

Abstract
The tutorial introduces the readers to the fundamentals of antibody pharmacokinetics (PK) in the context of drug development. Topics covered include an overview of antibody development, PK characteristics, and the application of antibody PK/pharmacodynamics (PD) in research and development decision‐making. We also discuss the general considerations for planning a nonclinical PK program and describe the types of PK studies that should be performed during early development of monoclonal antibodies.
BRIEF HISTORY OF ANTIBODY DEVELOPMENT
Endogenous antibodies are mainly produced by differentiated plasma B‐cells. They function to neutralize pathogens such as bacteria and viruses. As the largest class of biopharmaceuticals, therapeutic antibodies have been developed for the treatment of a broad range of diseases including cancer, immunological disorders, and infectious diseases.1, 2 Over 60 antibodies have been marketed in the United States, and ∼350 new antibody entities are in active clinical development.3 Of the five antibody immunoglobulin (Ig) subtypes (IgA, IgD, IgE, IgG, and IgM), the IgGs are the most abundant and most frequently explored as therapeutics.4
Development of monoclonal antibodies (mAbs) as therapeutics was initiated by the introduction of mouse hybridoma technology 40 years ago.5 However, the use of mouse mAbs as therapeutics was handicapped by their ubiquitous induction of antidrug antibodies (ADA), their short half‐life, and the lack of an effector function.6 Efforts to reduce immunogenicity led to the development of chimeric and humanized antibodies, which constitute the majority of marketed antibodies today.7 Moreover, phage‐display technology, which utilizes bacteriophage expressing recombinant human antigen‐binding fragments for high‐affinity binder selection, led to the development of fully human antibodies with enhanced diversity and potency.8 Human antibodies can also be generated from transgenic mice engineered with human immunoglobulin genes, human hybridomas, and patient‐derived lymphocytes.9
IgGs are Y‐shaped 150 kDa immunoglobulins consisting of two pairs of identical heavy and light chains linked by disulphide bonds10 (Figure 1). The two arms of the Y constitute the antigen‐binding region (Fab) and are formed by the variable domains from both the heavy and light chains (Fv). The selective binding of antibody to antigen through variable domains serves crucial pharmacological functions, such as blockage of cytokines and growth factors, as shown by antagonistic antibodies against the TNF family (e.g., infliximab)11 and receptor blockade and/or receptor modulation, as shown by antibodies against programmed death 1 (PD‐1) receptor (e.g., pembrolizumab).12 The stem region of the Y constitutes the so called fragment crystallizable region (Fc) and is composed of only the heavy chains. This region is responsible for mAb binding to Fc receptors for IgG and proteins of complement system, i.e., Fc gamma receptors (FcγRs), complement (C1q) protein, and neonatal FcR (FcRn).13 The main therapeutic functions of IgG are dictated by their interactions with several classes of binding partners: antigen, complement, Fc receptor for IgG (FcγRs), and the neonatal FcR (FcRn). Among them, the selective binding of antibody to antigen through variable domains serves crucial pharmacological functions, such as blockage of cytokines and growth factors, as shown by antagonistic antibodies against the TNF family (e.g., infliximab)12 and receptor blockade and/or receptor modulation, as shown by antibodies against programmed death 1 (PD‐1) receptor (e.g., pembrolizumab).13 Other functions of IgG are dependent on the interaction of the Fc region with other proteins. Binding of mAb Fc to FcγRs and complement protein leads to cellular depletion through both Fcγ‐mediated antibody‐dependent cytotoxicity (ADCC) and C1q‐mediated complement protein‐dependent cytotoxicity (CDC), as exemplified by anti‐CD20 antibodies14 and binding to FcRn leads to long half‐life of mAb in circulation. Similar to its biological functions, the pharmacokinetic (PK) characteristics of antibody are also driven by interactions with their binding partners (antigen, FcγRs, and FcRn).
Figure 1.
