

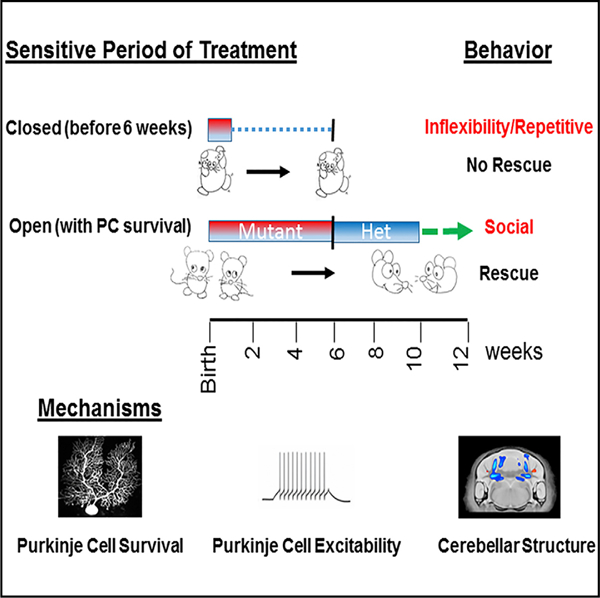
SUMMARY
Despite a prevalence exceeding 1%, mechanisms underlying autism spectrum disorders (ASDs) are poorly understood, and targeted therapies and guiding parameters are urgently needed. We recently demonstrated that cerebellar dysfunction is sufficient to generate autistic-like behaviors in a mouse model of tuberous sclerosis complex (TSC). Here, using the mechanistic target of rapamycin (mTOR)-specific inhibitor rapamycin, we define distinct sensitive periods for treatment of autistic-like behaviors with sensitive periods extending into adulthood for social behaviors. We identify cellular and electro-physiological parameters that may contribute to behavioral rescue, with rescue of Purkinje cell survival and excitability corresponding to social behavioral rescue. In addition, using anatomic and diffusion-based MRI, we identify structural changes in cerebellar domains implicated in ASD that correlate with sensitive periods of specific autism-like behaviors. These findings thus not only define treatment parameters into adulthood, but also support a mechanistic basis for the targeted rescue of autism-related behaviors.
In Brief
A mechanistic understanding of and establishment of time windows for effective therapy (sensitive periods) for autism-related behaviors remain unknown. Tsai et al. delineate specific time windows for treatment of specific autism-relevant behaviors and evaluate underlying cellular, electrophysiological, and anatomic mechanisms for these sensitive periods.
Graphical Abstract
INTRODUCTION
Autism spectrum disorder (ASD) is a prevalent neurodevelopmental disorder characterized by social impairment, restrictive interests, and cognitive inflexibility. Recently, the cerebellum has been implicated in the pathogenesis of these disorders, with cerebellar pathology the most consistently identified finding in postmortem studies of individuals with ASD (Bauman and Kemper, 1985; Ritvo et al., 1986; Skefos et al., 2014; Whitney et al., 2008). In these studies, significant reductions in Purkinje cell (PC) number have been consistently identified (reviewed in Fatemi et al., 2012). Moreover, isolated cerebellar injury during prematurity results in a >30-fold increase in the risk for ASD (Limperopoulos et al., 2007). However, the precise contribution of cerebellar dysfunction to ASD pathogenesis remains unclear.
Tuberous sclerosis complex (TSC) is a neurogenetic disorder with high rates of neurobehavioral dysfunction, including high rates of ASD, with 25%–60% of patients with TSC carrying ASD diagnoses (Capal et al., 2017; Jeste et al., 2008; McDonald et al., 2017). TSC results from mutation in either TSC1 or TSC2, which together act to negatively regulate mTOR signaling (reviewed in Lipton and Sahin, 2014, and Tsai and Sahin, 2011). Cerebellar dysfunction and lesions also both associate with ASD in TSC (Eluvathingal et al., 2006; Weber et al., 2000), and evidence of reduced PC density in the brains of individuals with TSC has been identified (Reith et al., 2011). For these reasons, we developed a mutant mouse model wherein Tsc1 was deleted in cerebellar PCs. In this model, we demonstrated that cerebellar dysfunction is sufficient to generate autistic-like behavior and that PC-specific loss of Tsc1 was sufficient to generate pathology consistent with ASD clinical pathology (Tsai et al., 2012). Moreover, PC-Tsc1 mice demonstrated electrophysiological dysfunction, with reductions in spontaneous PC firing rates and impaired PC excitability, findings consistent with previous studies demonstrating cerebellar function in ASD (Asano et al., 2001; Ryu et al., 1999). Similar pathologic findings and behavioral deficits have been demonstrated in a cerebellar model in which Tsc2 is lost in PCs (Reith et al., 2011, 2013), and ASD behaviors have subsequently been observed in rodent models with cerebellar-specific loss of the ASD-associated genes, Shank2 (Peter et al., 2016) or PTEN (Cupolillo et al., 2016). We further demonstrated that early treatment with the mechanistic target of rapamycin (mTOR)-specific inhibitor rapamycin, initiated at postnatal day (P) 7, was sufficient to prevent the development of behavioral, anatomic, and physiologic abnormalities in PC Tsc1-mutant mice (Tsai et al., 2012).
Although early treatment has been demonstrated to prevent the development of ASD-related pathology, whether treatment initiated at later stages can also rescue ASD behaviors and whether sensitive periods of therapeutic benefit exist remain undefined. A sensitive period for therapy defines a time window during which therapy is effective for the treatment of a specific phenotype, in this model, autistic-like behaviors. Existence of these periods has been supported by superior efficacy of early therapeutic interventions (LeBlanc and Fagiolini, 2011; Nelson et al., 2007) compared with children receiving delayed access to care. We hypothesized that a sensitive period for autistic behavior exists and that cellular and circuit-based mechanisms underlie continued access to these periods while also mediating closure of these periods.
In this study, we used rapamycin therapy, combined with a multimodal approach, to define the sensitive periods of treatment of ASD-related behaviors and their underlying structural and molecular mechanisms. We found that treatment initiated in young adulthood (at 6 weeks) rescued social behaviors but not repetitive behaviors or behavioral inflexibility. Moreover, we determined that rescue of social behaviors correlates with rescue of specific MRI-based structural changes, cellular pathology, and PC excitability, while motor learning rescue appears to be independent of PC survival or rescue of cellular excitability. Using this integrated approach, we provide mechanistic insight into the underlying basis of ASD while also delineating mechanistic and temporal parameters for treatment of these disorders.
RESULTS
Rapamycin Therapy Initiated in Young Adulthood Rescues Social and Motor Behavior
To characterize the sensitive periods of treatment efficacy, we initiated rapamycin treatment starting at 6 weeks of age, equivalent to young adulthood in the mouse. At this time, behavioral, anatomic, and electrophysiological abnormalities are already present in untreated mutant mice (Figure 1A) (Tsai et al., 2012). We continued treatment for 4 weeks prior to the commencement of evaluation of behavior, anatomy, or physiology consistent with the delay in evaluation after our previously performed, P7-onset rapamycin therapy (Figure 1A). We then performed behavioral evaluation in the setting of continued rapamycin treatment (Figure S1). We examined social function in the three-chambered apparatus (Figures 1B–1D; Table S1) (Yang et al., 2011) and identified social impairment in vehicle-treated mutants (L7Cre;Tsc1flox/flox) in tests of social approach (Figure 1C). How-ever, unlike mice treated with vehicle, mutants treated with rapamycin initiated at 6 weeks showed a significant social preference, comparable with that of their control littermates (Figure 1C), suggesting a behavioral rescue of social behaviors with rapamycin treatment initiated even during adulthood. Similarly, in a social novelty paradigm (Figures 1B and 1D), rapamycin treatment initiated at 6 weeks rescued the deficits in social novelty seen in vehicle-treated mutants (Figure 1D). Because rodent social behavior is guided largely by olfactory cues, we assessed olfactory function and found intact responses in all genotypes to nonsocial olfactory cues (Figure 1E). However, when animals were exposed to social olfactory cues, vehicle-treated mutant mice demonstrated reduced responses, whereas rapamycin-treated mutants responded to social olfactory stimuli similarly to control littermates (Figure 1E), consistent with rescue of social impairment with rapamycin therapy.
Figure 1. Sensitive Period Remains Open at 6 Weeks for Treatment of Social and Motor Behaviors.

(A) Schema for treatment paradigm.
(B) Social testing in three-chambered apparatus.
(C–F) Rapamycin initiated at 6 weeks rescues (C and D) social behaviors in three chambered apparatus, (E) social olfaction, and (F) motor function on rotarod.
(G and H) No rescue is achieved for (G) repetitive grooming nor (H) reversal learning on water T maze.
Two-way ANOVA, Bonferroni post hoc analysis. *p < 0.05, **p < 0.01, and ***p < 0.001. FA, familiar animal; NA, novel animal; NO, novel object; Ns, not significant. Rapa, rapamycin; RevDay, reversal day; VEH, vehicle. Data are reported as mean ± SEM.
