EMT.
Activation of RhoA-mediated invasion in prostate cancer
Activation of the androgen receptor (AR) by dihydroxytestosterone (DHT) promotes the growth, migration, and invasion of prostate cancer (PCa) cells (Culig and Santer 2014). A 2015 study (Iglesias-Gato et al. 2015) demonstrated that OTUB1 potentiates androgen-dependent and androgen-independent PCa cell invasion in vitro in a canonical manner. Interestingly, OTUB1 did not affect proliferation. Focusing on OTUB1 in the androgen-dependent setting, the authors performed a proteomic screen and a phospho-protein array with and without OTUB1 knockdown. The proteomic screen revealed that OTUB1 silencing reduced the expression of several AR regulated proteins, while the array demonstrated that OTUB1 silencing opposed AR signaling. In particular, the data suggests that OTUB1 facilitates the androgen-mediated reduction in S392 phosphorylation of P53. Phosphorylation tightly regulates P53 tumor suppressive activity and reduced P53 levels have been associated with increased cell migration via RhoA activation (Gadea et al. 2007, Du et al. 2016), indicating that OTUB1 may promote PCa cell invasion by altering P53/RhoA activity. Strikingly, the authors demonstrate that DHT treatment caused a reduction in P53 and activation of RhoA in a manner dependent on OTUB1 DUB activity. In addition, OTUB1 also stimulated RhoA activation in a DHT/AR-independent manner. Collectively, these data suggests that, while AR signaling can modulate OTUB1/RhoA mediated PCa cell invasion, AR signaling is not required.
In the same study, immunohistochemical analysis of human PCa tissue demonstrated significantly higher OTUB1 expression in luminal epithelial tumor tissue compared to unmatched, normal tissue independent of tumor grade. To investigate the in vivo effects of OTUB1, mouse xenografts were performed with OTUB1-knockdown PCa cells. The results demonstrated that OTUB1 silencing reduced primary tumor growth and that OTUB1-knockdown tumors displayed lower Ki67 positivity relative to their controls. This contrasts with the in vitro assays, where OTUB1 did not affect proliferation. OTUB1 silencing also significantly reduced the number of metastatic foci in the lymph nodes and completely blocked metastasis to the liver and kidneys. Taken together, this study suggests that OTUB1 contributes to PCa growth and metastatic potential.
Potentiation of TGFb signaling and migration
The TGFb signaling pathway has contrasting effects on tumor biology. In the early stages of tumor development TGFb signaling exerts anti-proliferative effects by inhibiting cell cycle progression via cyclin-dependent kinase inhibition, which downregulates the expression of c-myc and promotes apoptosis (Lin & Zhao 2015). However, in advanced tumors, TGFb signaling promotes malignancy by stimulating angiogenesis, immune system evasion, and invasiveness (Lin & Zhao 2015). TGFb signaling is initiated by extracellular ligands, such as TGFb, binding to their cognate cell surface receptors. This stimulates the phosphorylation of the SMAD2 and SMAD3 transcription factors, which complex with SMAD4 and translocate to the nucleus to stimulate the transcription of target genes (Massague 2008). Regulation of TGFb signaling occurs at multiple levels, including the ubiquitin-dependent degradation of activated, phosphorylated SMADs by the ubiquitin ligase NEDD4L (Gao et al. 2009). Recently, OTUB1 was identified as an inhibitor of NEDD4L-mediated ubiquitination of SMAD2 and SMAD3 (Herhaus et al. 2013). Interestingly, these investigators demonstrate that OTUB1 specifically interacts with phosphorylated SMAD2. Additionally, knockdown of OTUB1 reduced the expression of phosphorylated SMAD2 upon TGFb treatment and these effects were rescued with either proteasome inhibition or the expression of a knockdown resistant OTUB1 construct, suggesting that OTUB1 protects phospho-SMAD2 from proteasomal degradation. Effects on TGFb-induced transcription were also observed, as OTUB1 knockdown decreased luciferase activity from a TGFb responsive luciferase construct and reduced the expression of several TGFb responsive genes in a human keratinocyte (HaCAT) cell line. Using the same cell line, the group demonstrated that OTUB1 knockdown impaired cell migration in response to TGFb. Investigation into the biological mechanism revealed that OTUB1 inhibits NEDD4L-mediated ubiquitination of SMAD2 and SMAD3 in a manner dependent on lysine residue K17 and the N-terminus of OTUB1. Collectively, these data demonstrate that OTUB1 regulates TGFb signaling and downstream biological effects. Future studies should examine the effect of OTUB1 on TGFb signaling in the context of tumorigenesis and tumor cell migration.