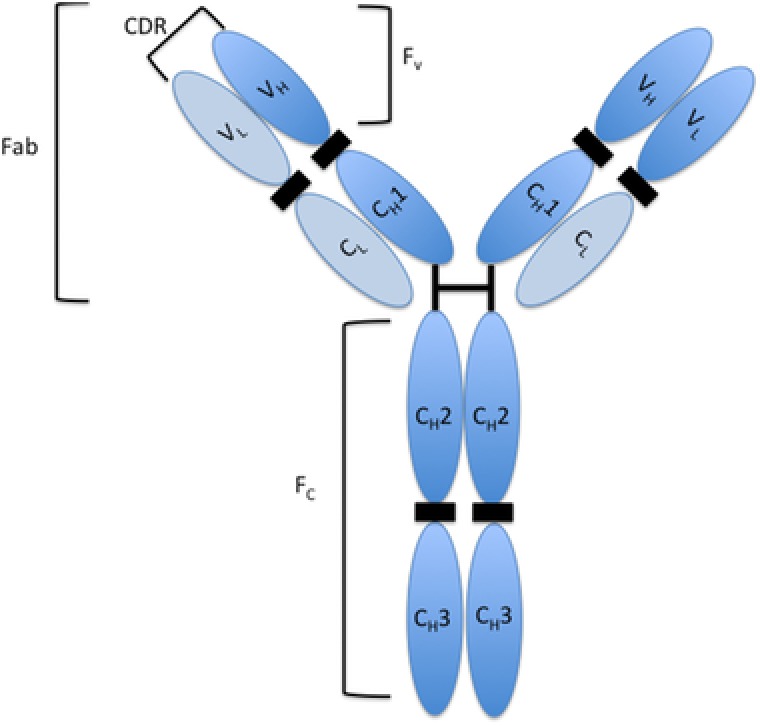
Schematic structure of IgG antibody: A simplified representation of IgG structure. VL = variable light chain; VH = variable heavy chain; CH1 = constant heavy chain domain 1; CH2 = constant heavy chain domain 2; CH3 = constant heavy chain domain 3; CDR = complementary determining region (responsible for specific antigen binding); Fab = fragment antigen‐binding (Fab); Fc = fragment crystallizable region; Fv = fragment variable.
mAb PHARMACOKINETICS
Typically, systemically administered mAbs exhibit biphasic PK profiles in circulation, i.e., a relatively fast distribution phase followed by a slower elimination phase. Other mAb‐specific PK characteristics include their confined distribution in vasculature and interstitial space because of their size and polarity, long half‐lives (∼11–30 days in humans, Table 3) from FcRn‐mediated recycling, and nonlinear PK due to target‐mediated clearance. A summary of the key features of mAb‐specific concepts and important PK parameters is presented in Table 1. While this tutorial focuses on mAb PK and its key determinants, a high‐level comparison of PK characteristics between small molecules and mAbs are shown in Table 2. A list of recently approved mAbs with their indications, dosing regimens, important PK parameters, and immunogenicity rates are presented in Table 3.
Table 3.