Children with ASD have significantly higher rates of motor dysfunction with dyspraxia, apraxia, hypotonia, and oculomotor abnormalities (Fournier et al., 2010; Ming et al., 2007; Mosconi et al., 2015). Previously, we had demonstrated that Tsc1 PC-mutant mice have impaired motor learning on the rotarod that is prevented with early (P7) rapamycin therapy (Tsai et al., 2012). With treatment initiation at 6 weeks, we found that vehicle-treated mutant mice demonstrated impaired rotarod performance. However, rapamycin therapy rescued these motor deficits (Figure 1F). We then evaluated whether social deficits might result from locomotor dysfunction. We evaluated locomotor performance in the open field and in the elevated plus maze and found that vehicle-treated mutants had increased exploration in the open field compared with controls. In mutants, exploration did not change with rapamycin therapy; however, in controls, rapamycin treatment resulted in increased activity (Figure S2A). Thus, differences in exploratory behaviors did not correlate with changes in social behaviors with rapamycin. Combined with no significant differences in exploratory behavior in elevated plus maze testing (Figure S2B), these data suggest that motor impairments were not responsible for observed social deficits. We further examined anxiety behaviors in the open field and elevated plus maze and found no differences in anxiety between vehicle-treated controls and mutants that would explain the observed social deficits (Figures S2C and S2D).
Repetitive Behaviors and Behavioral Flexibility Are Not Ameliorated with Adult-Onset Rapamycin Therapy
In addition to social impairments, ASD is characterized by deficits in repetitive behaviors and cognitive inflexibility. We examined repetitive behaviors and found that vehicle-treated mutants displayed increased repetitive grooming (Figure 1G). Unlike with social behaviors, upon rapamycin therapy initiated at 6 weeks, we observed no rescue of repetitive grooming behaviors (Figure 1G). Because individuals with autism also demonstrate behavioral inflexibility, we modeled this ‘‘insistence on sameness’’ and behavioral inflexibility with a reversal learning paradigm using the water T maze. All treatment groups displayed intact acquisition learning of the submerged escape platform during the 3 days of training (Figure 1H). Upon reversal of the platform location, vehicle-treated mutants displayed significant impairment in learning the location of the new platform location compared with littermate controls (Figure 1H). Unlike treatment initiated at 7 days of age, when rescue of reversal learning is achieved (Tsai et al., 2012), rapamycin therapy initiated at 6 weeks of age was unable to significantly ameliorate these reversal learning deficits (Figure 1H). Thus, in contrast with social and motor behaviors, repetitive and restrictive behaviors—two aspects of the second core deficit in individuals with ASD— were not rescued with rapamycin therapy initiated at 6 weeks of age. Taken together, these data suggest the presence of divergent sensitive periods for the treatment of abnormal autism-relevant behaviors.
Cellular Mechanisms Underlying Behavioral Rescue
To better understand the underlying mechanisms behind phenotypic rescue, we first examined anatomic and physiological function in the treatment cohorts at a cellular level. Previously, we had demonstrated that PC Tsc1-mutant mice display significant PC loss starting between 6 and 8 weeks of age, at the approximate onset of our present rapamycin treatment paradigm (Tsai et al., 2012). To examine whether rapamycin therapy at this juncture could rescue cell viability deficits in these mice, we quantified PC numbers in mutant and control cerebellum at time mirroring the aforementioned behavioral evaluations. PC loss observed in vehicle-treated mutants was rescued with rapamycin therapy initiated at 6 weeks (Figure 2A). To ensure that mTOR activity was altered with loss of Tsc1 and that rapamycin therapy abrogated these effects, cerebellar sections were also stained with anti-pS6 antibody. As expected, pS6 was increased in PCs in vehicle-treated mutants, consistent with loss of Tsc1, a negative regulator of mTOR, while rapamycin abrogated this effect (Figure 2A). Of note, PC numbers in rapamycin-treated controls were significantly reduced (p = 0.01) compared with vehicle-treated controls.
Figure 2. Treatment Initiated at 6 Weeks Rescues PC Survival and Impaired Spontaneous Firing and Excitability in PC Tsc1 Mice.
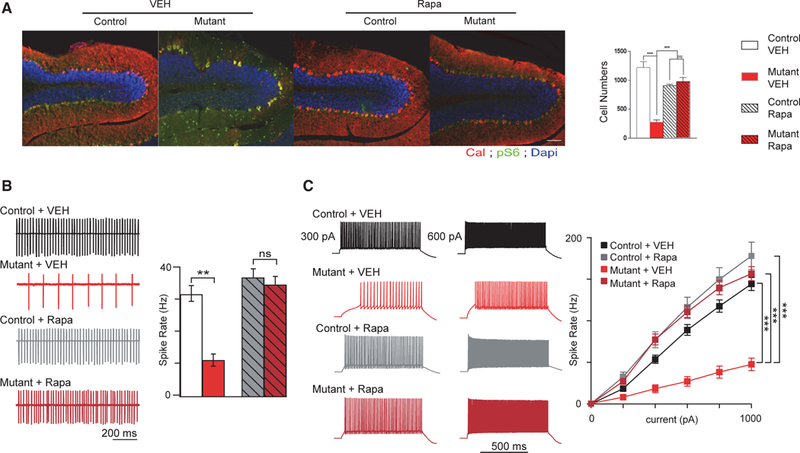
Recordings were performed at 11 weeks of age (5 weeks after treatment initiation at 6 weeks of age).
(A) PC loss is rescued with rapamycin therapy (quantification of PCs on left). Shown are midline vermis sagittal sections stained with calbindin (to identify PCs) and phosphoS6 (to evaluate mTOR activity) antibodies.
(B and C) Rapamycin initiated at 6 weeks rescues (B) intrinsic firing properties and (C) excitability deficits in mutant mice.
Two-way ANOVA, Bonferroni post hoc analysis. *p < 0.05, **p < 0.01, and ***p < 0.001. Ns, not significant; Rapa, rapamycin; VEH, vehicle. Data are reported as mean ± SEM.
We had also previously identified deficits in spontaneous PC firing rates in addition to impairments in PC excitability in mutant PCs (Tsai et al., 2012). We thus examined whether electrophysiological mechanisms might underlie differential behavioral sensitive periods. We first tested intrinsic PC firing rates using extracellular recordings at a time mirroring behavioral evaluations. Similar to untreated mutants, vehicle-treated mutant PCs demonstrated reduced frequency of spontaneous simple spike firing (Figure 2B). With rapamycin treatment at 6 weeks, we observed rescue of this reduction in spontaneous firing frequency (Figure 2B).
We then examined PC excitability in whole-cell recordings again at time mirroring the aforementioned behavioral evaluations. Again, similar to untreated PCs (Tsai et al., 2012), vehicle-treated mutant PCs demonstrated decreased neuronal excitability with significantly reduced firing compared with control PCs in response to comparable current input (Figure 2C). However, with rapamycin treatment, no significant differences could be identified in excitability between control and mutant PCs (Figure 2C). Together, these findings are consistent with social and motor behavioral rescue; however, the lack of behavioral rescue for repetitive and inflexibility behaviors suggests an alternative mechanism for the closed sensitive period for these behaviors.
Structural Changes in Mutant Cerebellum Rescued with Rapamycin Therapy
Whereas cellular and physiologic parameters appeared to be rescued coincident with the open sensitive period for rescue of motor and social impairments, mechanisms underlying the closed sensitive period for rescue of repetitive and inflexible behaviors remained unknown. We hypothesized that changes in specific cerebellar domains could mediate these differences in sensitive periods.
The circuitry underlying cerebellar regulation of these complex behaviors has been hypothesized with multiple connections to non-motor regions of the cerebral cortex identified (Buckner, 2013; Buckner et al., 2011; Voogd, 2014). Recent studies have also demonstrated connections between deep cerebellar nuclei and cerebral cortex that are abnormal in monogenetic models of autism (Rogers et al., 2013). To investigate whether alterations in cerebellar structures and/or cerebellar circuits might contribute to these distinct sensitive periods, we performed a MRI-based structural analysis on control and mutant mice with vehicle or rapamycin treatment initiated at 6 weeks. We performed a whole-brain regional analysis using MRI analysis of these four cohorts, examining volumetric changes and changes in cerebellar circuitry by deformation-based morphometry and diffusion tensor imaging (DTI).
Initial examination focused on the vehicle-treated animals to visualize the effect of PC Tsc1 loss. No significant differences were found in absolute volume (measured in cubic millimeters) throughout the brain at a false discovery rate (FDR) threshold of <10%. Relative volume differences (volume measured as a percentage of total brain volume) did highlight multiple regions within the cerebellum that were significantly different at an FDR of <10%, including white matter (WM) derived from regions implicated in clinical and basic autism studies, CrusI and CrusII (Figures 3A–3C; Table S2) (D’Mello et al., 2015; Stoodley, 2014; Stoodley et al., 2017). In addition, changes were observed in the cerebellar output nuclei (lateral, interposed, and medial; Figures 3A and 3D–3F) as well as several additional cerebellar regions implicated in autism, including anterior lobules 4 and 5 (Figures 3G and 3H) (D’Mello and Stoodley, 2015; Stoodley, 2016). Outside of the cerebellum and output nuclei, no volumetric changes were observed (Figure S3; Table S2).