Suppression of mTORC1: a role in autophagy
The multi-component mTORC1 kinase is a critical cellular nutrient sensor that, under optimal nutrient conditions, stimulates cell growth and inhibits autophagy (Bar-Peled & Sabatini 2014). DEPTOR, an mTORC1 component, suppresses mTORC1 activity but is rapidly degraded by the SCFβ-TRCP ubiquitin ligase following mTORC1 activation (Gao et al. 2011). In a recent study (Zhao et al. 2018), OTUB1 overexpression was found to post-translationally stabilize DEPTOR and rescue SCFβ-TRCP-mediated reduction in DEPTOR expression, suggesting that OTUB1 blocks SCFβ-TRCP-mediated DEPTOR ubiquitination. Importantly, OTUB1−/− Hela cells displayed a striking reduction in endogenous DEPTOR expression. Further analysis determined that ubiquitination of ectopic DEPTOR was inhibited non-canonically; as OTUB1 WT and C91S, but not D88 (a residue required for non-canonical activity), suppressed DEPTOR ubiquitination. In addition, amino acid-induced mTORC1 signaling was enhanced by OTUB1 knockdown. Examination of the biological consequences of OTUB1’s suppression of mTORC1 demonstrated that OTUB1 silencing inhibited starvation-induced autophagy in lung carcinoma cells, as determined by a decrease in the number of LC3II- positive puncta, an autophagy marker. OTUB1 knockdown also increased cell viability and size, two biological outputs inversely associated with autophagy (Neufeld 2012). As autophagy can promote tumor progression by enhancing metastasis (White 2015), future studies should focus on whether OTUB1-enhanced autophagy supports tumor metastasis. In addition, the ability of OTUB1 to regulate other forms of autophagy should be investigated.
Regulation of P53, MDMX, and c-IAP: a role in apoptosis
Although the majority of studies point to a tumorigenic role for OTUB1, the laboratory of Mu-Shui Dai has published studies indicating that OTUB1 promotes apoptosis in some contexts. A study from 2012 found that OTUB1 non-canonically inhibits MDM2-mediated ubiquitination of P53 to promote its stabilization (Sun et al. 2012). OTUB1 overexpression was demonstrated to increase the expression of P53 transcriptional targets and induce apoptosis of osteosarcoma and lung cancer cell lines in a P53-dependent manner. Mutational analysis of OTUB1 found that D88, a catalytic triad residue, abrogated OTUB1’s ability to inhibit P53 ubiquitination, induce P53 transcriptional targets, and potentiate apoptosis. Interestingly, OTUB1 was also found to directly bind and weakly deubiquitinate P53, which may further enhance OTUB1-mediated P53 stabilization. Biochemical interrogation into the OTUB1-P53 interaction revealed that OTUB1 binds to the DNA binding domain of P53 and P53 interacts with the N- and C-termini of OTUB1. To investigate the biological implications of OTUB1-mediated P53 stabilization, studies investigating P53 activity in response to DNA damage were conducted. Expression of the OTUB1 D88 mutant or OTUB1 knockdown attenuated the induction of P53-induced transcripts following DNA damage of osteosarcoma cells. Importantly, these effects were rescued by a knockdown resistant OTUB1 construct.