Summary of PK parameters for marketed mAb (2011–2017) as indicated in their labels
| mAb and company | MOA | Indication | Dosing regimen, bioavailability | Clearance+ (mL/day*) | Vss, (L) | Half‐life (Days) | Cmax, (μg/mL) & Tmax, | Immunogenicity (%)* |
|---|---|---|---|---|---|---|---|---|
|
PD‐1 blocker | Merkel cell carcinoma | i.v. infusion (60 minutes) 10 mg/kg every 2 weeks | 590 | 4.72 | 6.1 | 4.1 | |
|
CD20‐directed cytolytic antibody | Relapsing and primary progressive forms of multiple sclerosis | i.v. infusion 300 mg every 2 weeks for a month, then 600 mg every 6 months | 170 | 2.78 | 26 | 141‐ 212 | |
|
IL‐4Rα antagonist | Moderate‐to‐severe atopic dermatitis | Two 300 mg s.c. injections, followed by one 300 mg every other week, 64 % | 4.8 ± 1.3 |
|
|||
|
PD‐L1blocking antibody | Urothelial carcinoma | i.v. infusion (60 minutes) 10 mg/kg every 2 weeks | 342 | 5.6 | 17 | 3.3 | |
|
IL‐6 receptor antagonist | Rheumatoid arthritis | 200 mg s.c. injections every 2 weeks | 7.3 | 8‐10 |
|
9.8 | |
|
IL‐17RA antagonist | Moderate to severe plaque psoriasis | 210 mg s.c. injections at weeks 0, 1, 2 and followed every 2 weeks, 55 % | 3000 ± 3500 | 8.9±9.4 |
|
3 | |
|
PDGFR‐ α blocker | Soft tissue sarcoma |
|
560 | 7.7 | 11 | 3.5 | |
|
mAb against Clostridium difficile toxin B | Reduce recurrence of Clostridium difficile infection in 18+ years patients |
|
317 | 7.33 | 19 | 185 | 0 |
|
IL‐17 antagonist | Moderate to severe plaque psoriasis in adult candidates |
|
140 | 7.10‐ 8.60 | 22‐31 |
|
<1 |
|
GD2‐binding mAb | Pediatric neuroblastoma |
|
210 | 5.4 ± 1.5 | 10 | 11.5 ± 2.3 | 13‐18 |
|
PCSK9 inhibitor | Treatment of adults with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease |
|
40‐50 L/kg | 17‐20 | 3‐7 days | 4.8 | |
|
PCSK9 inhibitor mAb | heFH, Clinical atherosclerotic CVD, HoFH |
|
288 | 3.3 | 11‐17 |
|
0.1 |
|
IL‐5 antagonist mAb | Severe asthma patients with an eosinophilic phenotype |
|
280 | 3.6 | 16‐22 | 6 | |
|
CD38‐directed monoclonal antibody | Multiple Myeloma |
|
171.4 ± 95.3 | 4.7 ± 1.3 | 18 ± 9 | 915 ± 410 | Not reported due to Daratumumab interference in assay |
|
EGFR antagonist | Squamous non‐small cell lung cancer |
|
338.4 | 7.0 | 14 days | 4.1 | |
|
SLAMF7‐directed immunostimulant | Multiple myeloma |
|
5.8 – 17.5 (20 mg‐ 0.05 mg/kg doses) | 18.5 | |||
|
VEGFR2 antagonist | Gastric cancer or gastro‐esophageal junction adenocarcinoma |
|
7.4 | ||||
|
IL‐6 antagonist | Multicentric Castelman's disease (HIV negative and (HHV negative) |
|
230 | 4.5 | 20.6 | 332 ± 139 | 0.2 |
|
Integrin Receptor Antagonist | Adult UC & Adult Crohn's Disease |
|
157 | 5 | 25 | 4 (during treatment) & 13 (end of study) | |
|
PD‐1 blocking Ab | Unresectable or metastatic melanoma |
|
220 | 26 | 0 | ||
|
PD‐1 blocker Ab | Unresectable or metastatic melanoma | 3 mg/kg | 228 | 8 | 26.7 | 8.5 | |
|
TNF blocker | Moderate to severe RA & Active PsA & Active AS |
|
4.9‐6.7 mL/day/kg | 58 to 126 mL/kg | 14 |
|
4 |
|
IL‐6 receptor inhibitor | RA |
|
300 | 13 | 183 ± 85.6 | 2 | |
|
CD20‐directed cytolytic mAb | Chronic lymphocytic leukemia | i.v. infusion | 90 | 3.8 | 28.4 | 13 | |
|
Her2/neu receptor antagonist | Her2 + metastatic breast cancer patients |
|
240 | 18 | 2.8 | ||
|
Antibody that binds to the Protective Antigen of B. anthracis | Inhalation anthrax due to Bacillus anthracis |
|
1020.3 ± 140.6 | 0 | |||
|
B‐lymphocyte stimulator specific inhibitor | Systemic lupus erythematosus |
|
215 | 5.29 | 19.4 | 313 | 0.7 |
|
Human CTLA‐4 blocking antibody | Melanoma |
|
367.2 | 7.21 | 14.7 | 21.8 | 1.1 |
*Unless stated otherwise; + Clearance values describe the elimination rate observed in clinic at the approved dose. For some mAbs, multiple clearance values were reported. Different clearance values address nonlinear clearance at given doses. *Immunogenicity incidence rate is described as percentage value. The percentage value is the percentagewise ratio of the number of patients where immunogenicity was confirmed to the patients dosed in the trials.