Figure 3. Brain Alterations in PC Tsc1-Mutant Mice Are Partially Rescued with Rapamycin Therapy.
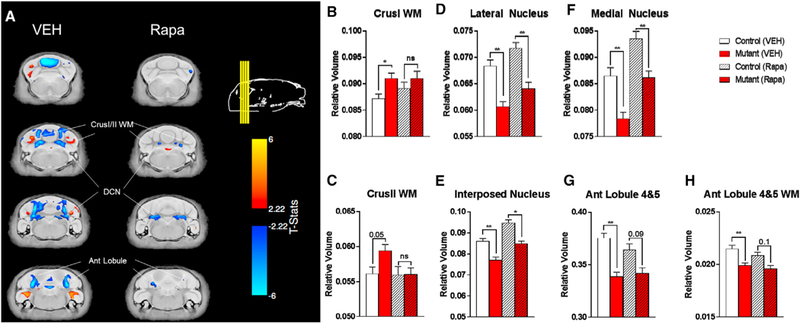
(A) Voxelwise differences in relative volume between control and mutant mice are partially rescued with rapamycin therapy.
(B–F) Bar plots summarizing data showing partial rescue of (B) CrusI white matter (WM), (C) CrusII WM, (D) lateral CN, (E) interposed CN, and (F) medial CN.
(G and H) Bar plots also summarize data demonstrating areas altered in PC Tsc1-mutants that are not rescued with rapamycin treatment, including anterior (Ant) lobules 4 and 5 (G) gray and (H) WM.
*FDR < 0.05; **FDR < 0.01; and ns, FDR > 0.1. CON, control; MUT, PC Tsc1 mutant; Rapa, rapamycin; VEH, vehicle. Data are reported as mean ± SEM.
To examine whether changes in specific areas might mediate specific behavioral rescue, we then examined whether rapamycin might rescue the aforementioned, identified volumetric changes. Rapamycin treatment initiated at 6 weeks of life did have significant effects on the brain. In all mouse cohorts, rapamycin therapy resulted in a 10% decrease in total brain size and alterations in multiple extra-cerebellar brain structures (Figure S3; Table S2). Rapamycin therapy, however, also resulted in at least a partial rescue of numerous volumetric changes identified in PC Tsc1 mutants. In CrusI/II WM, no significant differences in volume were observed after rapamycin therapy with a significant rescue of volumetric change noted in CrusII WM with rapamycin therapy (Figures 3A–3C; Table S2). In the lateral, medial, and interposed deep cerebellar nuclei, continued significant differences in volume remained even with rapamycin therapy, although a significant change was observed upon rapamycin therapy (FDR < 10%) (Figures 3A and 3D–3F; Table S2). In contrast, in PC Tsc1-mutant mice, after rapamycin therapy, anterior lobule 4 and 5 volumes continued to differ between controls and mutants, without significant changes in volume noted in mutants after rapamycin therapy (Figures 3G and 3H; Table S2). As CrusI and CrusII have been implicated in social behaviors in ASD, while anterior lobule volumetric abnormalities correlate with repetitive and restricted behaviors in ASD, these data raise the possibility that anatomic changes may contribute to the observed differential sensitive periods (D’Mello et al., 2015; Stoodley et al., 2017).
To identify whether disruptions in WM integrity might also be identified in PC Tsc1 mutants, we additionally performed DTI. We evaluated both control and PC Tsc1 mutants treated with vehicle and rapamycin. In mutant mice, although we observed trends toward reduced fractional anisotropy and elevated diffusivity measures in the cerebellar nuclei (Figure S4; Table S3) and in the cerebellar WM compared with control littermates, no differences in these measures achieved statistical significance (FDR < 0.1) from our whole-brain analyses. Rapamycin treatment eliminated these trends in all diffusion measures (Figure S4; Table S3).
Rapamycin Therapy Initiated at 10 Weeks Does Not Rescue Social Behaviors in Mutant Mice
Because the sensitive period for treatment appeared to remain open at 6 weeks for therapeutic amelioration of social impairments and motor deficits seen in PC Tsc1-mutant mice, we asked whether it remains open even further into adulthood. We examined a cohort of animals in which we delayed the initiation of rapamycin therapy until 10 weeks of age, a period corresponding to later adulthood in the mouse. Four weeks after the initiation of treatment, we initiated behavioral evaluation in conjunction with ongoing rapamycin or vehicle treatment. We first evaluated social interaction, and consistent with the 6 week treatment paradigm, vehicle-treated PC Tsc1-mutant mice demonstrated impairments in social interaction in both social approach and social novelty assays. However, unlike treatments initiated at P7 or at 6 weeks, rapamycin therapy initiated at 10 weeks of age was unable to rescue social deficits in mutant mice in both social approach and novelty paradigms (Figures 4A, S5A, and S5B; Table S4). Similar to mutant mice with therapy initiated at 6 weeks, rapamycin therapy initiated at 10 weeks was also unable to rescue repetitive grooming or behavioral inflexibility in the water T maze (Figures 4B and 4C). Similar to findings with the three-chambered apparatus, delayed rapamycin therapy starting at 10 weeks was unable to rescue social olfactory deficits in mutant mice (Figure 4D). Because these animals are substantially older and more severely affected, vehicle-treated mutants displayed significant motor deficits on the rotarod (Figure 4E) in addition to locomotor deficits in the open field and elevated plus maze (Figures S5C and S5D). Despite these deficits, late rapamycin therapy initiated at 10 weeks partially rescued motor learning of mutant animals in rotarod testing and rescued completely locomotor performance in the open field and elevated plus maze (Figures 4E, S5C, and S5D). Similar to the 6 week treatment paradigm, rapamycin treatment also increased time spent in the center of the open field but not in open arms of the elevated plus maze (Figures S5E and S5F).
Figure 4. At 10 Weeks, Sensitive Period Is Open for Social Behaviors in the Presence of PC Survival.

(A–C) Rapamycin initiated at 10 weeks is able to rescue social behaviors in (A) social approach, (B) social novelty, and (C) social olfaction behavioral paradigms only in heterozygous (het), not homozygous (mutant), PC Tsc1 mutants.
(D and E) No rescue is seen for either genotype in (D) reversal learning in water T maze or (E) with repetitive grooming.
(F) Despite these findings, persistent rescue of motor learning on the rotarod was observed in homozygous PC Tsc1-mutant mice.
(G) PC viability is not rescued in homozygous PC Tsc1 mutants with rapamycin initiated at 10 weeks of age.
Two-way ANOVA, Bonferroni post hoc analysis. *p < 0.05, **p < 0.01, and ***p < 0.001. FA: familiar animal; NA, novel animal; NO, novel object; Ns, not significant; Rapa, rapamycin; Rev, reversal; VEH, vehicle. Data are reported as mean ± SEM.
PC Loss Is Not Rescued with 10 Week Rapamycin Treatment
To attain a better understanding of the mechanisms underlying the behavioral impairments and the regulation of the sensitive period, we again examined PC survival. We assessed PC numbers and found that PC survival in mutant mice is not rescued with treatment initiated at 10 weeks (Figures 4F and S6), consistent with loss of PCs starting at about 6 weeks of life in untreated mutants (Tsai et al., 2012). As in 6 week cohort, pS6 staining was elevated in vehicle-treated mutants, an elevation that was abrogated with rapamycin treatment (Figure S6). We then asked whether electrophysiological parameters could be modified by this late therapy. We first examined intrinsic firing rates using extracellular recordings. We found that unlike with PC loss, decreased spontaneous simple spike firing rates were rescued to control levels even with delayed rapamycin therapy (Figure 5A). However, similar to PC loss, PC excitability continued to be reduced in rapamycin-treated mutants, at comparable levels with vehicle-treated mutants (Figure 5B). Taken together, rapamycin treatment initiated at 10 weeks was able to rescue PC spontaneous activity deficits in mutant mice, whereas PC cell loss and deficits in PC excitability remained despite rapamycin therapy initiated at this time.
Figure 5. Electrophysiology Phenotypes with 10 Week Treatment Paradigm.

Recordings were performed at 15 weeks of age (5 weeks after treatment initiation at 10 weeks of age).
(A and B) Intrinsic PC firing properties are rescued in mutants with rapamycin therapy (A), while PC excitability is not rescued in mutant mice even with rapamycin therapy (B).
Two-way ANOVA, Bonferroni post hoc analysis. **p < 0.01 and ***p < 0.001. Ns, not significant; Rapa, rapamycin; VEH, vehicle. Data are reported as mean ± SEM.