A subsequent study by the Dai laboratory determined that OTUB1 promotes apoptosis by stabilizing MDMX (Chen et al. 2017), a regulator of P53 levels and activity (Wade et al. 2010). In this study, OTUB1 was shown to non-canonically inhibit MDM2-mediated ubiquitination of MDMX. Interestingly, previous studies have demonstrated that MDMX can recruit phosphorylated P53 to the mitochondria to potentiate the intrinsic apoptotic pathway. This study demonstrated that OTUB1 not only stabilized cytosolic MDMX but also increased its abundance at the mitochondria. Taken together, these investigators suggest a model where OTUB1 plays a multifaceted role in promoting P53-induced apoptosis where it 1) directly stabilizes P53 by inhibiting MDM2 and 2) stabilizes MDMX to allow phosphorylated P53 to localize to the mitochondria and activate the intrinsic apoptotic pathway. Future studies should examine whether MDMX depletion attenuates OTUB1-induced apoptosis.
A role for OTUB1 in the extrinsic apoptotic pathway has also been suggested (Goncharov et al. 2013). This study found that OTUB1 deubiquitinates and stabilizes c-IAP1 upon TNFR activation. These investigators demonstrate that activation of TNFR in the presence of OTUB1 knockdown accelerates c-IAP1 degradation and alters TNFR signaling to promote caspase-8 activation and apoptosis. Importantly, this enhanced apoptosis was dependent on TNFR and caspase activation, as inhibition of either rescued cell viability. In addition, OTUB1 was shown to be recruited to TNFR complexes upon ligand activation, further supporting a role for OTUB1 in regulating ligand-activated TNFR signaling. In Zebrafish, depletion of OTUB1 lead to blood vessel narrowing and reduced blood flow, a phenotype similar to c-IAP1 loss in this study.
Development of an OTUB1 inhibitor
As OTUB1 potentiates several cancer-promoting pathways, development of an OTUB1 inhibitor may offer substantial clinical benefit for some patients. Discussed below are methods and properties of OTUB1 that could be exploited for the successful development of an inhibitor.
DUBs are considered druggable targets due to their active site cysteines which readily react with electrophiles (D’Arcy et al. 2015). Indeed, several compounds demonstrate broad or narrow specificity towards DUBs, with some entering the clinical setting (D’Arcy et al. 2015, Wang et al. 2016). Although most of these target the active site residue to inhibit catalytic activity, OTUB1 represents a unique target due to its canonical and non-canonical activities. In theory, therapeutic agents could be identified that inhibit its canonical activity, non-canonical activity, or both. Compounds that inhibit the catalytic activity of a broad range of cysteine-dependent DUBs, such as N-ethylmaleimide, have been demonstrated to inhibit OTUB1 catalytic activity (Balakirev et al. 2003). In addition, inhibitors that demonstrate some specificity for OTUB1 have been characterized (Ernst et al. 2013). In this study, ubiquitin point mutants were synthesized and screened for binding to various DUBs. Ubv.B1.1 was identified as an ubiquitin mutant that bound to the distal ubiquitin binding site of OTUB1 and was demonstrated to inhibit OTUB1 catalytic activity and interfere with OTUB1-E2 complex formation. Thus, Ubv.B1.1 likely inhibits both canonical and non-canonical OTUB1 activity. In addition, the small molecule PR-619 is predicted to inhibit OTUB1 DUB activity based on its ability to block OTUB1 binding to an active site probe (Altun et al. 2011). Although promising, PR-619 has broad activity and would likely need modification to narrow its specificity.