i.v. = intravenous; s.c. = subcutaneous; IL = Interleukin; IL‐17RA = IL‐17 Receptor A
PSA = Psoriatic Arthritis; AS = Ankylosing Spondylitis; RA = Rheumatoid Arthritis; UC = Ulcerative Colitis; CTLA‐4 = cytotoxic T‐lymphocyte antigen 4; TNF = Tumor Necrosis factor; PD‐1= Programmed death receptor‐1; HoFH = homozygous familial hypercholesterolemia; HHV = human herpes virus 8; VEGFR2 = Vascular endothelial growth factor receptor 2; EGFR= Epidermal growth factor receptor; CVD = cardiovascular disease; PDGFR‐ α = Platelet derived growth factor receptor alpha; heFH = Heterozygous familial hypercholesterolemia; PCSK9 = Proprotein convertase subtilisin kexin type 9, SLAMF7= Self‐ligand receptor of the signaling lymphocytic activation molecule
Table 1.
Key mAb‐specific concepts and PK parameters
| Concepts/parameters | Determinants |
|---|---|
| Target mediated drug disposition (TMDD) | TMDD is the phenomenon when the interaction between the mAb and its target contributes significantly to the kinetics of mAb distribution and clearance. Key determinants are binding affinity of mAb, antigen density, turnover rate, internalization rate, and dose levels.33 |
| Specific or dose‐dependent clearance | As the result of TMDD, interactions between mAbs and their targets may lead to fast removal of mAbs from circulation at non‐saturable dose range. Rate and extent of dose‐dependent clearance depend on internalization rate, antigen density, binding affinity and turnover kinetics of antigen. |
| Nonspecific clearance | Nonspecific clearance of mAb refers to target independent, nonspecific cellular uptake of mAbs via pinocytosis/proteolysis and subsequent removal from circulation. Recently marketed mAbs exhibit clearance values between 90–560 mL/ day and half‐lives between 11–30 days (Table 3), comparable to endogenous IgG half‐life (∼21 days).31 |
| Volume of distribution | mAbs generally exhibit a low volume of distribution of 3–8 L at steady state reflecting the volume of vascular and interstitial spaces.23 |
| Immunogenicity and ADA | Immunogenicity refers to the immune response of the host against the therapeutic protein. These responses include anaphylaxis, cytokine release syndrome, and ADA formation, may affect exposure, efficacy and safety and should be closely monitored. ADA formation may contribute to accelerated mAb clearance. In some cases, higher ADA titers were associated with lower therapeutic trough concentrations.62 |
Table 2.