Social Behaviors Are Rescued with 10 Week Rapamycin Treatment in Heterozygous Tsc Mutants
We have previously demonstrated that heterozygous cerebellar Tsc1-mutant mice display social impairment and repetitive behaviors in the absence of PC loss (Tsai et al., 2012). To examine whether the closure of the sensitive period for rescue of social behaviors in homozygous mutants is related to PC demise, we treated heterozygous mutants (hets) with vehicle or rapamycin initiated at 10 weeks of age. After 4 weeks of treatment, we performed behavioral analysis in setting of concurrent rapamycin treatment. Consistent with our previous studies, vehicle-treated hets showed social impairments (Figure 4A). However, unlike what we observed with homozygous mutant mice, rapamycin treatment initiated at 10 weeks of age was sufficient to rescue social impairments in het mutant mice in social approach (Figure 4A), social novelty (Figures 4B and S5B), and social olfactory (Figure 4C) paradigms. To evaluate whether repetitive or inflexibility behaviors would be modifiable by rapamycin therapy in this het model, we examined grooming and reversal learning performance in the water T maze. Vehicle-treated hets displayed impaired reversal learning (Figure 4D) and repetitive grooming behaviors (Figure 4E). However, similar to homozygous mutants, rapamycin therapy was insufficient to rescue these behaviors suggesting a closure of this sensitive period for these behaviors. Rotarod performance in het mice remained unchanged in these het mice consistent with the lack of ataxia in the het model (Figure 4F). We then examined PC survival in hets, and, consistent with our previous studies (Tsai et al., 2012), we observed no PC loss in either vehicle-or rapamycin-treated het mice (Figures 4G and S6). These studies thus support a model in which the sensitive period for repetitive and inflexible behaviors is independent of PC survival, while the sensitive period remains open well into adulthood in the presence of continued PC survival.
DISCUSSION
Despite the high societal cost of autism (Leigh and Du, 2015), targeted therapies remain unavailable, and the timing during which those therapies will reap benefits is poorly understood. Sensitive periods, defined as periods when treatment paradigms will demonstrate therapeutic benefit, have been postulated for the treatment of many complex neuropsychiatric disorders, including autism (Tsai, 2016). In addition, the mechanisms underlying ASDs remain poorly understood. Recent studies have implicated the cerebellum in the pathogenesis of these disorders, but how the cerebellum regulates complex cognitive and neuropsychiatric behaviors remains poorly understood.
In this study, we used a model of TSC, a neurodevelopmental disorder associated with high rates of autism, and took advantage of the efficacy of rapamycin therapy to rescue the molecular signaling deficits in this model (Tsai et al., 2012). Using this model, we were able to interrogate the mechanistic contributions of cerebellar dysfunction to autistic-like behaviors while also defining the sensitive period for treatment of various ASD behavioral deficits and their underlying mechanisms. We demonstrated that distinct, therapeutic sensitive periods exist for individual autistic-like behaviors. Thus, despite correcting the downstream mTOR signaling defect resulting from Tsc1 loss, only certain behaviors can be ameliorated, while other behavioral dysregulation persists. We show that at a time when the therapeutic window for repetitive and inflexibility behaviors appears closed, the sensitive period for amelioration of motor and social deficits remains open, even into young adulthood in our murine model. These data demonstrate that correction of the molecular or signaling deficits in neurodevelopmental disorders may be insufficient to fix every abnormal behavior associated with the disorder, because of the presence of specific sensitive periods of treatment efficacy. These findings are summarized in Table 1. One caveat we feel is important to address here is that although these data suggest the potential to improve social behaviors into young adulthood in this rodent model, further study will be needed to assess whether parallel time windows are present in humans as well.
Table 1.
Impact of Treatment on Behavior and Cellular Phenotypes
| Rapa 6 Weeks |
Rapa 10 Weeks |
||
|---|---|---|---|
| Mutant | Het | Mutant | |
| Social approach | rescue | rescue | no |
| Social novelty | rescue | rescue | no |
| Social olfaction | rescue | rescue | no |
| Grooming | no | no | no |
| Water T maze | no | no | no |
| Rotarod | rescue | no deficit | rescue |
| PC survival | rescue | no deficit | no |
‘‘Rescue’’ denotes rescue with rapamycin treatment. ‘‘No’’ denotes lack of rescue with rapamycin treatment. ‘‘No deficit’’ indicates an absence of deficit in this model. PC, Purkinje cell; Rapa, rapamycin.
To better evaluate the mechanisms underlying these sensitive periods, we examined cellular and functional phenotypes with vehicle and rapamycin therapy. We found that the sensitive period for treatment of social behaviors continued to be accessible, correlating with PC survival and maintenance of PC excitability. Whereas social behaviors in homozygous mutants could no longer be rescued when treatment was initiated at 10 weeks of age, those same social deficits in heterozygous mutant mice could be rescued even at this later time point, correlating with the maintenance of PCs in heterozygous mutants compared with the widespread PC loss observed in homozygous Tsc1-mutant mice. These data thus suggest that although PC loss is not required for ASD-related social impairments (PC Tsc1 hets have social deficits in absence of PC loss), maintenance of PC survival is likely a critical factor for molecular-based rescue of social impairments. Intriguingly, although ASD is a very heterogeneous disorder, a large proportion of individuals with ASD have been identified to have significant PC loss (Skefos et al., 2014; Whitney et al., 2008), although the developmental window during which this occurs is not well defined. These data support further investigation into the timing of PC loss in patients with ASD while also suggesting that interventions supporting PC survival may offer additional benefit to strategies targeting social impairments in ASD. Taken together, these data suggest that the presence or absence of PC loss may modify sensitive periods of therapy and that PC loss may provide a marker that helps delineate the sensitive periods of treatment of social impairment.
On the flip side, these data also suggest that despite restoration of cellular and physiologic phenotypes and, independent of PC loss, the sensitive period for treatment of repetitive and inflexibility behaviors remained closed, suggesting an alternative mechanism to differentiate the sensitive periods of therapy for these behaviors. Potential mechanisms that may contribute to these differential sensitive periods emerge from our electrophysiology studies.
We have previously identified impairment in both spontaneous firing rates and excitability in PC Tsc1-mutant PCs (Tsai et al., 2012), and we have also identified reductions in spontaneous firing in TSC patient-derived PC precursors (Sundberg et al., 2018). Rescue of spontaneous firing rate deficits independent of excitability deficits appears to correlate best with rescue of motor deficits, which although not a canonical deficit in ASD, is a frequent comorbid finding (Fournier et al., 2010; Ming et al., 2007; Mosconi et al., 2015). Association of motor deficits with spontaneous firing deficits, independent of excitability deficits, is consistent with previous studies (Chopra and Shakkottai, 2014), while rescue of this specific electrophysiological phenotype supports a possible mechanism that mTOR-related regulation of specific ion channels (Häusser et al., 2004; Raman and Bean, 1997) at specific developmental periods may be driving these observed effects. Studies further characterizing the molecular basis for these findings will be an important avenue of future study.
In addition, social impairments and rescue of these social domains may relate to the decreased excitability observed in mutant PCs, consistent with our data supporting the rescue of PC excitability upon treatment with rapamycin. Disruption in activity-dependent neuronal signaling networks has been implicated in ASD (Ebert and Greenberg, 2013), and experience-dependent neuronal activity affects critical-period plasticity (Hensch, 2004; Hubel and Wiesel, 1970; Yamamuro et al., 2018). As excitability is critical for ongoing neural function, rescue of excitability deficits in PC-Tsc1 mutant mice may contribute to the improvement in social behaviors observed with rapamycin therapy. However, because neuronal activity is also required both for the establishment of neural circuits and for refining neural connections within those networks, disruption of intrinsic excitability during development can negatively affect the establishment of proper circuit networks (Contractor et al., 2015; Sim et al., 2013; Wiesel and Hubel, 1963; Yamamuro et al., 2018). After refining those networks during specific developmental time windows, subsequent restoration of excitability may thus be insufficient to alter network properties, which may contribute to the existence of and closure of sensitive periods for treatment (Hensch, 2004; LeBlanc and Fagiolini, 2011). Thus, differential impact on specific circuits that mediate repetitive and/or inflexibility behaviors compared with circuits that mediate social behavior may provide a potential mechanism for the inability to rescue repetitive and inflexible behaviors while the sensitive period remains open for rescue of social behaviors.
