

Abstract
Apixaban is metabolized by cytochrome P450 (CYP) 3A4 in the liver and intestine, undergoes direct intestinal excretion, and is a substrate to permeability glycoprotein (P‐gp) and breast cancer resistance protein (BCRP) transporters. We examined the drug interactions between cyclosporine and tacrolimus (combined inhibitors of CYP3A4, P‐gp, and BCRP) with apixaban in 12 healthy adult male volunteers. Apixaban 10 mg was administered orally alone, in combination with 100 mg cyclosporine or 5 mg tacrolimus. Co‐administration with cyclosporine resulted in increase in apixaban maximum plasma concentration (C max) and area under the plasma concentration‐time curve from time zero to the last quantifiable concentration (AUC (0‐tlast)) with associated geometric mean ratios (GMRs) and 90% confidence intervals (CIs) of 143% (112, 183) and 120% (97, 148), respectively. Co‐administration with tacrolimus resulted in reduction in apixaban C max and AUC (0‐tlast) with associated GMRs (90% CI) of 87% (69, 112) and 78% (63, 97), respectively. The observed changes in apixaban exposure margins with cyclosporine or tacrolimus are within the range of the historical clinical development program, therefore, apixaban dose adjustments are not warranted.
Study Highlights.
What is the current knowledge on the topic?
Apixaban is a commonly used anticoagulant that undergoes metabolism by CYP3A4 and direct intestinal excretion by P‐gp and BCRP transporters. Apixaban exposure doubles in the presence of a strong dual inhibitor of CYP3A4 and P‐gp that requires dose reduction or avoidance. Solid organ transplant recipients commonly require anticoagulation and are universally maintained on calcineurin inhibitors (CNIs), which are weak CYP3A4 as well as potent P‐gp and BCRP inhibitors.
What question did this study address?
The study sought to evaluate the DDI between apixaban and both CNIs, cyclosporine and tacrolimus.
What does this study add to our knowledge?
The results indicate that both cyclosporine and tacrolimus caused a small change in apixaban exposure. Overall, the change in apixaban exposure is not clinically relevant.
How might this change clinical pharmacology or translational science?
This phase I study provides evidence of no significant DDI between apixaban and CNIs. Based upon clinically accepted apixaban exposure margins, no dose adjustment is suggested for patients co‐administered CNIs.
The use of organ transplantation expanded after the approval of the calcineurin inhibitors (CNIs) cyclosporine and tacrolimus.1, 2 With improved survival and increased life expectancy, many solid organ transplantation recipients develop venous thromboembolism (VTE) and nonvalvular atrial fibrillation (NVAF). There is an incidence of 7–9.1% of first VTE event in renal transplant recipients, with a much higher rate of recurrence after stopping anticoagulation than matched controls without renal disease.3, 4, 5, 6, 7 VTE incidence is similarly elevated in liver, lung, and heart transplant recipients.8, 9, 10, 11 Similarly, NVAF is common after solid organ transplantation, with a cumulative incidence of 3.6–7.3% at 12 and 36 months post‐kidney transplant.12 The incidence of NVAF after lung transplantation is 33–39%13 and between 0.3% and 24% following heart transplant.14
Apixaban is a direct oral anticoagulant (DOAC) that inhibits factor Xa.15 The use of DOACs, including apixaban, has substantially increased due to ease of administration, lack of coagulation monitoring, and fixed dosage schedule in contrast to warfarin.16 Like warfarin, however, the concern remains for increased risk of bleeding with increased drug exposure, particularly in certain clinical scenarios. Special patient populations, such as solid organ transplantation recipients, are often excluded from participation in the initial drug approval process. The routine use of CNIs in transplant recipients provides concern for potential drug‐drug interaction (DDI) with the use of apixaban.