High-throughput screening techniques that rely on ubiquitin chain cleavage have identified compounds that inhibit DUB catalytic activity (Arnst et al. 2013). While similar methods may identify OTUB1 canonical inhibitors, identification of non-canonical inhibitors is a more difficult challenge. Inhibition of OTUB1’s non-canonical activity would require prevention of the OTUB1-E2 interaction. Thus, these compounds would be protein-protein interaction inhibitors, targeting one of the critical OTUB1-E2 interaction surfaces mentioned above. Although progress has been made in their identification, such compounds are considered difficult to synthesize (Arkin et al. 2014). An alternative approach could take advantage of the “product inhibition mimicry” model described above which proposes the non-canonical property of OTUB1 relies on the formation of a ternary complex between the E2 conjugated to a donor ubiquitin, a free ubiquitin, and OTUB1. This complex positions the epsilon-amino group of K48 from the donor ubiquitin and the C-terminus of the free ubiquitin over the catalytic cysteine, mimicking the cleavage product of a deubiquitinase reaction. Theoretically, compounds that bind to OTUB1’s catalytic cysteine and sterically interfere with “product inhibition mimicry” complex formation would inhibit the OTUB1-E2 interaction and, thus, the non-canonical activity of OTUB1. An unanswered but interesting question is whether treatment of OTUB1 with a general DUB inhibitor that binds catalytic cysteines will inhibit OTUB1 non-canonical activity. This mechanism would likely involve inhibitor-mediated steric hindrance between the inhibitor and the donor and free ubiquitins mentioned above. Of course, these compounds would also inhibit DUB activity and be considered dual inhibitors. High-throughput screening for non-canonical inhibitors could take advantage of the unique property of the OTUB1 interacting E2 UBC13, which can synthesize polyubiquitin chains in the absence of an ubiquitin ligase (Juang et al. 2012). Upon binding to UCB13, OTUB1 caps the elongating ubiquitin chain and inhibits polyubiquitin chain synthesis (Juang et al. 2012). Indeed, several groups have used this property to screen for OTUB1 mutations that abolish non-canonical activity, albeit with low-throughput techniques (Juang et al. 2012, Sato et al. 2012). With modification, this activity could be used for high-throughput screening of non-canonical inhibitors. Thus, high-throughput screening for canonical, non-canonical, and dual OTUB1 inhibitors is possible.
Conclusion
A majority of the studies described above suggest a protumorigenic role for OTUB1, particularly with regard to tumor progression. However, its effects on tumor growth are mixed. Some studies demonstrate that OTUB1 associates with or potentiates tumor growth (lung, breast, ovarian), while others demonstrate no involvement or a suppression of growth by OTUB1 (osteosarcoma, glioma, colon, gastric). Although assay variability could account for these discrepancies, the effects of OTUB1 are likely tumor type and/or stage dependent. In particular, the mutational and/or activation status of P53 and TGFb are likely significant determinants of OTUB1’s effects. TGFb and P53 exert opposing effects on tumor biology based on mutational status and/or tumor stage. In the early stages of tumorigenesis, P53 and TGFb suppress tumor growth by thwarting oncogenic cellular changes and inhibiting proliferation, respectively (Massague 2008, Sun et al. 2012). However, during tumor progression, TGFb signaling switches from antiproliferative to promigratory (Massague 2008) and P53 mutation results in the expression of an oncogenic form of P53 (Soussi & Wiman 2015). Thus, OTUB1 may be tumor suppressive early in tumorigenesis, where it promotes WT P53 function and suppresses growth through TGFb. However, once WT P53 function is lost and TGFb signaling becomes promigratory, OTUB1 can stimulate the oncogenic pathways described above, while bypassing the tumor suppressive effects of WT P53 and TGFb. Although OTUB1 inhibition in the clinical setting may be difficult, due to OTUB1’s broad tissue distribution and high cellular expression, therapeutic targeting of this DUB may offer substantial benefit to individuals diagnosed with a wide range of tumors.
Acknowledgments
Funding
The authors were supported by NIH predoctoral fellowship CA210467 (K VanderVorst), NIH predoctoral training program GM099608 (A Berg), and by NIH grants CA123541 and CA166412 (KL Carraway).
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the review.