To evaluate this possibility and to identify potential contributing cerebellar regions, we examined the structural integrity of brain regions in whole-brain, volumetric, and diffusion MRI-based studies. We demonstrated significant alterations in volumes of specific cerebellar domains in PC Tsc1-mutant mice. It has been hypothesized that a regional, functional topography exists for the cerebellum beyond the duplicated cerebellar homunculus for motor functions (Klein et al., 2016). Recent studies point to distinct non-motor roles for specific cerebellar regions, while studies in autism point to associations between specific cerebellar domains and social versus repetitive and inflexible behavioral abnormalities (D’Mello et al., 2015; Stoodley, 2016; Stoodley et al., 2017; Tsai, 2016). Consistent with these findings, in our mutant mice, we identified altered cerebellar volumes in specific domains that have been associated with social dysfunction in ASD that are rescued with rapamycin therapy (D’Mello et al., 2015; Mosconi et al., 2015; Tsai, 2016). In addition, we also identified volumetric changes in cerebellar domains that are not rescued with rapamycin therapy and have been implicated in repetitive behaviors associated with ASD, consistent with the lack of behavioral rescue of repetitive and inflexible behaviors with rapamycin therapy. Further evaluation of a functional contribution of these domains to specific ASD behaviors will be an important avenue of future study. Of note, we also found that rapamycin treatment resulted in reduction in brain volumes and PC numbers in control animals, consistent with inhibition of mTOR-mediated translation, a potential impact that should be monitored in future clinical trials of mTOR inhibition. Last, alterations in the WM of TSC patients have also been reported and respond to treatment with mTOR inhibitors (Lewis et al., 2013; Peters et al., 2012; Tillema et al., 2012). Although we identify a trend toward altered WM integrity in PC Tsc1 mutants that is rescued with rapamycin treatment, statistical significance is not reached in the whole-brain analyses conducted for this study. Further study using specific region of interest-based analyses may yield additional insights into potential WM alterations in this model.
Our findings implicate altered cerebellar topography as a possible mediator of both behavioral dysfunction in PC Tsc1-mutant mice and behavioral rescue with rapamycin therapy. These findings further raise the question whether differential plasticity of cerebellar-regulated circuits may mediate rescue of social behavior, whereas for repetitive and restricted behaviors, plasticity of the specific circuit pathways is such that despite molecular-based rescue, behavior can no longer be ameliorated. These results mirror findings from Shank3- and Ube3A-mutant mice, in which molecular rescue during adulthood normalizes certain behaviors but other behaviors cannot be rescued (Mei et al., 2016; Silva-Santos et al., 2015), while studies of Syngap1 mutants revealed no improvement on behavior and cognitive phenotypes when correction of genetic deficit was initiated during adulthood (Clement et al., 2012). Taken together with findings from this study, these data point to potential limitations of molecular therapies while also pointing to a complex contribution of circuit network properties and anatomic-based mechanisms (PC loss, for example) that likely contribute to defining the sensitive periods of therapy for ASD behaviors. Moreover, these data also point to the possibility that targeted circuit modulation might provide a therapeutic strategy (Selimbeyoglu et al., 2017; Stoodley et al., 2017) that may have the potential to overcome closure of sensitive periods as defined by molecular-based therapeutic strategies.
The cerebellum has recently been demonstrated to have extensive connections to diverse cortical regions, while cerebro-cerebellar connectivity has been demonstrated to be altered in individuals with ASD (Buckner et al., 2011; D’Mello et al., 2015; D’Mello and Stoodley, 2015; Stoodley et al., 2017). Investigation of the specific cerebellar domains and downstream cerebellar-regulated circuit-based mechanisms underlying these ASD-related behaviors and leveraging these findings into potential circuit-based therapeutic modalities are important avenues of further research. Thus, although this study informs both timing and benefit of therapeutic intervention, our data also point to mechanisms that may contribute to behavioral dysfunction in these disorders. Moreover, these data lay the framework for subsequent functional evaluation of cerebellar domains and circuits to delineate the precise distributed brain circuits regulated by the cerebellum that mediate critical behaviors seen in individuals with ASD.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Calbindin | Sigma | RRID: AB_2313712 |
| Phospho-S6 | Cell Signaling | 2211 |
|
Chemicals | ||
| Rapamycin | LC Labs | R-5000 |
| NBQX | Sigma | N183 |
| R-CPP | Tocris | 247 |
| Picrotoxin | Tocris | 1128 |
| Biocytin | Tocris | 3349 |
| Almond extract | McCormick | UPC: 052100070643 |
| Banana extract | McCormick | UPC: 052100070674 |
| Multihance Contrast Agent | Bracco Imaging | 0270-5164-15 |
|
Software and Algorithms | ||
| Ethovision | Noldus | https://www.noldus.com/animal-behavior-research/products/ethovision-xt |
| Three Chambered automated crossing | National Instruments | cFP-2000 |
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
|
Experimental Models: Organisms/Strains | ||
| Tsc1 floxed mice (Tsc1tm1Djk/J) | Generous gift from Dr. David Kwiatkowski | IMSR_JAX:005680 |
| L7Cre mice (B6.129-Tg(Pcp2-cre)2Mpin/J) | JAX | IMSR_JAX:004146 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Tsai (peter.tsai@utsouthwestern.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Mice
L7cre; Tsc1flox/flox (mutant) animals were generated by crossing L7/Pcp2-Cre (L7Cre) (Barski et al., 2000); floxed Tsc1 (Tsc1flox/+[het]) mice (Kwiatkowski et al., 2002) to generate het mice, (L7Cre;Tsc1flox/flox [mutant]) mice, and control mice (floxed Tsc1 mice (Tsc1flox/flox), L7Cre+ or – Tsc1+/+mice). We have shown previously (Tsai et al., 2012) that control animals of these various genotypes act similarly on the behavioral paradigms examined in this study. Only male animals were used for behavioral experiments. Mice were of mixed genetic backgrounds (C57BL/6J, 129 SvJae, BALB/cJ). Ages of mice are as specified in behavioral analysis methods and illustrated in Figure S1. All animals were group housed and maintained on a 12hr:12hr light dark cycle. All experimental protocols were approved by the Boston Children’s Hospital Animal Research at Children’s Hospital Committee and Animal Resource Committee at University of Texas Southwestern.
METHOD DETAILS
Rapamycin Treatment
Rapamycin was dissolved in 0.25% polyethylene glycol and 0.25% tween prior to usage. Vehicle or rapamycin was administered intraperitoneally every Monday, Wednesday, and Friday with rapamycin dosed at 6 mg/kg per injection starting at 6 weeks (6 week cohort) or 10 weeks (10 week cohort) of age. As shown in Figure 1A and Figure S1, rapamycin was administered for 4 weeks prior to onset of behavioral or electrophysiology testing, and rapamycin administration was continued throughout the period of behavioral testing. Electrophysiology and/or perfusions for Purkinje cell quantification were performed 5 weeks after initiation of treatment to approximate behavioral testing timeline.
Behavioral Analysis
Behavioral testing was performed in the following order (Figure S1): 4 weeks after treatment initiation: Rotarod. 5–7 weeks after treatment initiation: Open Field, Elevated Plus, Three Chambered Social Interaction, Grooming. 6–8 weeks after treatment initiation: Water T Maze, Olfactory testing. Differences in number for behavioral testing cohorts resulted from variation between genotypes generated from crosses, and new cohorts were not utilized for each behavioral paradigm. In ordering and grouping these tests, every attempt has been made to refer to previous literature regarding behavioral test order in mice (Crawley, 2008; McIlwain et al., 2001; Võikar et al., 2004).
Accelerating Rotarod
Animals were tested using the Accelerating Rotarod as described over 5 consecutive days (Buitrago et al., 2004). Animals were tested 4 weeks after initiation of treatment paradigm (rapamycin or vehicle initiated at either 6 or 10 weeks of age).
Open Field
Open field testing was performed as described for 15 minute period (Holmes et al., 2001). Movement and time spent in center quadrants were recorded by video camera, and automated analysis was performed using Noldus (Virginia) Ethovision software. Animals were tested 5–7 weeks after initiation of treatment paradigm (rapamycin or vehicle initiated at either 6 or 10 weeks of age).
Elevated Plus Maze
Elevated Plus Maze was performed as previously described for a 5 minute period (Tsai et al., 2012; Yuan et al., 2012). Distance traveled and time in open arms were recorded by video camera and automated analysis was performed using Noldus Ethovision software. Light in the open arms was 15 lux. Animals were tested between 7–9 weeks of age. Animals were tested between 5–7 weeks after initiation of treatment paradigm (rapamycin or vehicle initiated at either 6 or 10 weeks of age).
Social Interaction
Animals were tested for social interaction in the three chambered apparatus (Dold Labs) as previously described (Yang et al., 2011). Time in chambers and number of crossings between chambers was recorded in an automated manner (National Instruments). Time spent interacting with the novel animal and object was recorded by the examiner with stopwatch. Animals were tested between 5–7 weeks after initiation of treatment paradigm (rapamycin or vehicle initiated at either 6 or 10 weeks of age). All behavioral assays (including social interaction) were performed by examiner blinded to genotype.
Grooming
After habituation, animals were observed for 10 minutes, and time spent grooming was recorded as described (Silverman et al., 2011). Animals were tested between 5–7 weeks after initiation of treatment paradigm (rapamycin or vehicle initiated at either 6 or 10 weeks of age).
Water T-Maze
Reversal learning was tested using the water T maze as described (Bednar et al., 2002). On Days 1–3, mice were given 15 trials and tasked to locate a submerged platform placed in one of the maze arms. After 15 trials on Day 3, the platform was changed to the other T arm. Mice were then tested for 15 additional trials (Reversal Day1). Then for 2 subsequent days (Reversal Day 2–3), mice were given 15 trials/day. Number of correct trials and number of trials required to achieve 5 consecutive correct trials were recorded. Animals were tested between 6–8 weeks after initiation of treatment paradigm (rapamycin or vehicle initiated at either 6 or 10 weeks of age).
