

More than 2 decades after the initial concept of chimeric antigen receptor T (CAR‐T) cell was described, incremental improvements in molecular biology, virology, T‐cell immunology, and manufacturing process led to regulatory approval of the first CAR‐T cell product for the treatment of B‐cell malignancies. Here, we detail the unique translational pathway of CAR‐T cell therapy, highlighting challenges and facilitators of its translation that may be applicable to future cell and gene therapies in development.
Cancer and the need for novel therapeutics
Cancer remains a major international public health concern, with a predicted 1.7 million new cases and 609,640 cancer‐related deaths in the United States during 2018.1
Current treatment options include surgery, chemotherapy, and radiotherapy. Despite advances in cancer detection, surgical technologies, radiation precision, chemotherapeutic agents, and the emergence of targeted therapy, metastatic cancer remains largely incurable. The rare but durable responses after immunotherapy have highlighted the potential for this approach as a new pillar in the treatment of cancer.
History and development of cellular immunotherapy
Allogeneic bone marrow transplantation represents the oldest form of cellular immunotherapy. The pivotal experiments were performed by Jacobson and Ford in 1948 (Figure 1). In 1977, the initial report of 100 patients with leukemia who received an allogenic transplantation highlighted a few novel observations: (i) patients can engraft after bone marrow transplantation; (ii) this technique is an effective therapy for the treatment of otherwise fatal leukemia; and (iii) patients develop a “secondary disease,” graft‐vs.‐host disease.
Figure 1.
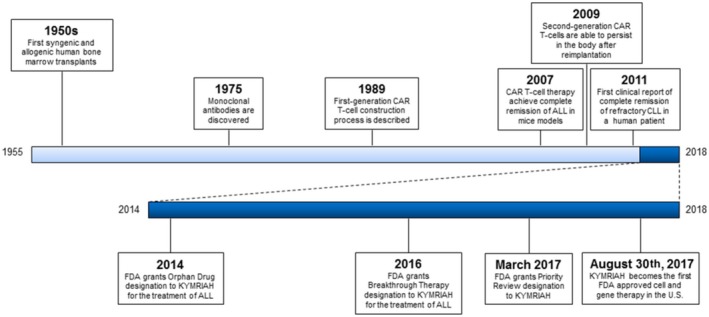
Historical timeline of the clinical translation of chimeric antigen receptor T (CAR‐T) cell therapy. ALL, acute lymphocytic leukemia; CLL, chronic lymphocytic leukemia; FDA, US Food and Drug Administration.
These findings prompted scientists to explore new ways of using human T cells to target and destroy cancer cells. The initial concept consisted of harvesting T cells from a patient, genetically modifying them ex vivo to confer them anticancer activity, in vitro expansion in sufficient numbers to reach therapeutic levels, and, reimplantation of cells back into the patient.
In 1989, Gross and colleagues2 designed and detailed the construction process of what later became known as first‐generation CAR‐T. Gross et al.2 engineered genetically modified human cytotoxic T cells in which expression of chimeric surface receptors was promoted, conferring antibody‐like specificity upon the cells. Although first‐generation CAR‐T cells were innovative, existing tissue‐culture techniques failed to produce cells in sufficient numbers for therapeutic reimplantation. Despite overcoming production limitations, first‐generation CAR‐T cells remained incapable of achieving clinical effectiveness due to their inability to undergo robust proliferation or persist inside the body. These challenges stalled further progress until 2007, when Brentjens and colleagues3 demonstrated complete eradication of acute lymphocytic leukemia (ALL) in mouse models using first‐generation CAR‐T cells designed to target CD19. Results pictured CD19 as a promising target for B‐cell malignancies; however, levels of CAR‐T cells persisting inside the body remained low. In 2009, Milone and colleagues4 showed that by adding an additional CD‐137 co‐stimulatory signal to a first‐generation CD19‐targeted CAR‐T cell not only conferred the ability to persist inside the body, but also increased its antileukemic efficacy in mouse models (known as second‐generation CAR‐T cells). Two years later, investigators from the University of Pennsylvania reported complete remissions in three patients with refractory chronic lymphocytic leukemia after therapy with a second‐generation CAR‐T cell (CTL019).5 CAR‐T cells persisted at high levels for 6 months; in follow‐up reports, CAR‐T cells persisted for up to 5 years.6, 7 Based on this success, investigators decided to extend these findings to patients with ALL and, in 2013, reported complete remission in two patients with relapsed and refractory B‐cell ALL after infusion with CTL019. These studies showed highly promising results that ignited interest and research in the field from major pharmaceutical companies, academic institutions, and regulatory bodies.