References
- Ali SH, O’Donnell AL, Mohamed S, Mousa S & Dandona P 2004. Overexpression of estrogen receptor-alpha in the endometrial carcinoma cell line Ishikawa: inhibition of growth and angiogenic factors. Gynecol Oncol 95 637–645. [DOI] [PubMed] [Google Scholar]
- Altun M, Kramer HB, Willems LI, McDermott JL, Leach CA, Goldenberg SJ, Kumar KG, Konietzny R, Fischer R, Kogan E, et al. 2011. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem Biol 18 1401–1412. [DOI] [PubMed] [Google Scholar]
- Alvarez-Fernandez M & Medema RH 2013. Novel functions of FoxM1: from molecular mechanisms to cancer therapy. Front Oncol 3 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin MR, Tang Y & Wells JA 2014. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol 21 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnst JL, Davies CW, Raja SM, Das C & Natarajan A 2013. High-throughput compatible fluorescence resonance energy transfer-based assay to identify small molecule inhibitors of AMSH deubiquitinase activity. Anal Biochem 440 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baietti MF, Simicek M, Abbasi Asbagh L, Radaelli E, Lievens S, Crowther J, Steklov M, Aushev VN, Martinez Garcia D, Tavernier J, et al. 2016. OTUB1 triggers lung cancer development by inhibiting RAS monoubiquitination. EMBO Mol Med 8 288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakirev MY, Tcherniuk SO, Jaquinod M & Chroboczek J 2003. Otubains: a new family of cysteine proteases in the ubiquitin pathway. EMBO Rep 4 517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L & Sabatini DM 2014. Regulation of mTORC1 by amino acids. Trends Cell Biol 24 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates RC, DeLeo MJ 3rd & Mercurio AM 2004. The epithelial-mesenchymal transition of colon carcinoma involves expression of IL-8 and CXCR-1-mediated chemotaxis. Exp Cell Res 299 315–324. [DOI] [PubMed] [Google Scholar]
- Borodovsky A, Ovaa H, Kolli N, Gan-Erdene T, Wilkinson KD, Ploegh HL & Kessler BM 2002. Chemistry-based functional proteomics reveals novel members of the deubiquitinating enzyme family. Chem Biol 9 1149–1159. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang YG, Li Y, Sun XX & Dai MS 2017. Otub1 stabilizes MDMX and promotes its proapoptotic function at the mitochondria. Oncotarget 8 11053–11062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ & Urbe S 2013. Deubiquitylases from genes to organism. Physiol Rev 93 1289–1315. [DOI] [PubMed] [Google Scholar]
- Culig Z & Santer FR 2014. Androgen receptor signaling in prostate cancer. Cancer Metastasis Rev 33 413–427. [DOI] [PubMed] [Google Scholar]
- D’Arcy P, Wang X & Linder S 2015. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacology & Therapeutics 147 32–54. [DOI] [PubMed] [Google Scholar]
- Du C, Wu H & Leng RP 2016. UBE4B targets phosphorylated p53 at serines 15 and 392 for degradation. Oncotarget 7 2823–2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann MJ, Iphofer A, Akutsu M, Altun M, di Gleria K, Kramer HB, Fiebiger E, Dhe-Paganon S & Kessler BM 2009. Structural basis and specificity of human otubain 1-mediated deubiquitination. Biochem J 418 379–390. [DOI] [PubMed] [Google Scholar]
- Edelmann MJ, Kramer HB, Altun M & Kessler BM 2010. Post-translational modification of the deubiquitinating enzyme otubain 1 modulates active RhoA levels and susceptibility to Yersinia invasion. FEBS J 277 2515–2530. [DOI] [PubMed] [Google Scholar]