Olfaction
Olfaction was tested as previously described (Yang and Crawley, 2009). Animals were presented sequentially with odors on cotton tipped applicators: first non-social, then social odors. Odors were presented in 3 consecutive trials per odorant stimulus (2 minutes/ trial) in the following order: water, almond extract, banana extract, social odor 1, and lastly social odor 2. Social odors were swipes from cages containing unfamiliar, gender (male), and age matched animals. Animals were tested between 6–8 weeks after initiation of treatment paradigm (rapamycin or vehicle initiated at either 6 or 10 weeks of age).
Slices
Acute sagittal slices (250–300 µm thick) were prepared from the cerebellar vermis of 4 and 6 week old mutant and control littermates. Slices were cut in an ice cold artificial cerebrospinal fluid (ACSF) solution consisting of (mM): 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1 MgCl2, 2 CaCl2, and 25 glucose (pH 7.3, osmolarity 310) equilibrated with 95% O2 and 5% CO2. Slices were initially incubated at 34ºC for 25 minutes, and then at room temperature prior to recording in the same ACSF.
Recordings
Visually guided (infrared DIC videomicroscopy and water-immersion 40x objective) whole-cell recordings were obtained with patch pipettes (2–4 MU) pulled from borosilicate capillary glass (World Precision Instruments) with a Sutter P-97 horizontal puller. Electro-physiological recordings were performed at 31–33 C. For current-clamp recordings, the internal solution contained (in mM): 150 K-gluconate, 3 KCl, 10 HEPES, 0.5 EGTA, 3 MgATP, 0.5 GTP, 5 phosphocreatine-tris2, and 5 phosphocreatine-Na2. pH was adjusted to 7.2 with NaOH. Current-clamp and extracellular recordings were performed in NBQX (5 µM), R-CPP (2.5 µM), and picrotoxin (20 µM) to block AMPA receptors, NMDA receptors, and GABAA receptors respectively. All drugs were purchased from Sigma-Al-drich or Tocris.
Data Acquisition and Analysis
Electrophysiological data were acquired using a Multiclamp 700B amplifier (Axon Instruments), digitized at 20 kHz with either a National Instruments USB-6229 or PCI-MIO 16E-4 board, and filtered at 2 kHz. Acquisition was controlled both with custom software written in either MATLAB (generously provided by Bernardo Sabatini, HMS, Boston, MA), or IgorPro (generously provided by Matthew Xu-Friedman, SUNY Buffalo). Series resistance was monitored in voltage-clamp recordings with a 5 mV hyperpolarizing pulse, and only recordings that remained stable over the period of data collection were used. Glass monopolar electrodes (1–2 MU) filled with ACSF in conjunction with a stimulus isolation unit (WPI, A360) were used for extracellular stimulation of CFs and PFs.
Immunohistochemistry/Quantification of Purkinje Cells
Mice were perfused and post-fixed with 4% paraformaldehyde. Sections were prepared by cryostat sectioning and were stained with the following antibodies: PhosphoS6 (Cell Signaling, #2211) and calbindin (Sigma, #CB955). Anti-Calbindin staining was performed to identify Purkinje cells while PhosphoS6 staining was performed as a marker for mTOR signaling (increased with loss of Tsc1; reduced with rapamycin administration). Purkinje cell numbers were quantified by totaling all Purkinje neurons from midline vermis sections from mice that had been treated for 5 weeks with treatment initiation commencing at either 6 or 10 weeks of age depending on cohort in order to mirror behavioral testing paradigms.
Microscopy
Intracellular labeling of Purkinje cells was accomplished using recording pipettes filled with 0.05% biocytin (Tocris). Neurons in deeper portions of the Purkinje cell layer were targeted and filled for 3 min, and then the pipette was slowly withdrawn so that the cell membrane could reseal. Slices (250 µm thick) were then fixed in 4% paraformaldehyde in 0.1 M phosphate buffer for 24 hours, rinsed thoroughly in PBS, and incubated for 90 min in a PBS solution containing 0.5% Triton-X, 10% normal goat serum and streptavidin Alexa Fluor 488 conjugate (1:500, Life Technologies). Slices were then rinsed in PBS, mounted on Superfrost Plus slides (VWR International), airdried, and coverslipped in Vectashield mounting media (Vector Labs). Immunohistochemical studies were captured using Zeiss Confocal LSM710. Images were processed and morphology quantified using ImageJ software with studies performed by examiner blinded to genotypes.
Magnetic Resonance Imaging
A total of 40 male mice were used for these studies. Total numbers included 16 mutant mice (8 vehicle and 8 rapamycin) and 23 litter-mate controls (13 vehicle and 10 rapamycin). One mouse was excluded due to extremely large ventricles making our cohort 39 mice. A multichannel 7.0 Tesla MRI scanner (Agilent Inc. Palo Alto, CA) was used to image the brains, left within their skulls. Sixteen custom built solenoid coils were used to image the brains in parallel (Bock et al., 2005).
Anatomical Scan (40um)
Parameters for the anatomical MRI scans: T2-weighted, 3D fast spin echo sequence, with a cylindrical acquisition of k-space, and with a TR of 350 ms, and TEs of 12 ms per echo for 6 echoes, field of view of 20 × 20 × 25 mm3 and a matrix size of 504 × 504 × 630 giving an image with 0.040 mm isotropic voxels (Spencer Noakes et al., 2017). Total imaging time 14 hr.
Diffusion Tensor Imaging (DTI)
DTI sequence parameters: 3D diffusion weighted fast spin echo sequence with an echo train length of 6, TR of 275 ms, first TE of 32 ms, and TEs of 10 ms for the remaining 5 echoes, field of view is 14 × 14 × 25 mm3 and a matrix size of 180 × 180 × 324 yielding an image with 0.078 mm isotropic voxels. Five b = 0 s/mm2 images and 30 high b-value images (b = 1917 s/mm2) in 30 directions were acquired with one average per direction. The 30 directions were distributed following the Jones30 scheme (Jones et al., 1999). Total imaging time 14 hours.
Registration and Deformation Based Analysis
To visualize and compare any differences in the mouse brains the anatomical images, or b = 0 s/mm2 images from the DTI study, were separately linearly (6 parameter followed by 12 parameter) and nonlinearly registered together. All scans are then resampled with appropriate transform and averaged to create a population atlas for each scan type representing the average anatomy of the study sample. All registrations were performed with a combination of mni_autoreg tools (Collins et al., 1994) and advanced normalization tools (ANTs) (Avants et al., 2008; 2011). The result of the registration was to have all scans deformed into exact alignment with each other in an unbiased fashion (Lerch et al., 2008; Nieman et al., 2006). The Jacobian determinants of the deformation fields were then used as measures of volume at each voxel. For the diffusion measurements, the images were analyzed using the FSL software package (FMRIB, Oxford UK), which was used to create Fractional Anisotropy (FA), Mean Diffusivity (MD), Axial Diffusivity (AD), and Radial Diffusivity (RD) maps for each of the individual brains. Then the same transformation that was used on the b = 0 s/mm2 images was applied to the diffusion maps to align them. The intensity differences were then calculated between groups. Significant regional volume or diffusion changes could then be calculated by warping a pre-existing classified MRI atlas onto the population atlas, which allows for the volume of 159 segmented structures encompassing cortical lobes, large WM structures (i.e., corpus callosum), cerebellum, and brainstem. This atlas built upon the Dorr atlas (Dorr et al., 2008) with additional delineations of the cerebellum according to Steadman et al. and the cortex according to Ullmann et al. (Steadman et al., 2014; Ullmann et al., 2013). As differences were expected within the cerebellum, voxelwise analyses for volume and diffusion metrics were restricted to the cerebellum. Multiple comparisons were controlled for using the false discovery rate (Genovese et al., 2002).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Data are reported as mean ± SEM, and statistical analysis was carried out with GraphPad Prism software using two-way ANOVA with Bonferroni’s multiple comparison tests for post hoc analysis. Significance was defined as p < 0.05 or FDR of < 0.05 (for MRI studies). Numbers of animals and statistical results utilized for all studies is included in Tables S1–S4. Sample sizes were based on the following formula: n = 1+2C(s/d)2 (with C calculated for p = 0.05, power 0.9). All testing was done blinded to animal genotype. ROUT methodology in Graphpad Prism was utilized to determine the presence of outliers. Behavioral Testing results can be found in Table S1 (6 week Rapa treatment cohort) and S4 (10 week Rapa treatment cohort). MRI results can be found in Supplemental Tables S2 (Volume Studie) and S3 (Diffusion studies). A total of 40 male mice were used for MRI studies including 16 mutant mice (8 vehicle and 8 rapamycin) and 23 littermate controls (13 vehicle and 10 rapamycin).
Supplementary Material
Highlights.