Apixaban is metabolized by the cytochrome P450 (CYP)3A4 enzyme in the human intestine and liver with minor contributions from 1A2, 2C8, 2C9, 2C19, and 2J2.17, 18 In addition, apixaban undergoes direct intestinal excretion and is a substrate to both permeability glycoprotein (P‐gp) and breast cancer resistance protein (BCRP) transporters.18, 19 Apixaban undergoes active transport with efflux ratios (ERs) in LLC‐PK1‐P‐gp cell monolayers of 23–38 compared with 1.4–4.4 in control cells. Efflux is extensively inhibited by ketoconazole (a potent inhibitor of P‐gp and CYP3A4).19 Similarly, apixaban is actively transported in BCRP‐cDNA‐transfected cells with ERs of 8–12 compared with ERs of 1.4–2.4 in control cells.19 In humans, apixaban co‐administration with ketoconazole results in a twofold increase in apixaban area under the plasma concentration‐time curve (AUC) and a 1.6‐fold increase in maximum plasma concentration (C max). This change drives a suggestion to reduce the dose by 50% or avoiding co‐administration in the US package insert, whereas the co‐administration is not recommended in the European label.18, 20 Conversely, administration with diltiazem (moderate inhibitor of CYP3A4 alone) results in a 1.4‐fold increase in AUC and 1.3‐fold increase in C max that does not require dose reduction.18, 20
Cyclosporine and tacrolimus are potent immunosuppressive agents approved for prevention of organ rejection as well as for treatment of severe rheumatoid arthritis and plaque psoriasis.21, 22 Both are extensively metabolized by the CYP3A4/5 in the liver, and to a lesser extent in the gastrointestinal tract and the kidneys.21, 22, 23 Cyclosporine is itself a weak inhibitor of CYP3A4, as well as a potent inhibitor of both P‐gp and BCRP.21, 23, 24 Although both cyclosporine and tacrolimus demonstrate in vitro CYP3A4‐mediated inhibition of midazolam metabolism, in vitro in vivo extrapolations suggest that the interaction is only clinically relevant for cyclosporine.25 Simeprevir is a combined substrate of CYP3A4, P‐gp, BCRP, and organic anion‐transporting polypeptide 1B1 and 1B3.26 Cyclosporine caused a 4.7‐fold and 5.8‐fold increase in C max and AUC(0‐24) of simeprevir, whereas tacrolimus caused a 79% and 85% increase in Cmax and AUC(0‐24) of simeprevir.26 Systemic exposure to atorvastatin (CYP3A4, P‐gp, and organic anion‐transporting polypeptide 1B1 substrate) and its metabolites was increased 15‐fold with cyclosporine, whereas tacrolimus had no impact on its pharmacokinetics (PKs).27 Co‐administration of cyclosporine with colchicine (CYP3A4, P‐gp substrate) increased colchicine C max by 224% and AUC(0–∞) by 215%, indicating a clinically important DDI.28 On the whole, this suggests that the potential for drug interactions are similar for cyclosporine and tacrolimus, but do not fully overlap.
We hypothesized that cyclosporine would alter apixaban exposure due to combined inhibition of CYP3A4, P‐gp, and BCRP. Tacrolimus is often considered to share overlapping inhibitory activity on these pathways based upon a review of known clinical drug interactions of cyclosporine and tacrolimus.29 Because tacrolimus is more frequently prescribed (>90%) in transplant recipients,30, 31 we also aimed to investigate its impact on apixaban exposure.
Methods
Patients and study design
This was a phase I, investigator‐initiated, open‐label, randomized, two‐sequence, three‐period, single‐site, crossover trial performed in healthy volunteers at the Clinical Research Unit of Thomas Jefferson University Hospital (Philadelphia, PA). There was 1:1 randomization to one of the two treatment sequences (Figure 1). Nonsmoking men and women aged 18–55 years with a body mass index of 19–33 kg/m2 were allowed to participate in this study. The main exclusion criteria included history of venous or arterial thromboembolic disease, major bleeding event, cardiac, gastrointestinal, hepatic, or renal disease. Subjects who underwent major surgery within 6 months before starting study treatment were excluded. The study was approved by the Institutional Review Board of Thomas Jefferson University and registered with Clinicaltrials.gov (NCT 03083782). The study was conducted in accordance with the applicable regulatory requirements and International Conference on Harmonization Good Clinical Practice. All subjects provided written informed consent prior to initiation of study‐specific procedures.
Figure 1.
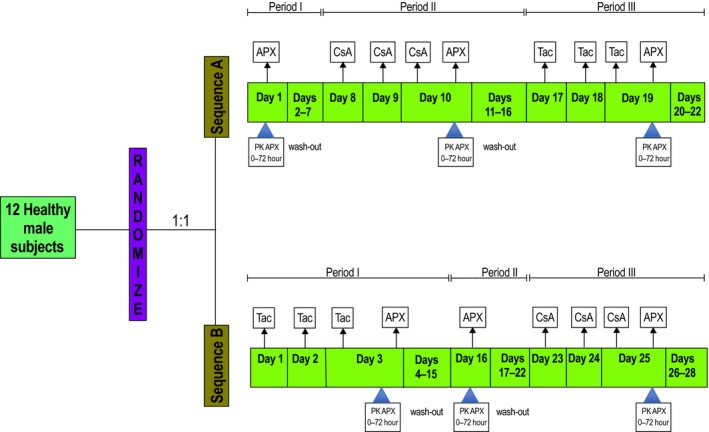
Study design and treatment schedules. Apixaban (APX) was administered as a single 10 mg oral dose in three treatment periods in both sequences, alone; with 100 mg oral cyclosporine (CsA) administered daily for 3 days; with 5 mg oral tacrolimus (Tac) administered daily for 3 days. APX pharmacokinetic (PK) blood samples were collected predose through 72 hours after each administration. CsA and Tac trough concentrations were measured before their respective third dose.