Comparison of PK characteristics between small molecules and monoclonal antibodies
| Features | Small Molecules | mAbs |
|---|---|---|
| Molecular weight | <500 Da preferred | ∼150 kDa |
| Drug substance | Chemical entities | Proteins |
| Administration routes | Oral administration feasible and preferred | i.v. or s.c. /i.m. |
| Absorption | Through passive diffusion/permeability and active transporters | Mainly through lymphatic uptake due to their large molecular size |
| Distribution | High volume of distribution to tissues, often exceed the biological volume except acidic chemical compounds. Volume of distribution of small molecules depend on plasma and tissue protein binding. | Distribution is mainly limited to vascular and interstitial fluids |
| Metabolism | Metabolized through CYP P450 enzymes followed by conjugation reactions with transferase enzymes | Metabolized /catabolized through proteolysis to small peptides and amino acids |
| Excretion | Biliary and renal | Recycled through FcRn receptor |
| Clearance and half‐life |
|
|
| Bioanalysis | LC‐MS/MS | Ligand based ELISA and recently LC‐MS/MS |
| Selectivity and toxicity | Off‐target toxicity is observed as well as on‐target toxicity | Highly specific, mostly on target toxicities or exaggerated pharmacology |
| Target | Both intracellular and surface targets | Membrane proteins or soluble proteins in circulation |
| Drug–drug interaction | Expected and need to be investigated for CYP P450 and transporter interactions | Rarely observed with some exceptions (e.g., mAbs modulating cytokine pathway may interact with CYP3A4‐mediated clearance of small molecule drugs)100 |
| Pharmacodynamics and PK/PD interactions | Short acting, in line with PK properties; PK is usually not affected by PD | Long acting PD effect and direct impact on PK; PK/PD are mechanistically linked |
| Immunogenicity | Not commonly observed | Expected and need to be monitored |
ABSORPTION AND ROUTES OF mAb ADMINISTRATION
mAbs have limited oral bioavailability (typically <1–2%)15 and are therefore not usually administered orally. The limited bioavailability of orally administered mAbs is due to their limited penetration across the intestinal epithelium16 and susceptibility to enzymatic degradation by proteases and peptidases in the intestinal lumen.17 The administration routes of choice for mAbs are intravenous (i.v.), subcutaneous (s.c.), and occasionally intramuscular (i.m.).
While the i.v. route may result in higher systemic exposures, s.c. administration is an established modality for almost all disease indications given its convenience and option of self‐administration.18 Since blood capillaries only allow substances with less than 16 kDa to reach systemic circulation, s.c. or i.m. administered mAbs presumably enter the circulation through lymphatic fluid drainage.19 The maximum plasma concentrations of mAbs are often reached around 3–7 days postdosing and bioavailability varies between 50–90% following s.c. dosing in humans (Table 3). Low circulating concentrations and/or variable bioavailability of mAbs can be attributed to: 1) physiological factors such as blood flow and skin morphology, etc. At the site of injection, 2) biological interactions (target expression, binding, degradation at the site of injection, etc.),20 and 3) molecule and product specific characteristics (molecule charge, glycosylation, formulation, and volume of the injection, etc.).
Non‐human primate (NHP) models are often used for early assessment of bioavailability for their physiological and biochemical proximity to humans.21 Rodents and mini pig have also been explored to further understand the mechanism and variables in mAbs absorption.22 Despite efforts in predicting mAbs bioavailability in humans using preclinical models, the translatability remains uncertain.20
DISTRIBUTION MECHANISMS OF mAbs
At steady state, mAbs generally exhibit a low volume of distribution of 3–8 L (Table 3), reflecting their confined distribution to the vascular and interstitial spaces because of their size and polarity.23 The primary mechanism of antibody diffusion in tissues is through the convective transport through paracellular pores in vascular endothelial cell membranes.19 Additional factors influencing mAbs distribution to individual organs or tissues include: drug‐specific features such as their binding affinity to specific target antigens, targets internalization rate,24 mAb hydrophilicity, charge,25 and tissue‐specific features such as membrane structure and blood flow.26 Optimizing these determinants to improve the distribution to relevant organs or tissues is currently being explored. For example, Rudnick et al. showed that when targeting high‐density and rapidly internalized antigens such as HER2, a lower‐affinity antibody could penetrate tumors more effectively, while an ultrahigh‐affinity antibody could limit the distribution of the antibody to tumor as a result of “binding site barrier.”27 Another effective utility of target‐specific binding is to deliver antibodies to “off‐limit” sites such as the central nervous system (CNS), which is discussed in a later section (see “Next‐generation antibody”).