Social behaviors in a cerebellar autism mouse model can be rescued into adulthood
Sensitive period for repetitive behaviors closes earlier than for social behavior
Adult rescue of social behaviors in this model requires Purkinje cell survival
Rescue of cerebellar domain volumes correlates with rescue of specific ASD behaviors
ACKNOWLEDGMENTS
P.T.T. acknowledges support from the Nancy Lurie Marks Foundation, the Hearst Foundation, and the National Institute of Neurologic Disorders and Stroke of the NIH (K08 NS083733). We also thank Dr. David Kwiatkowski for generous gift of Tsc1 floxed mice. M.S. acknowledges support from the Nancy Lurie Marks Family Foundation, the Boston Children’s Hospital Boston Translational Research Program, and the Intellectual and Developmental Disabilities Research Center (U54 HD090255). J.P.L. acknowledges support from the Canadian Institutes of Health Research, the Ontario Brain Institute, and Brain Canada.
Footnotes
DECLARATION OF INTERESTS
M.S. received research funding from Roche, Novartis, Pfizer, and LAM Therapeutics unrelated to this project and served on the Scientific Advisory Boards of Sage Therapeutics, Takeda, Roche, and the PTEN Research Foundation and on the Professional Advisory Board of the Tuberous Sclerosis Alliance.
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.039.
REFERENCES
- Asano E, Chugani DC, Muzik O, Behen M, Janisse J, Rothermel R, Mangner TJ, Chakraborty PK, and Chugani HT (2001). Autism in tuberous sclerosis complex is related to both cortical and subcortical dysfunction. Neurology 57, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Avants BB, Epstein CL, Grossman M, and Gee JC (2008). Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med. Image Anal 12, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, and Gee JC (2011). A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 54, 2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski JJ, Dethleffsen K, and Meyer M (2000). Cre recombinase expression in cerebellar Purkinje cells. Genesis 28, 93–98. [PubMed] [Google Scholar]
- Bauman M, and Kemper TL (1985). Histoanatomic observations of the brain in early infantile autism. Neurology 35, 866–874. [DOI] [PubMed] [Google Scholar]
- Bednar I, Paterson D, Marutle A, Pham TM, Svedberg M, Hellström-Lindahl E, Mousavi M, Court J, Morris C, Perry E, et al. (2002). Selective nicotinic receptor consequences in APP(SWE) transgenic mice. Mol. Cell. Neurosci 20, 354–365. [DOI] [PubMed] [Google Scholar]
- Bock NA, Nieman BJ, Bishop JB, and Mark Henkelman R (2005). In vivo multiple-mouse MRI at 7 tesla. Magn. Reson. Med 54, 1311–1316. [DOI] [PubMed] [Google Scholar]
- Buckner RL (2013). The cerebellum and cognitive function: 25 years of insight from anatomy and neuroimaging. Neuron 80, 807–815. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Krienen FM, Castellanos A, Diaz JC, and Yeo BT (2011). The organization of the human cerebellum estimated by intrinsic functional connectivity. J. Neurophysiol 106, 2322–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buitrago MM, Schulz JB, Dichgans J, and Luft AR (2004). Short and long-term motor skill learning in an accelerated rotarod training paradigm. Neurobiol. Learn. Mem 81, 211–216. [DOI] [PubMed] [Google Scholar]
- Capal JK, Horn PS, Murray DS, Byars AW, Bing NM, Kent B, Bucher LA, Williams ME, O’Kelley S, Pearson DA, et al. ; TACERN Study Group (2017). Utility of the Autism Observation Scale for infants in early identification of autism in tuberous sclerosis complex. Pediatr. Neurol 75, 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra R, and Shakkottai VG (2014). Translating cerebellar Purkinje neuron physiology to progress in dominantly inherited ataxia. Future Neurol 9, 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, Aceti M, Creson TK, Ozkan ED, Shi Y, Reish NJ, Al-monte AG, Miller BH, Wiltgen BJ, Miller CA, et al. (2012). Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DL, Neelin P, Peters TM, and Evans AC (1994). Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J. Comput. Assist. Tomogr 18, 192–205. [PubMed] [Google Scholar]
- Contractor A, Klyachko VA, and Portera-Cailliau C (2015). Altered neuronal and circuit excitability in fragile X syndrome. Neuron 87, 699–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley JN (2008). Behavioral phenotyping strategies for mutant mice. Neuron 57, 809–818. [DOI] [PubMed] [Google Scholar]
- Cupolillo D, Hoxha E, Faralli A, De Luca A, Rossi F, Tempia F, and Carulli D (2016). Autistic-like traits and cerebellar dysfunction in Purkinje cell PTEN knock-out mice. Neuropsychopharmacology 41, 1457–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello AM, and Stoodley CJ (2015). Cerebro-cerebellar circuits in autism spectrum disorder. Front. Neurosci 9, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello AM, Crocetti D, Mostofsky SH, and Stoodley CJ (2015). Cerebellar gray matter and lobular volumes correlate with core autism symptoms. Neuroimage Clin 7, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorr AE, Lerch JP, Spring S, Kabani N, and Henkelman RM (2008). High resolution three-dimensional brain atlas using an average magnetic resonance image of 40 adult C57Bl/6J mice. Neuroimage 42, 60–69. [DOI] [PubMed] [Google Scholar]
- Ebert DH, and Greenberg ME (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eluvathingal TJ, Behen ME, Chugani HT, Janisse J, Bernardi B, Chakraborty P, Juhasz C, Muzik O, and Chugani DC (2006). Cerebellar lesions in tuberous sclerosis complex: neurobehavioral and neuroimaging correlates. J. Child Neurol 21, 846–851. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Aldinger KA, Ashwood P, Bauman ML, Blaha CD, Blatt GJ, Chauhan A, Chauhan V, Dager SR, Dickson PE, et al. (2012). Consensus paper: pathological role of the cerebellum in autism. Cerebellum 11, 777–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier KA, Hass CJ, Naik SK, Lodha N, and Cauraugh JH (2010). Motor coordination in autism spectrum disorders: a synthesis and meta-analysis. J. Autism Dev. Disord 40, 1227–1240. [DOI] [PubMed] [Google Scholar]
- Genovese CR, Lazar NA, and Nichols T (2002). Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage 15, 870–878. [DOI] [PubMed] [Google Scholar]
- Häusser M, Raman IM, Otis T, Smith SL, Nelson A, du Lac S, Loewenstein Y, Mahon S, Pennartz C, Cohen I, and Yarom Y (2004). The beat goes on: spontaneous firing in mammalian neuronal microcircuits. J. Neurosci 24, 9215–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK (2004). Critical period regulation. Annu. Rev. Neurosci 27, 549–579. [DOI] [PubMed] [Google Scholar]
- Holmes A, Hollon TR, Gleason TC, Liu Z, Dreiling J, Sibley DR, and Crawley JN (2001). Behavioral characterization of dopamine D5 receptor null mutant mice. Behav. Neurosci 115, 1129–1144. [PubMed] [Google Scholar]
- Hubel DH, and Wiesel TN (1970). The period of susceptibility to the physiological effects of unilateral eye closure in kittens. J. Physiol 206, 419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeste SS, Sahin M, Bolton P, Ploubidis GB, and Humphrey A (2008). Characterization of autism in young children with tuberous sclerosis complex. J. Child Neurol 23, 520–525. [DOI] [PubMed] [Google Scholar]
- Jones DK, Simmons A, Williams SC, and Horsfield MA (1999). Noninvasive assessment of axonal fiber connectivity in the human brain via diffusion tensor MRI. Magn. Reson. Med 42, 37–41. [DOI] [PubMed] [Google Scholar]
- Klein AP, Ulmer JL, Quinet SA, Mathews V, and Mark LP (2016). Non-motor functions of the cerebellum: an introduction. AJNR Am. J. Neuroradiol 37, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, and Onda H (2002). A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum. Mol. Genet 11, 525–534. [DOI] [PubMed] [Google Scholar]
- LeBlanc JJ, and Fagiolini M (2011). Autism: a ‘‘critical period’’ disorder? Neural Plast 2011, 921680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh JP, and Du J (2015). Brief report: forecasting the economic burden of autism in 2015 and 2025 in the United States. J. Autism Dev. Disord 45, 4135–4139. [DOI] [PubMed] [Google Scholar]
- Lerch JP, Pruessner J, Zijdenbos AP, Collins DL, Teipel SJ, Hampel H, and Evans AC (2008). Automated cortical thickness measurements from MRI can accurately separate Alzheimer’s patients from normal elderly controls. Neurobiol. Aging 29, 23–30. [DOI] [PubMed] [Google Scholar]
- Lewis WW, Sahin M, Scherrer B, Peters JM, Suarez RO, Vogel-Farley VK, Jeste SS, Gregas MC, Prabhu SP, Nelson CA 3rd, and Warfield SK (2013). Impaired language pathways in tuberous sclerosis complex patients with autism spectrum disorders. Cereb. Cortex 23, 1526–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limperopoulos C, Bassan H, Gauvreau K, Robertson RL Jr., Sullivan NR, Benson CB, Avery L, Stewart J, Soul JS, Ringer SA, et al. (2007). Does cerebellar injury in premature infants contribute to the high prevalence of longterm cognitive, learning, and behavioral disability in survivors? Pediatrics 120, 584–593. [DOI] [PubMed] [Google Scholar]