Study treatments
The study design and trial treatments are summarized in Figure 1. All subjects were admitted to the Clinical Research Unit 12 hours before receipt of apixaban (Eliquis, Bristol‐Myers Squibb Company, Princeton, NJ, and Pfizer, New York, NY) in all treatment periods and remained under direct observation until 24 hours after study procedures were complete. The remainder of study procedures was performed on an outpatient basis. All treatments were orally administered, and subjects received 10 mg dose of apixaban in all treatment periods. Subjects were required to fast (except for water) from 10 hours before until 2 hours after treatment administration. Subjects were also required to refrain from concomitant medications that affect CYP enzymes. Subjects were not permitted to consume grapefruit‐containing products, caffeine, or ethanol during the study treatments. All treatment administrations were performed under direct observation, including a mouth check to confirm swallowing of each dose.
Six subjects randomized to sequence A received apixaban alone in period I. The subjects then received cyclosporine 100 mg (Neoral soft gelatin capsules, Novartis Pharmaceuticals, East Hanover, NJ) once daily for 3 days followed by another dose of apixaban in period II. To achieve effective immunosuppression in transplant patients, the mean initial cyclosporine dose ranges from 7–9 mg/kg per day in two divided doses and is adjusted based upon plasma concentration. Our trial selection of 100 mg dose was based upon precedent drug interaction studies that demonstrated PK interaction with acceptable safety profile and is the highest strength capsule dosage form available.21, 27, 28 In period III, subjects received 5 mg tacrolimus (Prograf; Astellas Pharma US, Northbrook, IL) once daily followed by 10 mg apixaban. We selected the 5 mg dose of tacrolimus because it is the highest strength capsule dosage form available and is commonly used in clinical drug interaction studies.22, 27 Subjects randomized to sequence B received tacrolimus 5 mg once daily for 3 days followed by apixaban in period I. In treatment period II, subjects received 10 mg apixaban alone. The subjects then received cyclosporine 100 mg once daily for 3 days followed by apixaban. There was a washout period of at least 7 days between all treatment periods except period I in sequence B, in which washout was extended to 13 days.
Sample collection
Blood samples for apixaban PK analysis were collected immediately predose and at 1, 2, 3, 4, 6, 12, 24, 48, and 72 hours after apixaban administration in each treatment period. Blood samples for cyclosporine and tacrolimus trough concentrations were collected immediately predose on day 3 of respective treatment periods in both sequences.
Sample analysis
Following collection, blood for clinical laboratory safety values, cyclosporine, and tacrolimus concentrations was processed by the Jefferson Hospital CLIA/CAP certified clinical laboratory. Cyclosporine and tacrolimus plasma concentrations were determined by validated liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) assays. Whole blood for apixaban PK analysis was collected into 4 mL dipotassium EDTA tubes and immediately subjected to plasma separation by centrifugation at 1,000 g for 15 minutes. Once separated, plasma was stored at –20°C until ready for processing. Samples were analyzed in batches at the end of each treatment period. The plasma concentration of apixaban was determined using a validated LC‐MS/MS assay (AB Sciex API 3200MD).32 The method is based on existing literature for direct oral anticoagulant measurement by LC‐MS/MS, using commercial calibrators (Hyphen Biomed) with d4‐rivaroxaban as an internal standard (Santa Cruz Biotechnology, Dallas, TX). The calibration curve in plasma was linear over the range of 6.0–600 ng/mL. The between‐run precision for all levels of quality control samples was below 10% coefficient of variation; accuracy/recovery centered on 100%. No analytical interferences or ion suppression effects were observed in the assays.
Pharmacokinetic analysis
Single‐dose PK parameters for apixaban were determined based on plasma concentrations over time. The analyses were performed in R version 3.3 (Vienna, Austria) with noncompartmental analysis by the PKNCA package (version 0.8.1). The area under the plasma concentration‐time curve from time zero to the last quantifiable concentration (AUC(0‐tlast)) and extrapolated to infinity (AUC(0‐∞)) were calculated using the linear up/log down trapezoidal method. The maximum observed plasma concentration (C max) and the time required to achieve the C max (T max) were directly determined from the apixaban concentration data. The terminal half‐life (t 1/2) was estimated as log(ln)2/λz, in which the slope of the terminal phase of the plasma concentration‐time curve (λz) was determined by the least squares method (log linear regression of at least three data points) with a weighting factor of 1. The oral clearance (CL/F, where F is oral bioavailability) was calculated by dividing the dose by AUC(0‐∞). The apparent volume of distribution (Vd/F) was calculated by dividing the dose by λz*AUC(0‐∞).
