

Abstract
TiC14 promoted coupling of ethyl glyoxylate and dihydrofuran or dihydropyran provided oxonium ion intermediates which upon reaction with a nucleophilic trapping agent such as allyltrimethylsilane provided 2,3-disubstituted tetrahydrofurans and pyrans in good yields. The overall protocol constitutes an efficient three component coupling reaction in one pot.
Functionalized tetrahydrofurans and tetrahydropyrans are embedded in many biologically important natural products.1 Consequently, a number of methods for the syntheses of 2,5-disubstituted tetrahydrofurans and 2,6-disubstituted tetrahydropyrans have been developed and utilized in the synthesis of a variety of bioactive compounds.2 However, the synthesis of 3-substituted or 2,3-disubstituted tetrahydrofurans and tetrahydropyrans thus far, has received very little attention. Of particular interest, there is no general method for the synthesis of 3-(β-carboxy-α-hydroxymethyl)-tetrahydrofuran, 3-(β-carboxy-α-hydroxymethyl)-tetrahydropyran or their corresponding 2-substituted derivatives. A number of previous reports deal with Lewis acid catalyzed reactions of dihydropyran and aldehyde or acetal. Mukaiyama and co-workers have investigated condensation of dihydropyran and acetal in the presence of TMSCI and tin (II) chloride.3 Sugimura and Osumi have reported BF3.OEt2 catalyzed cycloaddition of vinyl ethers and isopropylidene D-aldose aldehyde.4 Based on these reports, we envisioned that the reaction of dihydrofuran and bidentate aldehyde such as ethyl glyoxylate in the presence of an appropriate Lewis acid would provide a chelated oxonium ion intermediate (A) as illustrated in Scheme 1. Therefore, trapping of this oxonium ion by carbon nucleophiles may provide convenient access to 3-(β-carboethoxy-α-hydroxymethyl)-2-substituted tetrahydrofuran or tetrahydropyran derivatives. Herein we report that the initial treatment of ethyl glyoxylate and 2,3-dihydro-2H-furan or 3,4-dihydro-2H-pyran in the presence of TiCl4 followed by exposure of the resulting reaction mixture to nucleophiles provided a number of 3-(β-carboethoxy-α-hydroxymethyl)-2-substituted tetrahydrofuran or tetrahydropyran derivatives in excellent yields. The overall process not only results in a three component carbon-carbon bond formation, the reaction also led to the creation of three contiguous chiral centers in the condensation product.
Scheme 1.

Initial attempts of the TMSCI and tin (II) chloride promoted reaction of dihydrofuran and ethyl glyoxylate failed to provide the desired condensation product. After surveying a number of Lewis acids and reaction conditions, we found that the coupling reaction can be best carried out in the presence of TiC14. Thus, freshly distilled ethyl glyoxylate (1 equiv)5 and dihydrofuran (1.1 equiv) in CH2Cl2 were treated with commercial6 TiC14 (1 M in CH2Cl2, 1.1 equiv) at −78°C for 1 h. After this period, triethylsilane (3 equiv) was added and the resulting reaction mixture was continued to stir at −78° to 23°C for 1 h before quenching with saturated aqueous NaHCO3 solution. Standard workup and flash chromatography over silica gel provided 3-(β-carboethoxy-α-hydroxymethyl)-tetrahydrofuran 2 in 88% yield (1:1 mixture of diastereomers by 1H NMR). The feasibility of this protocol was examined with dihydropyran and ethyl vinyl ether in the presence of a number of nucleophiles. The results are summarized in Table 1.
Table 1.
TiC14 promoted coupling reactions of ethyl glyoxylate with vinyl ethers.a
| Entry | Vinyl ether | Nucleophile | Product | Yield (%)b | Ratio c |
|---|---|---|---|---|---|
| 1 | 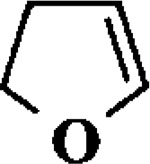 |
Et3SiH | 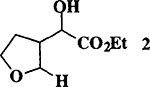 |
88 | 1 : 1 |
| 2 | 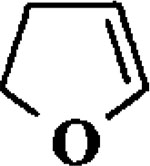 |
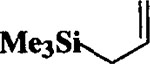 |
89 | (4 : l)d | |
| 3 | 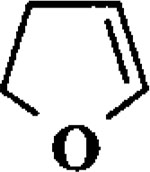 |
MeOH | 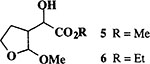 |
36 | 1 : 1.5 : 1 : 1 |
| 36 | 1 : 2 : 2 : 2 | ||||
| 4 | 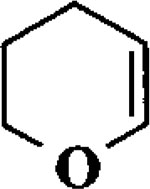 |
Et3SiH | 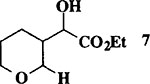 |
95 | 1 : 1 |
| 5 | 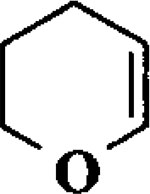 |
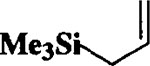 |
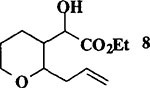 |
85 | (1 : 1d) |
| 6 | 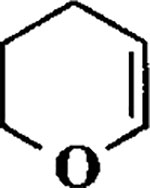 |
MeOH | 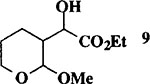 |
65 | (2: 1)e |
| 7 | 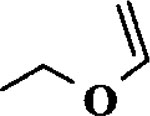 |
Et3SiH | 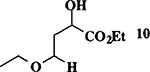 |
78 | — |
| 8 | 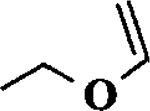 |
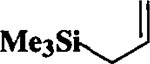 |
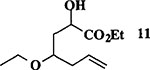 |
83 | 1 : 1 |
All reactions were carried out as described in the text.