- Lipton JO, and Sahin M (2014). The neurology of mTOR. Neuron 84, 275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald NM, Varcin KJ, Bhatt R, Wu JY, Sahin M, Nelson CA 3rd, and Jeste SS (2017). Early autism symptoms in infants with tuberous sclerosis complex. Autism Res 10, 1981–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain KL, Merriweather MY, Yuva-Paylor LA, and Paylor R (2001). The use of behavioral test batteries: effects of training history. Physiol. Behav 73, 705–717. [DOI] [PubMed] [Google Scholar]
- Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, and Feng G (2016). Adult restoration of Shank3 expression rescues selective autistic-like pheno-types. Nature 530, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X, Brimacombe M, and Wagner GC (2007). Prevalence of motor impairment in autism spectrum disorders. Brain Dev 29, 565–570. [DOI] [PubMed] [Google Scholar]
- Mosconi MW, Wang Z, Schmitt LM, Tsai P, and Sweeney JA (2015). The role of cerebellar circuitry alterations in the pathophysiology of autism spectrum disorders. Front. Neurosci 9, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CA 3rd, Zeanah CH, Fox NA, Marshall PJ, Smyke AT, and Guthrie D (2007). Cognitive recovery in socially deprived young children: the Bucharest Early Intervention Project. Science 318, 1937–1940. [DOI] [PubMed] [Google Scholar]
- Nieman BJ, Flenniken AM, Adamson SL, Henkelman RM, and Sled JG (2006). Anatomical phenotyping in the brain and skull of a mutant mouse by magnetic resonance imaging and computed tomography. Physiol. Genomics 24, 154–162. [DOI] [PubMed] [Google Scholar]
- Peter S, Ten Brinke MM, Stedehouder J, Reinelt CM, Wu B, Zhou H, Zhou K, Boele HJ, Kushner SA, Lee MG, et al. (2016). Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun 7, 12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Sahin M, Vogel-Farley VK, Jeste SS, Nelson CA 3rd, Gregas MC, Prabhu SP, Scherrer B, and Warfield SK (2012). Loss of white matter microstructural integrity is associated with adverse neurological outcome in tuberous sclerosis complex. Acad. Radiol 19, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, and Bean BP (1997). Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J. Neurosci 17, 4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith RM, Way S, McKenna J 3rd, Haines K, and Gambello MJ (2011). Loss of the tuberous sclerosis complex protein tuberin causes Purkinje cell degeneration. Neurobiol. Dis 43, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith RM, McKenna J, Wu H, Hashmi SS, Cho SH, Dash PK, and Gambello MJ (2013). Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neuro-biol. Dis 51, 93–103. [DOI] [PubMed] [Google Scholar]
- Ritvo ER, Freeman BJ, Scheibel AB, Duong T, Robinson H, Guthrie D, and Ritvo A (1986). Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA-NSAC Autopsy Research Report. Am. J. Psychiatry 143, 862–866. [DOI] [PubMed] [Google Scholar]
- Rogers TD, Dickson PE, McKimm E, Heck DH, Goldowitz D, Blaha CD, and Mittleman G (2013). Reorganization of circuits underlying cerebellar modulation of prefrontal cortical dopamine in mouse models of autism spectrum disorder. Cerebellum 12, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu YH, Lee JD, Yoon PH, Kim DI, Lee HB, and Shin YJ (1999). Perfusion impairments in infantile autism on technetium-99m ethyl cysteinate dimer brain single-photon emission tomography: comparison with findings on magnetic resonance imaging. Eur. J. Nucl. Med 26, 253–259. [DOI] [PubMed] [Google Scholar]
- Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, Ramakrishnan C, Fenno LE, Davidson TJ, Wright M, and Deisseroth K (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci. Transl. Med 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, Kushner SA, and Elgersma Y (2015). Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J. Clin. Invest 125, 2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Turner SM, Barkan CL, Tolu SS, Saxena R, Hung AY, Sheng M, and Crawley JN (2011). Sociability and motor functions in Shank1 mutant mice. Brain Res 1380, 120–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim S, Antolin S, Lin CW, Lin Y, and Lois C (2013). Increased cell-intrinsic excitability induces synaptic changes in new neurons in the adult dentate gyrus that require Npas4. J. Neurosci 33, 7928–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skefos J, Cummings C, Enzer K, Holiday J, Weed K, Levy E, Yuce T, Kemper T, and Bauman M (2014). Regional alterations in Purkinje cell density in patients with autism. PLoS ONE 9, e81255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer Noakes TL, Henkelman RM, and Nieman BJ (2017). Partitioning k-space for cylindrical three-dimensional rapid acquisition with relaxation enhancement imaging in the mouse brain. NMR Biomed 30, 30. [DOI] [PubMed] [Google Scholar]
- Steadman PE, Ellegood J, Szulc KU, Turnbull DH, Joyner AL, Henkel-man RM, and Lerch JP (2014). Genetic effects on cerebellar structure across mouse models of autism using a magnetic resonance imaging atlas. Autism Res 7, 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley CJ (2014). Distinct regions of the cerebellum show gray matter decreases in autism, ADHD, and developmental dyslexia. Front. Syst. Neurosci 8, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley CJ (2016). The cerebellum and neurodevelopmental disorders. Cerebellum 15, 34–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley CJ, D’Mello AM, Ellegood J, Jakkamsetti V, Liu P, Nebel MB, Gibson JM, Kelly E, Meng F, Cano CA, et al. (2017). Altered cerebellar connectivity in autism and cerebellar-mediated rescue of autism-related behaviors in mice. Nat. Neurosci 20, 1744–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg M, Tochitsky I, Buchholz DE, Winden K, Kujala V, Kapur K, Cataltepe D, Turner D, Han MJ, Woolf CJ, et al. (2018). Purkinje cells derived from TSC patients display hypoexcitability and synaptic deficits associated with reduced FMRP levels and reversed by rapamycin. Mol. Psychiatry, Published online February 15, 2018 10.1038/s41380-018-0018-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillema JM, Leach JL, Krueger DA, and Franz DN (2012). Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology 78, 526–531. [DOI] [PubMed] [Google Scholar]
- Tsai PT (2016). Autism and cerebellar dysfunction: Evidence from animal models. Semin. Fetal Neonatal Med 21, 349–355. [DOI] [PubMed] [Google Scholar]
- Tsai P, and Sahin M (2011). Mechanisms of neurocognitive dysfunction and therapeutic considerations in tuberous sclerosis complex. Curr. Opin. Neurol 24, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, Steinberg J, Crawley JN, Regehr WG, and Sahin M (2012). Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmann JF, Watson C, Janke AL, Kurniawan ND, and Reutens DC (2013). A segmentation protocol and MRI atlas of the C57BL/6J mouse neocortex. Neuroimage 78, 196–203. [DOI] [PubMed] [Google Scholar]
- Võikar V, Vasar E, and Rauvala H (2004). Behavioral alterations induced by repeated testing in C57BL/6J and 129S2/Sv mice: implications for phenotyping screens. Genes Brain Behav 3, 27–38. [DOI] [PubMed] [Google Scholar]
- Voogd J (2014). What we do not know about cerebellar systems neuroscience. Front. Syst. Neurosci 8, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber AM, Egelhoff JC, McKellop JM, and Franz DN (2000). Autism and the cerebellum: evidence from tuberous sclerosis. J. Autism Dev. Disord 30, 511–517. [DOI] [PubMed] [Google Scholar]
- Whitney ER, Kemper TL, Bauman ML, Rosene DL, and Blatt GJ (2008). Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: a stereological experiment using calbindin-D28k. Cerebellum 7, 406–416. [DOI] [PubMed] [Google Scholar]
- Wiesel TN, and Hubel DH (1963). Effects of visual deprivation on morphology and physiology of cells in the cats lateral geniculate body. J. Neurophysiol 26, 978–993. [DOI] [PubMed] [Google Scholar]
- Yamamuro K, Yoshino H, Ogawa Y, Makinodan M, Toritsuka M, Yamashita M, Corfas G, and Kishimoto T (2018). Social isolation during the critical period reduces synaptic and intrinsic excitability of a subtype of pyramidal cell in mouse prefrontal cortex. Cereb. Cortex 28, 998–1010. [DOI] [PubMed] [Google Scholar]
- Yang M, and Crawley JN (2009). Simple behavioral assessment of mouse olfaction. Curr. Protoc. Neurosci Chapter 8, Unit 8, 24. [DOI] [PMC free article] [PubMed]
- Yang M, Silverman JL, and Crawley JN (2011). Automated three-chambered social approach task for mice. Curr. Protoc. Neurosci Chapter 8, Unit 8, 26. [DOI] [PMC free article] [PubMed]
- Yuan E, Tsai PT, Greene-Colozzi E, Sahin M, Kwiatkowski DJ, and Malinowska IA (2012). Graded loss of tuberin in an allelic series of brain models of TSC correlates with survival, and biochemical, histological and behavioral features. Hum. Mol. Genet 21, 4286–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.