Safety determinations
History, vital signs, physical examination, and 12‐lead electrocardiograms were performed at screening, baseline, discharge, and post‐treatment visits. Adverse event (AE) data were collected from subjects and from investigator's review of clinical data.
Statistical analysis
The study sample size was determined a priori by using previously reported DDI for apixaban and diltiazem.20 Twelve subjects were anticipated to provide 98% power to detect a mean difference of 0.329 for AUC(0–tlast) and 85% power to detect a mean difference of 0.262 for C max between apixaban alone and apixaban with cyclosporine using a one‐sided t‐test with a significance (alpha) level of 0.05. Statistical analyses were carried out using SAS/STAT version 9.4 (SAS Institute, Cary, NC). The log‐transformed C max, AUC(0–tlast), and AUC(0–∞) were analyzed in separate linear mixed effects models adjusting for the random effect of subject and for the fixed effect of the sequence (sequence A vs. B). With the chosen design, the period effect was confounded with treatment effects, and, therefore, was not included in the model. The fitted models were used to compute geometric least square means for each treatment (apixaban alone, apixaban with cyclosporine, and apixaban with tacrolimus) and 90% confidence intervals (CIs) for the geometric least square mean ratios (GMRs) corresponding to testing the treatment differences. Kenward and Rodger33 estimated denominator degrees of freedom were used for computing the CIs and model‐based type 3 tests of the fixed effects. The marginal residuals from all fitted linear mixed effects models were evaluated for adequacy of the normal distribution assumptions. The GMRs and 90% CIs of C max, AUC(0–tlast), and AUC(0–∞) between the test and reference treatments were estimated and the generally accepted limits of equivalence (80–125%) were used as the criteria to detect differences between treatments. We used one‐way analysis of variance model on t 1/2, Vd/F, and CL/F data to estimate statistically significant differences using a significance level of <0.05. The P values were corrected for multiple comparisons using the Dunnett test.
Results
All 12 enrolled subjects completed the study. The demographic characteristics of subjects are outlined in Table 1. Mean apixaban plasma concentration‐time profiles with and without cyclosporine are shown in Figure 2, and summary PK parameters are outlined in Table 2. In the presence of cyclosporine, the GMR (90% CI) for apixaban C max, AUC(0–tlast), and AUC(0–∞) were 143% (112, 183), 120% (97, 148), and 119% (99, 144), respectively. The 90% CI for only C max, but not AUC(0–tlast) or AUC(0–∞), lay completely above 1. The mean CL/F of apixaban decreased from 5.8 to 4.6 L/h (P = 0.2), whereas the mean Vd/F decreased from 89 to 45 L (P = 0.002). The mean t 1/2 of apixaban decreased from 12.1 to 6.8 hours (P = 0.03) in the presence of cyclosporine. The mean (SD) cyclosporine trough concentration was 22.4 (3.9) ng/mL, whereas clinically accepted therapeutic blood levels range between 100 and 400 ng/mL.
Table 1.
Subject demographics
| No. of subjects (n = 12) | n (%) |
|---|---|
| Male | 12 (100.0) |
| Age (years) | 41 |
| Range | 25–54 |
| BMI (Kg/M2), range | 24–33 |
| Race | |
| Black or African American | 9 (75) |
| White | 3 (25) |
BMI, body mass index.
Figure 2.
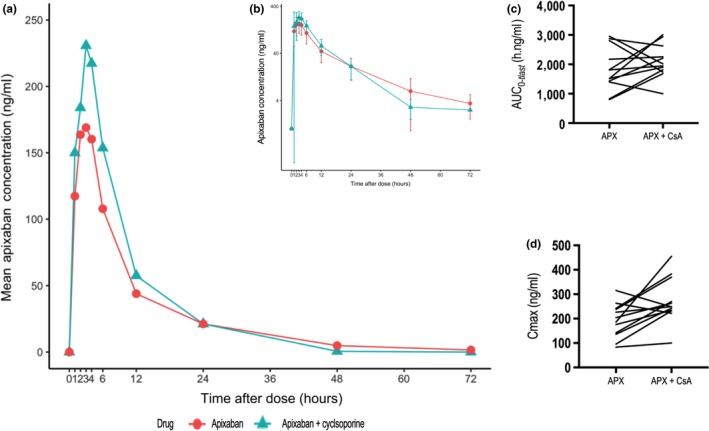
Plasma concentration‐time profiles and pharmacokinetic parameters of apixaban (APX) with and without cyclosporine (CsA). Mean plasma concentration‐time profiles of apixaban in 12 healthy subjects following a single 10 mg oral dose of apixaban alone or in the presence of 3 daily doses of 100 mg cyclosporine; apixaban plasma concentration is presented on a linear scale (a) and log‐transformed scale (b), error bars show SD; comparison of area under the plasma concentration‐time curve from time zero to the last quantifiable concentration (AUC (0–tlast)) (c) and maximum plasma concentration (C max) (d) with and without cyclosporine.