It is worth noting that insights on mAbs distribution are often revealed through physiologically based PK modeling28, 29 integrated analytical tools including enzyme‐linked immunosorbent assay (ELISA), radioisotope quantification, imaging, and liquid chromatography‐mass spectrometry (LC‐MS)19 along with physiologically based PK modeling28, 29 using tissue specific exposure data.
METABOLISM/CATABOLISM AND CLEARANCE MECHANISMS OF mAbs
Elimination of mAbs through the kidney is considered insignificant, since typical mAb molecular weight (150 kDa) is higher than the glomerular filtration threshold (∼55 kDa).23 Instead, mAbs are mainly metabolized and eliminated through proteolytic degradation that results in smaller peptides and amino acids. Clearance pathways for the metabolism and elimination of therapeutic antibodies from circulation include nonspecific clearance through pinocytosis and proteolysis, target‐mediated specific clearance, and other mechanisms such as ADA‐mediated clearance (Figure 2).
Figure 2.
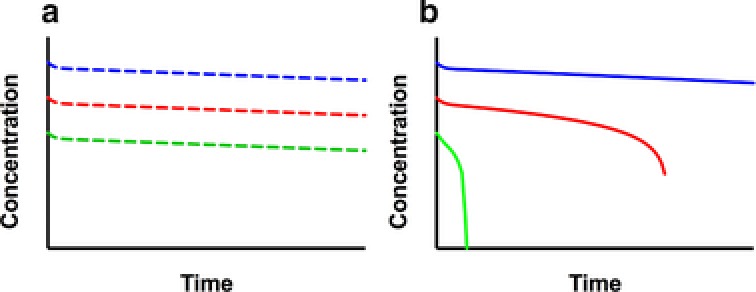
Representative PK profiles for linear and nonlinear clearance at the same doses. (a) The linear PK profiles with parallel elimination slopes. (b) The nonlinear PK profiles: the slope of the terminal phase decreases as the dose increases and approaches linear PK range as the dose reaches saturation dose level.
Nonspecific clearance
There are at least two distinct mechanisms that contribute to nonspecific clearance of mAbs: nonspecific endocytosis in cells, and proteolysis in liver and in reticuloendothelial system (RES). Both processes are employed as clearing mechanisms for endogenous antibodies, and provide the biological basis for a predictable range of clearance and half‐life of well‐behaved antibody therapeutics (Table 3). Nonspecific endocytosis refers to target‐independent, nonspecific uptake of mAbs by the cells via pinocytosis (e.g., fluid‐phase endocytosis), and subsequent removal of mAbs from the circulation. One key mediator of this process is the FcRn, which protects the internalized antibody from rapid intracellular catabolism.30 FcRn binds to the Fc region of the IgG in a pH‐dependent manner, i.e., the binding between IgG and FcRn is strong at acidic pH of 6.0–6.5 and weak at neutral pH of 7.0–7.5. When early endocytic vesicles are acidified, IgG‐FcRn binding is enhanced. The IgG‐FcRn complex is protected from lysosomal pathway and transported back to the cell membrane, where pH is neutral and the antibody is released back into the circulation. This recycling process is most likely responsible for the observed relatively long half‐life of mAbs of 11–30 days in humans (Table 3), which is similar to the half‐life of endogenous IgG (∼21 days).31 Nonspecific clearance is considered dose‐independent since most therapeutic antibodies at clinically relevant dose levels would fall far below the circulating endogenous IgG levels of ∼10 g/ml.23 Improved understanding of the IgG‐FcRn interaction along with advances in protein engineering has led to opportunities to engineer mAbs with extended half‐life.32
Nonspecific clearance of mAbs also occurs through proteolysis in the liver and the reticuloendothelial system (RES) located in reticular connective tissue.33 This elimination pathway of mAbs is mediated through binding of the Fc region of the antibody to FcγR‐expressing cells, such as Kupffer cells in liver, monocytes and/or macrophages of the RES, followed by receptor‐mediated endocytosis and degradation in lysosomes. The FcγR‐mediated elimination pathway is not saturable for mAbs therapeutics; therefore, FcγR binding alone is not expected to impact mAb PK when nonspecific clearance is dominant. However, it may have an impact on mAb PK through its pharmacodynamic (PD) interactions. For example, Fc‐mediated effector functions (ADCC or CDC) upon antibody–antigen interaction may contribute to observed clearance by depleting target cells, as shown for rituximab.34 The contribution of FcγR binding to mAb elimination is not fully understood and additional research is required to elucidate the mechanism.