Isolated yield.
Determined by 1H NMR.
Determined after Jones oxidation (product 4 X = CO).
Determined after TPAP oxidation.
When the oxonium ion intermediate resulting from the reaction of 2,3-dihydro-2H-furan and ethyl glyoxylate was reacted with allyltrimethylsilane, the 2,3-disubstituted tetrahydrofuran derivative 3 was obtained in 89% as a mixture of diastereomers (entry 2). Jones oxidation of this mixture gave the corresponding α-ketoester 4 as a 4:1 mixture of diastereomers. The relative ring stereochemistry of the major keto ester was assigned to be trans based upon 1H NMR NOE studies.7 When the oxonium ion was reacted with methanol, a mixture of methyl glycosides 5 and 6 (entry 3) were isolated in 72% combined yield. The reactions of 3,4-dihydro-2H-pyran and ethyl glyoxylate with triethylsilane, allyltrimethylsilane and methanol also provided variously substituted tetrahydropyran derivatives 7–9 in comparable yields (entries 4–6). Reaction with allyltrimethylsilane furnished tetrahydropyran derivatives 8 as a mixture of four diastereomers in 85% yield (entry 5). The ratio of cis and trans isomers at the C-2 and C-3 positions was determined to be 1:1 after Jones oxidation of 8 to the corresponding α-ketoesters. The reaction of the oxonium ion derived from dihydropyran and ethyl glyoxylate with methanol afforded a mixture of methyl glycosides 9 (entry 6) in 65% yield.9 Oxidation of the hydroxy group with TPAP exhibited a 2:1 mixture of anomers.10 Acyclic substrate ethyl vinyl ether and ethyl glyoxylate under the above reaction conditions also provides excellent yield of the corresponding coupling products (10 and 11; entries 7 and 8). The 1H NMR analysis of product 11 revealed the presence of a 1:1 mixture of diastereomers.11 It should be noted that the coupling in entry 4 with SnC14 in place of TiC14 afforded the coupling product 7 in only 19% isolated yield. Furthermore, attempted reaction of dihydrofuran, benzaldehyde and allylsilane in the presence of TiC14 as described above (entry 2) resulted in product derived from allylsilane addition to benzaldehyde (44% yield) and a complex mixture of unidentified products.
In summary, we have developed a novel three component coupling process for the synthesis of 3-(β-carboethoxy-α-hydroxymethyl)-2-substituted tetrahydrofuran and tetrahydropyran derivatives. In light of the utility of structurally related 3-tetrahydrofuranylglycine, this new class of heterocycles may have important biological potential.12 Further studies including diastereo- and enantioselective reactions are currently under investigation. The following example is representative of this procedure.