Table 2.
Summary pharmacokinetic parameters of apixaban
| Cyclosporine co‐administration (n = 12) | Tacrolimus co‐administration (n = 12) | ||||
|---|---|---|---|---|---|
| Pharmacokinetic parameter | Apixaban | Apixaban + cyclosporine | Point estimate of GMR (90% CI) | Apixaban + tacrolimus | Point estimate of GMR (90% CI) |
| C max ng ml‐1 | 179 [147, 219] | 257 [211, 313] | 1.43 (1.12, 1.83) | 157 [129, 191] | 0.87 (0.69, 1.12) |
| AUC(0–tlast) h ng ml−1 | 1684 [1427, 1987] | 2018 [1710, 2381] | 1.20 (0.97, 1.48) | 1318 [1117, 1555] | 0.78 (0.63, 0.97) |
| AUC(0–∞) h ng ml−1 | 1875 [1619, 2172] | 2237 [1931, 2591] | 1.19 (0.99, 1.44) | 1448 [1251, 1678] | 0.77 (0.64, 0.93) |
| T max (h) | 2.5 [1, 4] | 2.5 [1, 4] | ‐ | 2.5 [1, 4] | ‐ |
| t 1/2 (h) | 12.1 (7) [5, 23] | 6.8 (3.5) [4, 17] | ‐ | 7.0 (1.9) [5, 11] | ‐ |
| CL/F (L/h) | 5.8 (2.7) [3.1, 11.5] | 4.6 (0.9) [3.1, 5.8] | 7.3 (2.4) [4.3, 12.2] | ||
| Vd/F (L) | 89 (48) [45, 187] | 45 (32) [25, 144] | 72 (27) [31, 122] | ||
AUC(0‐tlast), area under the plasma concentration‐time curve from time zero to the last quantifiable concentration; AUC(0‐∞), area under the plasma concentration‐time curve from time zero extrapolated to infinity; CI, confidence interval; CL/F, apparent oral clearance (F is oral bioavailability) based on dose divided by AUC(0‐∞); C max, observed peak plasma concentration; GMR, geometric least square mean ratio; t 1/2, terminal elimination half‐life; T max, time taken to reach C max; Vd/F, apparent volume of distribution based on dose divided by the product of terminal elimination rate constant and AUC(0‐∞).Geometric least square mean [90% confidence intervals] for C max, AUC(0‐tlast), and AUC(0‐∞). Median [minimum, maximum] for T max. Arithmetic mean (SD) [minimum, maximum] for t 1/2, CL/F and Vd/F.
AUC is imputed by linear up/log down trapezoidal method.
Mean apixaban plasma concentration‐time profiles with and without tacrolimus are shown in Figure 3, and summary PK parameters are outlined in Table 2. In the presence of tacrolimus, the GMR (90% CI) for apixaban C max, AUC(0–tlast), and AUC(0–∞) were 87% (69, 112), 78% (63, 97), and 77% (64, 93), respectively. The 90% CI for both AUC(0–tlast) and AUC(0–∞), but not C max, lay completely below 1. Tacrolimus co‐administration increased the mean CL/F of apixaban from 5.8 to 7.3 L/h (P = 0.06) and the mean Vd/F decreased from 89 to 72 L (P = 0.4). The mean terminal t 1/2 of apixaban decreased from 12.1 to 7.0 hours (P = 0.04) in the presence of tacrolimus. The mean (SD) tacrolimus trough concentration was 3.8 (2.3) ng/mL, whereas clinically accepted therapeutic blood levels range between 5 and 8 ng/mL.
Figure 3.