Translation to the clinic
US Federal regulations define human cells, tissues, and cellular and tissue‐based products as “articles containing or consisting of human cells or tissues that are intended for implantation, transplantation, infusion, or transfer into a human recipient.” For cells or nonstructural tissues, “minimal manipulation” means that the processing of the HCT/P does not alter the relevant biological characteristics of cells or tissues. Due to the processes involved in the generation of CAR‐T cells, they do not meet the criteria for “minimal manipulation” set forth in these regulations and are, therefore, considered as “more than minimal manipulation” products. Such products and processes are the responsibility of the US Food and Drug Administration (FDA) Center for Biologics Evaluation and Research.
Biologics require the submission of an investigational new drug (IND) application to Center for Biologics Evaluation and Research. In an IND, the sponsor details information regarding the target subject population, justification for the use of the product, prior preclinical experience, known toxicities and their management, and manufacturing processes that ensure consistency of product quality. In the case of CAR‐T cell therapy, which involves multiple components, each with its own manufacturing challenges, one of the most challenging sections of the IND is the “Chemistry, Manufacturing, and Controls.” Here, the sponsor must show compliance with current Good Manufacturing Practice requirements, and outline the manufacturing process for each component involved. Compliance with the current Good Manufacturing Practice ensures the sterility and safety of the therapy's components throughout the manufacturing process, while also confirming that the final identity, purity, and potency of the manufactured CAR‐T cells are sufficient for clinical use.
Upon IND approval, sponsors are allowed to begin early, first‐in‐human trials; however, these can be halted at any moment if severe and/or fatal adverse events occur. Trials traditionally proceed through four phases; this pathway notoriously prolongs clinical translation of novel therapies. However, in the case of cutting‐edge products with highly demonstrated need in serious diseases, the FDA has created several newer regulatory pathways/programs in hopes of accelerating testing, review, and approval of these products. The FDA's expedited programs include: (i) “Fast Track,” which grants the sponsor access to frequent meetings and written communications with the FDA and Rolling Review status; (ii) “Breakthrough Therapy,” which grants all Fast Track features, plus intensive guidance on drug development programs for product testing and organizational commitment; (iii) “Accelerated Approval,” which reduces the standard waiting period to obtain clinically meaningful benefit from the drug; and (iv) “Priority Review,” in which the FDA commits to reviewing the application within 6 months.
The race for FDA approval
Based on the results of early phase clinical trials,6, 7, 8 Novartis Pharmaceuticals initiated collaborations with the University of Pennsylvania in 2012 that allowed investigators to expand CTL019 therapy to a larger number of patients. With this support, investigators conducted a seminal trial (n = 30) that provided evidence of complete remission of relapsed or refractory ALL in 90% of study participants.8 Although the trial demonstrated substantial evidence for clinical effectiveness, there were adverse reactions related to the therapy. However, the lack of effective treatments for ALL allowed the sponsors to receive “Orphan Drug” designation from the FDA, which provided additional regulatory assistance to speed translation.
In 2015, Novartis conducted a multicenter phase II trial (n = 75) to explore tisagenlecleucel's (formerly CTL019) clinical effectiveness in treating pediatric and young adult patients with CD19+ relapsed or refractory B‐cell ALL.9 Preliminary data led the FDA to grant KYMRIAH (tisagenlecleucel, Novartis, Basel, Switzerland) an additional “Breakthrough Therapy” designation. An interim analysis of trial data in 2016 demonstrated tisagenlecleucel efficacy, later earning it “Priority Review” designation.