Preparation of tetrahydrofuran 3:
To a mixture of freshly distilled ethyl glyoxylate5 (2.0 mmol) and 2,3-dihydrofuran (2.2 mmol) in dry CH2C12 (5 mL) was added a solution of TiC14 (1 M in CH2Cl2, 2.2 mmol) at −78°C, and the resulting mixture was stirred for 1 h. Allyltrimethylsilane (6.0 mmol) was added to the mixture at −78°C and the resulting mixture was stirred at −78° to 23°C for 1 h. Reaction was quenched with aqueous NaHCO3 solution (5 mL) and was extracted with EtOAc (2 × 30 mL). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. Flash column chromatography of the residue afforded the tetrahydrofuran 3 derivatives (381 mg) in 89% yield. Standard Jones oxidation of 3 provided the corresponding α-keto ester 4 (65% yield) as a mixture (4:1) of diastereomers: Major isomer: Rf = 0.19 (10% EtOAc in hexane); IR (neat) 1758 (shoulder), 1734, 1265 cm−1; 1H NMR (400 MHz, CDCl3) δ 1.37 (3H, t, J = 7.1 Hz), 2.14 (1H, m), 2.23 (1H, m), 2.34–2.47 (2H, m), 3.53 (1H, dt, J = 9.4 and 6.3 Hz), 3.82 (1H, m), 3.98 (1H, ddd, J = 8.6, 7.3, and 5.2 Hz), 4.15 (1H, q, J = 6.3 Hz), 4.33 (2H, q, J = 7.1 Hz), 5.08 (1H, dd, J = 10.3 and 2.0 Hz), 5.13 (1H, dd, J = 17.2 and 2.0 Hz), 5.78 (1H, ddd, J = 17.2, 10.3, and 7.1 Hz); 13C NMR (100 MHz, CDC13) δ 14.0, 29.7, 39.1, 51.6, 62.6, 67.7, 80.3, 118.0, 133.8, 161.0, 194.0; MS (FAB) m/z 213 (M++H). HRMS m/z Calcd for C11H17O4 (M++H) 213.1127. Found 213.1120.; Minor isomer: Rf = 0.16 (10% EtOAc in hexane); 1H NMR (400 MHz, CDC13) δ 1.37 (3H, t, J = 7.2 Hz), 2.05 (1H, m), 2.10–2.20 (2H, m), 2.32 (1H, m), 3.77 (1H, m), 4.05 (1H, m), 4.09 (1H, td, J = 8.4 and 4.4 Hz), 4.30 (1H, m), 4.31 (2H, q, J = 7.2 Hz), 5.03 (1H, dd, J = 16.0 and 1.5 Hz), 5.04 (1H, dd, J = 11.3 and 1.5 Hz), 5.74 (1H, m); 13C NMR (100 MHz, CDC13) δ 13.9, 28.2, 36.0, 49.5, 62.7, 67.1, 80.2, 117.9, 134.0, 161.0, 193.6.
Acknowledgment:
Financial support for this work was provided by the National Institutes of Health (GM 55600). We thank Professor David Crich for helpful discussions and Dr. P. Mathivanan for preliminary experimental assistance.
References and Notes
- 1.Westley JW Polyether Antibiotics: Naturally Occurring Acid Ionophores, Vol. I and II, Marcel Dekker: New York, 1982. [Google Scholar]
- 2.Katritzky AR; Rees CW; and Scriven EFV In Comprehensive Heterocyclic Chemistry II, Vol. 2 pp 386–392 and Vol. 5. pp 390–396, Pergamon Press: Oxford, 1996. [Google Scholar]; For excellent reviews see: [Google Scholar]; Boivin TLB Tetrahedron 1987, 43, 3309; [Google Scholar]; Harmange JC; Figadere B Tetrahedron: Asymm. 1993, 4, 1711. [Google Scholar]
- 3.Mukaiyama T; Wariishi K; Saito Y; Hayashi M; Kobayashi S Chem. Lett. 1988, 1101. [Google Scholar]
- 4.Sugimura H; Osumi K Tetrahedron Lett. 1989, 30, 1571. [Google Scholar]
- 5.Ethyl glyoxylate was purchased from Fluka Chemical Company as a 50% solution by weight in toluene and was distilled according to the literature procedures. See: Evans DA; MacMillan DWC; Campos KR J. Am. Chem. Soc. 1997, 119, 10859. [Google Scholar]
- 6. TiC14 was purchased from Aldrich Chemical Company, and used as received.
- 7. A 6% NOE was detected between the H(3) and the allylic protons of the major keto ester 4. Furthermore, consistent with the report of Clive et al, attempted radical cyclization of the thionoformates of 3, the major thionoformates (1:1 mixture) provided the deoxygenated product of 3 as a single trans isomer. The minor thionoformates, however, afforded the corresponding cis fused bicyclic systems (3:1 mixture).8.
- 8.Clive DLJ; Manning HW; Boivin TLB; Postema MHD J. Org. Chem. 1993, 58, 6857. [Google Scholar]
- 9. A small amount (15%) of a mixture of α,β-unsaturated ester resulting from dehydration of alcohol 9 was also isolated.
- 10.Ley SV; Norman J; Griffith WP; Marsden SP Synthesis 1994, 639. [Google Scholar]
- 11. All new compounds gave satisfactory spectroscopic and analytical results.
- 12.Ghosh AK; Thompson WJ; Holloway MK; McKee SP; Duong TT; Lee HY; Munson PM; Smith AM; Wai JM; Darke PL; Zugay JA; Emini EA; Schleif WA; Huff JR; Anderson PS J. Med. Chem. 1993, 36, 2300 and references cited therein. [DOI] [PubMed] [Google Scholar]