- Ernst A, Avvakumov G, Tong J, Fan Y, Zhao Y, Alberts P, Persaud A, Walker JR, Neculai AM, Neculai D, et al. 2013. A strategy for modulation of enzymes in the ubiquitin system. Science 339 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. 2014. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. 2014. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea G, de Toledo M, Anguille C & Roux P 2007. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol 178 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo LH, Ko J & Donoghue DJ 2017. The importance of regulatory ubiquitination in cancer and metastasis. Cell Cycle 16 634–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, et al. 2011. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell 44 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Alarcon C, Sapkota G, Rahman S, Chen PY, Goerner N, Macias MJ, Erdjument-Bromage H, Tempst P & Massague J 2009. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol Cell 36 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharov T, Niessen K, de Almagro MC, Izrael-Tomasevic A, Fedorova AV, Varfolomeev E, Arnott D, Deshayes K, Kirkpatrick DS & Vucic D 2013. OTUB1 modulates c-IAP1 stability to regulate signalling pathways. EMBO J 32 1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodenberger ML & Jenkins RB 2012. Genetics of adult glioma. Cancer Genet 205 613–621. [DOI] [PubMed] [Google Scholar]
- Hao M, Weng X, Wang Y, Sun X, Yan T, Li Y, Hou L, Meng X & Wang J 2018. Targeting CXCR7 improves the efficacy of breast cancer patients with tamoxifen therapy. Biochem Pharmacol 147 128–140. [DOI] [PubMed] [Google Scholar]
- Herhaus L, Al-Salihi M, Macartney T, Weidlich S & Sapkota GP 2013. OTUB1 enhances TGFbeta signalling by inhibiting the ubiquitylation and degradation of active SMAD2/3. Nat Commun 4 2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herhaus L, Perez-Oliva AB, Cozza G, Gourlay R, Weidlich S, Campbell DG, Pinna LA & Sapkota GP 2015. Casein kinase 2 (CK2) phosphorylates the deubiquitylase OTUB1 at Ser16 to trigger its nuclear localization. Sci Signal 8 ra35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu WC & Fuchs SY 2010. Ubiquitination-dependent regulation of signaling receptors in cancer. Genes Cancer 1 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias-Gato D, Chuan YC, Jiang N, Svensson C, Bao J, Paul I, Egevad L, Kessler BM, Wikstrom P, Niu Y, et al. 2015. OTUB1 de-ubiquitinating enzyme promotes prostate cancer cell invasion in vitro and tumorigenesis in vivo. Mol Cancer 14 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang YC, Landry MC, Sanches M, Vittal V, Leung CC, Ceccarelli DF, Mateo AR, Pruneda JN, Mao DY, Szilard RK, et al. 2012. OTUB1 co-opts Lys48-linked ubiquitin recognition to suppress E2 enzyme function. Mol Cell 45 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunarathna U, Kongsema M, Zona S, Gong C, Cabrera E, Gomes AR, Man EP, Khongkow P, Tsang JW, Khoo US, et al. 2016. OTUB1 inhibits the ubiquitination and degradation of FOXM1 in breast cancer and epirubicin resistance. Oncogene 35 1433–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D & Rape M 2012. The ubiquitin code. Annu Rev Biochem 81 203–229. [DOI] [PubMed] [Google Scholar]
- Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J & Gu W 2002. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416 648–653. [DOI] [PubMed] [Google Scholar]
- Li Y, Sun XX, Elferich J, Shinde U, David LL & Dai MS 2014. Monoubiquitination is critical for ovarian tumor domain-containing ubiquitin aldehyde binding protein 1 (Otub1) to suppress UbcH5 enzyme and stabilize p53 protein. J Biol Chem 289 5097–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RL & Zhao LJ 2015. Mechanistic basis and clinical relevance of the role of transforming growth factor-beta in cancer. Cancer Biol Med 12 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jiang WN, Wang JG & Chen H 2014. Colon cancer bears overexpression of OTUB1. Pathol Res Pract 210 770–773. [DOI] [PubMed] [Google Scholar]
- Liu B, Li J, Zheng M, Ge J, Li J & Yu P 2017. MiR-542–3p exerts tumor suppressive functions in non-small cell lung cancer cells by upregulating FTSJ2. Life Sci 188 87–95. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Aravind L & Koonin EV 2000. A novel superfamily of predicted cysteine proteases from eukaryotes, viruses and Chlamydia pneumoniae. Trends Biochem Sci 25 50–52. [DOI] [PubMed] [Google Scholar]