In 2017, the importance of the synergistic effects of “Orphan Drug,” “Breakthrough Therapy,” and “Priority Review” designations were highlighted by the FDA approval of tisagenlecleucel for the treatment of relapsed or refractory B‐cell ALL in patients 25 years of age or younger, making it the first FDA approved cell therapy available in the United States. Because of the risks of cytokine release syndrome and neurotoxicity, tisagenlecleucel was approved with a risk evaluation and mitigation strategy. This aims to reduce the risk of adverse reactions by (i) ensuring that those who prescribe, dispense, or administer tisagenlecleucel are aware of how to monitor for and manage side effects and (ii) that hospitals and associated clinics have on‐site immediate access to effective adverse reaction treatments. However, tisagenlecleucel status as the only FDA‐approved cell therapy available was short lived. Six weeks after tisagenlecleucel approval, YESCARTA (axicabtagene‐ciloleucel), a CAR‐T cell therapeutic from competing Kite Pharma (Los Angeles, California, USA), was approved by the FDA for the treatment of refractory diffuse large B‐cell lymphoma after appropriate testing in clinical trials. Similar to its competitor, axicabtagene‐ciloleucel received “Orphan Drug,” “Breakthrough Therapy,” and “Priority Review” designations that allowed for accelerated testing, review, and approval. Although these medications were approved independently for two different diseases and patient populations, both followed nearly identical regulatory pathways. Therefore, these two therapies may have set the standard and path to follow for developing gene and cell therapies.
Application and feasibility
Despite clinical success, FDA approval achievement, and overwhelming positive response internationally from physicians and the public, CAR‐T cell therapies still face many challenges in clinical practice. Due to their complex nature, CAR‐T cell therapies are only offered at institutions capable of providing the equipment, resources, and clinical expertise needed to successfully administer the therapy. Furthermore, this exclusivity may have played a role in CAR‐T cell therapy cost, which can be as high as $475,000 and $373,000 for a one‐time treatment of tisagenlecleucel or axicabtagene‐ciloleucel, respectively. Therefore, the combination of limited offering centers and excessively high costs may prevent most patients from receiving this therapy.
Lessons learned
The translational history of CAR‐T cell therapy can provide important lessons to help future researchers navigate the burgeoning regulatory environment for more than minimally manipulated cell‐based therapies. First, researchers need to be aware of regulatory bodies that oversee biologic therapies and show compliance with requirements. Second, investigators working on therapies intended for serious conditions with no available effective treatments should be aware of the various pathways provided by the FDA designed to help accelerate translation of such novel therapeutics. This is especially evident in the case of tisagenlecleucel and axicabtagene‐ciloleucel, which were able to take advantage of three of the four expedited programs. It is hoped that lessons learned from CAR‐T cell therapies will smooth the clinical translation of new gene and cell therapies in the dynamic regulatory environment surrounding these treatments. Last, developing gene and cell therapies must be mindful of circumstances that may limit the number of patients who can ultimately benefit from innovative technologies, such as patient's access to and potential costs associated with said therapies that may limit the number of patients who can ultimately benefit from them.
Funding
This publication was made possible by CTSA grant number UL1 TR002380 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Conflict of Interest
S.S.K. is an inventor on several patents in the field of CAR‐T cell immunotherapy, with licenses to Novartis (managed through an agreement between Mayo Clinic, University of Pennsylvania, and Novartis).
References
- 1. Siegel, R.L. , Miller, K.D , Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Gross, G. , Waks, T. & Eshhar, Z. Expression of immunoglobulin‐T‐cell receptor chimeric molecules as functional receptors with antibody‐type specificity. Proc. Natl. Acad. Sci. USA 86, 10024–10028 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brentjens, R.J. et al Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 13, 5426–5435 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Milone, M.C. et al Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 17, 1453–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Porter, D.L. , Levine, B.L. , Kalos, M. , Bagg, A. & June, C.H. Chimeric antigen receptor‐modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 365, 725–733 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kalos, M. et al T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 3, 95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Porter, D.L et al Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 7, 303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maude, S.L. et al Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maude, S.L. et al Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]