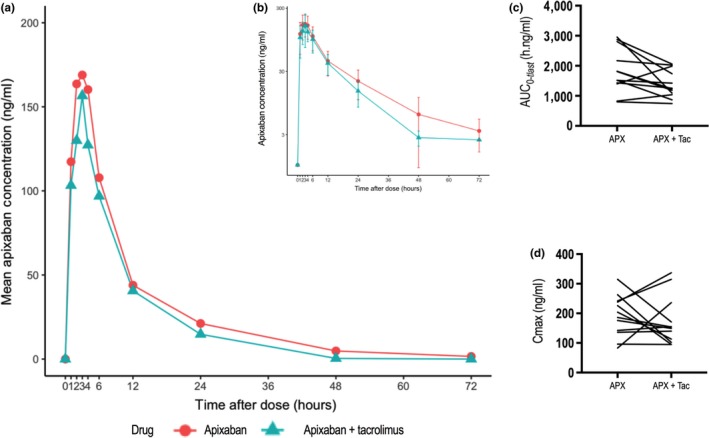
Plasma concentration‐time profiles and pharmacokinetic parameters of apixaban with and without tacrolimus. Mean plasma concentration‐time profiles of apixaban in 12 healthy subjects following a single 10 mg oral dose of apixaban alone or in the presence of 3 daily doses of 5 mg tacrolimus; apixaban plasma concentration is presented on a linear scale (a) and log‐transformed scale (b), error bars show SD; comparison of area under the plasma concentration‐time curve from time zero to the last quantifiable concentration (AUC (0–tlast)) (c) and maximum plasma concentration (C max) (d) with and without tacrolimus.
There were no serious SEs. All AEs were determined to be not related to study interventions. All events were considered mild and resolved without treatment. Overall, 50% of subjects experienced a total of 17 treatment‐emergent AEs and the most commonly encountered AE was headache (3 subjects).
Discussion
Traditionally, warfarin has been the mainstay of treatment of VTE and risk reduction of systemic embolism in NVAF. It is well‐established that the time‐in‐therapeutic range (TTR) for warfarin, as determined by international normalized ratio (INR), is the primary determinant of clinical outcome. The relative risk of recurrent VTE has been reported as 4.5 (95% CI: 3.1–6.6) when INR is subtherapeutic (<2.0).34 On the other hand, the relative risk of major bleed is 6.4 (95% CI: 2.5–16.1) when INR > 5.0.34 Because of multiple food‐drug interactions and intersubject variability, the TTR for warfarin varies considerably. Pokorney et al.35 reported that patients in US clinics had an overall mean and median TTR of 65% and 68% on warfarin, respectively. The use of warfarin, therefore, in transplant recipients is fraught with complications. DOACs, including apixaban, offer an attractive alternative but the potential for drug interaction with calcineurin inhibitors has not been determined.
Our study data indicate that, in the absence of interacting drugs, the PK parameters of apixaban are consistent with previously published reports of apixaban disposition.20, 36 Our study report is the first to address the clinical DDI of apixaban with CNIs. Co‐administration of apixaban with cyclosporine or tacrolimus resulted in small changes in apixaban PK parameters. Cyclosporine increased apixaban exposure by 20%, whereas tacrolimus decreased apixaban exposure by 22%, neither of which is clinically meaningful.
With co‐administration of cyclosporine, the mean CL/F of apixaban remained unchanged, whereas the mean Vd/F decreased from 89 to 45 L. The mean t 1/2 of apixaban decreased from 12.1 to 6.8 hours in the presence of cyclosporine. This observed decrease in mean Vd/F of apixaban with cyclosporine most likely reflects inhibition of intestinal P‐gp and/or BCRP transporters leading to an increased bioavailability. Cyclosporine increased apixaban exposure but decreased its t 1/2. With co‐administration of tacrolimus, the change in mean apixaban CL/F and Vd/F did not reach statistical significance, indicating lack of an inhibitory effect on intestinal P‐gp and/or BCRP transporters resulting in unaltered bioavailability. The mean t 1/2 of apixaban decreased from 12.1 to 7.0 hours. Tacrolimus decreased apixaban exposure as well as t 1/2. The observed decrease in apixaban exposure with tacrolimus was unexpected. Our results do not provide a mechanistic basis to explain the observed decrease in apixaban exposure with tacrolimus, therefore, it warrants further investigation.
Both cyclosporine 100 mg and tacrolimus 5 mg were administered once daily for 3 days and the trough levels indicate that both drugs did not reach steady‐state concentration. Because of an absence of steady‐state concentrations, suboptimal inhibition of CYP3A4 is possible. However, the P‐gp and BCRP transporter‐mediated interaction is not expected to change because the transporter interaction occurs at the intestinal luminal interface, independent of serum concentrations. The PK interaction results should be interpreted as a single‐dose interaction; however, additional change in apixaban exposure is not expected with cyclosporine or tacrolimus at clinically relevant steady‐state concentrations.
The single oral dose administration of apixaban demonstrated a 20% increase in exposure when concomitantly administered with cyclosporine and a 22% decrease with tacrolimus. The magnitude of change in apixaban plasma exposure in our study is within the range for a historical clinical development program and does not warrant dose modification. The findings reflect changes in apixaban exposure in healthy volunteers with CNIs; however, we expect similar exposure profile in solid organ transplant patients. Based upon changes in apixaban exposure described in the product label for other concomitant medications, body size, and organ function, no further dose adjustment is suggested for patients comedicated with cyclosporine or tacrolimus.