- Markson G, Kiel C, Hyde R, Brown S, Charalabous P, Bremm A, Semple J, Woodsmith J, Duley S, Salehi-Ashtiani K, et al. 2009. Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res 19 1905–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J 2008. TGFbeta in Cancer. Cell 134 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick F 2015. KRAS as a Therapeutic Target. Clinical Cancer Research 21 1797–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakada S, Tai I, Panier S, Al-Hakim A, Iemura S, Juang YC, O’Donnell L, Kumakubo A, Munro M, Sicheri F, et al. 2010. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 466 941–946. [DOI] [PubMed] [Google Scholar]
- Neufeld TP 2012. Autophagy and cell growth--the yin and yang of nutrient responses. J Cell Sci 125 2359–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Q, Chen J, Li X, Xu X, Zhang N, Zhou A, Zhou B, Lu Q & Chen Z 2017. Expression of OTUB1 in hepatocellular carcinoma and its effects on HCC cell migration and invasion. Acta Biochim Biophys Sin (Shanghai) 49 680–688. [DOI] [PubMed] [Google Scholar]
- Noroxe DS, Poulsen HS & Lassen U 2016. Hallmarks of glioblastoma: a systematic review. ESMO Open 1 e000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba R, Alsayed A, Zacny JP & Dudek AZ 2016. The Role of Forkhead Box Protein M1 in Breast Cancer Progression and Resistance to Therapy. Int J Breast Cancer 2016 9768183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Yamagata A, Goto-Ito S, Kubota K, Miyamoto R, Nakada S & Fukai S 2012. Molecular basis of Lys-63-linked polyubiquitination inhibition by the interaction between human deubiquitinating enzyme OTUB1 and ubiquitin-conjugating enzyme UBC13. J Biol Chem 287 25860–25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz CC, Cavadas MAS, Tambuwala MM, Hams E, Rodriguez J, von Kriegsheim A, Cotter P, Bruning U, Fallon PG, Cheong A, et al. 2013. Regulation of IL-1 beta-induced NF-kappa B by hydroxylases links key hypoxic and inflammatory signaling pathways. Proceedings of the National Academy of Sciences of the United States of America 110 18490–18495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz CC, Rodriguez J, Pickel C, Burr S, Fabrizio JA, Nolan KA, Spielmann P, Cavadas MA, Crifo B, Halligan DN, et al. 2016. FIH Regulates Cellular Metabolism through Hydroxylation of the Deubiquitinase OTUB1. PLoS Biol 14 e1002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W & Selbach M 2011. Global quantification of mammalian gene expression control. Nature 473 337–342. [DOI] [PubMed] [Google Scholar]
- Shen X, Xue Y, Si Y, Wang Q, Wang Z, Yuan J & Zhang X 2015. The unfolded protein response potentiates epithelial-to-mesenchymal transition (EMT) of gastric cancer cells under severe hypoxic conditions. Med Oncol 32 447. [DOI] [PubMed] [Google Scholar]
- Soussi T & Wiman KG 2015. TP53: an oncogene in disguise. Cell Death Differ 22 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP & Harper JW 2009. Defining the human deubiquitinating enzyme interaction landscape. Cell 138 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanisic V, Malovannaya A, Qin J, Lonard DM & O’Malley BW 2009. OTU Domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) deubiquitinates estrogen receptor (ER) alpha and affects ERalpha transcriptional activity. J Biol Chem 284 16135–16145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, Taichman RS, Pienta KJ & Wang J 2010. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev 29 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XX, Challagundla KB & Dai MS 2012. Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO J 31 576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam AB, Mercado EL, Hoffmann A & Niwa M 2012. ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS One 7 e45078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade M, Wang YV & Wahl GM 2010. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol 20 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mazurkiewicz M, Hillert EK, Olofsson MH, Pierrou S, Hillertz P, Gullbo J, Selvaraju K, Paulus A, Akhtar S, et al. 2016. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells (vol 6, 26979, 2016). Scientific Reports 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Shi J, Chai K, Ying X & Zhou BP 2013. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets 13 963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhou X, Xu M, Weng W, Zhang Q, Yang Y, Wei P & Du X 2016. OTUB1-catalyzed deubiquitination of FOXM1 facilitates tumor progression and predicts a poor prognosis in ovarian cancer. Oncotarget 7 36681–36697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng W, Zhang Q, Xu M, Wu Y, Zhang M, Shen C, Chen X, Wang Y & Sheng W 2016. OTUB1 promotes tumor invasion and predicts a poor prognosis in gastric adenocarcinoma. Am J Transl Res 8 2234–2244. [PMC free article] [PubMed] [Google Scholar]
- White E 2015. The role for autophagy in cancer. J Clin Invest 125 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener R, DiBello AT, Lombardi PM, Guzzo CM, Zhang X, Matunis MJ & Wolberger C 2013. E2 ubiquitin-conjugating enzymes regulate the deubiquitinating activity of OTUB1. Nat Struct Mol Biol 20 1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener R, Zhang X, Wang T & Wolberger C 2012. The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 483 618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HX, Wang GM, Lu X & Zhang L 2017. miR-542–3p targets sphingosine-1-phosphate receptor 1 and regulates cell proliferation and invasion of breast cancer cells. Eur Rev Med Pharmacol Sci 21 108–114. [PubMed] [Google Scholar]
- Xu L, Li J, Bao Z, Xu P, Chang H, Wu J, Bei Y, Xia L, Wu P, Yan K, et al. 2017. Silencing of OTUB1 inhibits migration of human glioma cells in vitro. Neuropathology 37 217–226. [DOI] [PubMed] [Google Scholar]
- Xu LZ, Lubkov V, Taylor LJ & Bar-Sagi D 2010. Feedback Regulation of Ras Signaling by Rabex-5-Mediated Ubiquitination. Current Biology 20 1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Yuan P, Yuan H, Wang Z, Run Z, Chen G, Zhao P & Xu B 2017. miR-542–3p inhibits colorectal cancer cell proliferation, migration and invasion by targeting OTUB1. Am J Cancer Res 7 159–172. [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Li H, Xue B, Jiang X, Huang K, Ge J, Zhang H & Chen B 2014. SDF-1/CXCR7 axis enhances ovarian cancer cell invasion by MMP-9 expression through p38 MAPK pathway. DNA Cell Biol 33 543–549. [DOI] [PubMed] [Google Scholar]
- Yue W, Wang JP, Li Y, Fan P, Liu G, Zhang N, Conaway M, Wang H, Korach KS, Bocchinfuso W, et al. 2010. Effects of estrogen on breast cancer development: Role of estrogen receptor independent mechanisms. Int J Cancer 127 1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang X, Yu Y, Deng L, Chen L, Peng X, Jiao C, Gao G, Tan X, Pan W, et al. 2018. OTUB1 protein suppresses mTOR complex 1 (mTORC1) activity by deubiquitinating the mTORC1 inhibitor DEPTOR. J Biol Chem 293 4883–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Zhou B, Ann DK, Minoo P, Liu Y, Banfalvi A, Krishnaveni MS, Dubourd M, Demaio L, Willis BC, et al. 2011. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am J Respir Cell Mol Biol 45 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Liu Y, Zhu R, Ding F, Cao X, Lin D & Liu Z 2018. OTUB1 promotes esophageal squamous cell carcinoma metastasis through modulating Snail stability. Oncogene [DOI] [PubMed]
- Zhou Y, Wu J, Fu X, Du W, Zhou L, Meng X, Yu H, Lin J, Ye W, Liu J, et al. 2014. OTUB1 promotes metastasis and serves as a marker of poor prognosis in colorectal cancer. Mol Cancer 13 258. [DOI] [PMC free article] [PubMed] [Google Scholar]