Funding
The funds to conduct this investigator‐initiated research were provided by an ARISTA‐USA grant administered by Bristol‐Myers Squibb and Pfizer. Babar Bashir was supported by an National Institutes of Health (NIH) institutional training grant T32GM008562.
Conflict of Interest
This research was sponsored by an ARISTA‐USA grant administered by Bristol‐Myers Squibb and Pfizer. Sponsors had no role in the writing of this manuscript or in the decision to publish it.
Author Contributions
B.B., D.F.S., I.C., and W.K.K. wrote the manuscript. B.B. and W.K.K. designed the research. B.B. and W.K.K. performed the research. B.B. and I.C. analyzed the data. D.F.S. contributed new reagents/analytical tools.
Acknowledgments
The authors would like to thank all the subjects who participated in this study; the clinical research unit staff, particularly Angela Pallatto, RN, for coordinating the study; Santhi Mantravadi, MD, for evaluating the subjects for safety; Benjamin Duy Tran, PharmD, for technical assistance with the pharmacokinetic analysis; and Scott Waldman, MD, PhD, for serving as the study medical monitor.
References
- 1. Starzl, T.E. et al The use of cyclosporin A and prednisone in cadaver kidney transplantation. Surg. Gynecol. Obstet. 1, 17–26 (1980). [PMC free article] [PubMed] [Google Scholar]
- 2. Scalea, J.R. , Levi, S.T. , Ally, W. & Brayman, K.L. Tacrolimus for the prevention and treatment of rejection of solid organ transplants. Exp. Rev. Clin. Immunol. 3, 333–342 (2016). [DOI] [PubMed] [Google Scholar]
- 3. Zanazzi, M. et al Venous thromboembolism in renal transplant recipients: High rate of recurrence. Transpl. Proc.. 6, 2493–2494. [DOI] [PubMed] [Google Scholar]
- 4. Allen, R.D. , Michie, C.A. , Murie, J.A. & Morris, P.J. Deep venous thrombosis after renal transplantation. Surg. Gynecol. Obstet. 2, 137–142 (1987). [PubMed] [Google Scholar]
- 5. Arnadottir, M. et al Thromboembolic complications after renal transplantation: a retrospective analysis. World J. Surg. 6, 757–761 (1983). [DOI] [PubMed] [Google Scholar]
- 6. Poli, D. et al Renal transplant recipients are at high risk for both symptomatic and asymptomatic deep vein thrombosis. J. Thromb. Haemost. 5, 988–992 (2006). [DOI] [PubMed] [Google Scholar]
- 7. Poli, D. et al High rate of recurrence in renal transplant recipients after a first episode of venous thromboembolism. Transplantation 6, 789–793 (2005). [DOI] [PubMed] [Google Scholar]
- 8. Yip, J. et al Deep vein thrombosis and pulmonary embolism in liver transplant patients: risks and prevention. Transplant. Direct. 4, e68 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alexander, B.R. et al Chemoprophylaxis use and risk of venous thromboembolism and death in adult patients following orthotopic liver transplantation. J. Pharm. Pract. 3, 218–223 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Alvarez Alvarez, R.J. et al Venous thromboembolism in heart transplant patients: incidence, clinical characteristics and risk factors. J. Heart Lung Transplant.. 4, S135–S136 (2015). [Google Scholar]
- 11. Aboagye, J.K. et al Venous thromboembolism in patients hospitalized for lung transplantation. Ann. Thorac. Surg.. 4, 1071–1076 (2018). [DOI] [PubMed] [Google Scholar]
- 12. Lentine, K.L. et al Incidence, predictors, and associated outcomes of atrial fibrillation after kidney transplantation. Clin. J. Am. Soc. Nephrol. 2, 288–296 (2006). [DOI] [PubMed] [Google Scholar]
- 13. Orrego, C.M. et al Atrial arrhythmias after lung transplant: underlying mechanisms, risk factors, and prognosis. J. Heart Lung Transplant.. 7, 734–740 (2014). [DOI] [PubMed] [Google Scholar]
- 14. Hamon, D. , Taleski, J. , Vaseghi, M. , Shivkumar, K. & Boyle, N.G. Arrhythmias in the heart transplant patient. Arrhythm. Electrophysiol. Rev. 3, 149–155 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wong, P.C. et al Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J. Thromb. Haemost. 5, 820–829 (2008). [DOI] [PubMed] [Google Scholar]
- 16. Barnes, G.D. , Lucas, E. , Alexander, G.C. & Goldberger, Z.D . National trends in ambulatory oral anticoagulant use. Am. J. Med.. 12, 1300–1305 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang, L. et al In vitro assessment of metabolic drug‐drug interaction potential of apixaban through cytochrome P450 phenotyping, inhibition, and induction studies. Drug Metab. Dispos. 3, 448–458 (2010). [DOI] [PubMed] [Google Scholar]
- 18. Apixaban (ELIQUIS®) tablets for oral use. U.S. Full prescribing information. (Bristol‐Myers Squibb Company, Princeton, NJ, and Pfizer Inc. New York, NY (2016) August 10, 2016.
- 19. Zhang, D. et al Characterization of efflux transporters involved in distribution and disposition of apixaban. Drug Metab. Dispos. 4, 827–835 (2013). [DOI] [PubMed] [Google Scholar]
- 20. Frost, C.E. et al Effect of ketoconazole and diltiazem on the pharmacokinetics of apixaban, an oral direct factor Xa inhibitor. Br. J. Clin. Pharmacol. 5, 838–846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cyclosporine (NEORAL®) soft gelatin capsules for oral use: U.S. Full prescribing information. (Novartis Pharmaceuticals Corporation, East Hanover, NJ. 2015).
- 22. Tacrolimus (PROGRAF®) capsules for oral use: U.S. full prescribing information. (Astellas Pharma US, Inc. Northbrook, IL. 2015); August 20, 2016.
- 23. Barbarino, J.M. , Staatz, C.E. , Venkataramanan, R. , Klein, T.E. & Altman, R.B. PharmGKB summary: cyclosporine and tacrolimus pathways. Pharmacogenet. Genomics 10, 563–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. The International Transporter Consortium, et al. Membrane transporters in drug development. Nat. Rev. Drug Discovery 3, 215–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amundsen, R. , Åsberg, A. , Ohm, I.K. & Christensen, H. Cyclosporine A‐ and tacrolimus‐mediated inhibition of CYP3A4 and CYP3A5 in vitro. Drug Metab. Dispos. 4, 655–661 (2012). [DOI] [PubMed] [Google Scholar]
- 26. Ouwerkerk‐Mahadevan, S. , Snoeys, J. , Peeters, M. , Beumont‐Mauviel, M. & Simion, A. Drug‐drug interactions with the NS3/4A protease inhibitor simeprevir. Clin. Pharmacokinet. 2, 197–208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lemahieu, W.P.D. et al Combined therapy with atorvastatin and calcineurin inhibitors: no interactions with tacrolimus. Am. J. Transplant. 9, 2236–2243 (2005). [DOI] [PubMed] [Google Scholar]
- 28. Wason, S. , DiGiacinto, J.L. & Davis, M.W. Effect of cyclosporine on the pharmacokinetics of colchicine in healthy subjects. Postgrad. Med. 4, 189–196 (2012). [DOI] [PubMed] [Google Scholar]
- 29. Christians, U. , Jacobsen, W. , Benet, L.Z. & Lampen, A. Mechanisms of clinically relevant drug interactions associated with tacrolimus. Clin. Pharmacokinet. 11, 813–851 (2002). [DOI] [PubMed] [Google Scholar]
- 30. Hart, A. et al Kidney. Am. J. Transplant. S2, 11–46 (2016). [Google Scholar]
- 31. Ekberg, H. et al Reduced exposure to calcineurin inhibitors in renal transplantation. N. Engl. J. Med. 25, 2562–2575 (2007). [DOI] [PubMed] [Google Scholar]
- 32. Korn, W.R. , Paziuk, T.M. & Stickle, D.F. Measurement of plasma Apixaban by LC‐MS/MS using the AB Sciex API 3200MD. Clin. Chem. 62, S214 (2016). [Google Scholar]
- 33. Kenward, M.G. & Roger, J.H. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics 3, 983–997 (1997). [PubMed] [Google Scholar]
- 34. Veeger, N.J.G.M. , Piersma‐Wichers, M. , Tijssen, J.G.P. , Hillege, H.L. & Meer, J. Individual time within target range in patients treated with vitamin K antagonists: main determinant of quality of anticoagulation and predictor of clinical outcome. A retrospective study of 2300 consecutive patients with venous thromboembolism. Br. J. Haematol. 4, 513–519 (2005). [DOI] [PubMed] [Google Scholar]
- 35. Pokorney, S.D. et al Patients’ time in therapeutic range on warfarin among US patients with atrial fibrillation: results from ORBIT‐AF registry. Am. Heart J.. 1, 141–148 (2015). [DOI] [PubMed] [Google Scholar]
- 36. Frost, C. et al Apixaban, an oral, direct factor Xa inhibitor: single dose safety, pharmacokinetics, pharmacodynamics and food effect in healthy subjects. Br. J. Clin. Pharmacol. 2, 476–487 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]