Target‐mediated or specific clearance
Target‐mediated clearance refers to the elimination of mAbs through its antigen‐specific interactions. After mAbs bind to a specific antigen (soluble or membrane bound) they may be internalized and catabolized through lysosomal degradation as an antibody–antigen complex.35 Target‐mediated clearance is usually identified and characterized with dose‐ranging studies in both preclinical and clinical settings. Target‐mediated clearance decreases with the saturation of the target, which in turn is dependent on dose. At and above the saturation dose level, the target‐mediated clearance becomes insignificant, and the clearance of antibody is largely mediated through nonspecific the FcRn pathway (discussed in “Nonspecific clearance” section) (Figure 2).
Multiple factors including binding affinity of mAbs, antigen density, turnover rate, and internalization rate, etc., affect the extent and depth of targeted mediated clearance. Its contribution to overall clearance diminishes with increased dose and elapse of time because of the gradual depletion of available targets. For example, alemtuzumab, an antibody targeting CD52‐expressing tumor cells, showed time‐dependent PK with decreased clearance and extended half‐life as tumor burden decreases after repeated dosing.36
It is important to characterize target‐mediated clearance and its relationship with dose and efficacy since PK in the nonlinear range often demonstrates significant variability, which may lead to variability in efficacy and safety. For example, Gibbs et al. demonstrated the critical importance of characterizing target‐mediated clearance in selecting an optimal dosing regimen for protein convertase substilisin kexin type 9 (PCSK9) antibody evolucumab.37 Their target‐mediated drug disposition PK/PD modeling suggested that doubling the dose and extending the dosing interval from biweekly to monthly did not adequately maintain the reduction in low‐density lipoprotein cholesterol (LDL‐C), and a higher dose was required to maintain stable levels of LDL‐C levels for a monthly dosing schedule.
Many other factors, such as antibody properties (hydrophobicity, charge,38 glycosylation patterns39), intersubject variability (disease status, body size, genetic polymorphisms, concomitant medication, comorbidities, etc.40), and immune‐mediated response generated by mAbs administration can also impact mAb clearance. Among these, the physicochemical characteristics of mAbs and ADA formation from immune‐mediated responses can have a major impact on mAb clearance and efficacy/safety.
Impact of physicochemical characteristics of mAbs on their PK
Physiochemical properties including hydrated size (Stokes radius), charge, hydrophobicity, electrophoretic mobility, isoelectric point (pI), and glycosylation can potentially influence mAb PK.38, 39 Igawa et al. showed that lowering the total pI by 1–2 units resulted in longer half‐lives and slower elimination rates.41 Yadav et al. also demonstrated that modifying Fv charge and hydrophobicity altered mAb PK and s.c. bioavailability42: more positive‐charged antibody variants showed faster nonspecific clearance than the less positively charged variants or the wildtype. In addition to charge and hydrophobicity, glycosylation is another factor that may significantly impact both the PK and PD of mAbs. IgGs contain a glycosylation site in the Fc region at amino acid position 297 and, in some cases, in the Fab region. Glycans that have a major impact on PK and PD of mAb include mannose, sialic acids, galactose, and fucose. For example, therapeutic mAbs with high terminal mannose glycans exhibit fast elimination from systemic circulation and lower efficacy.43, 44 The fast elimination of mAbs with high mannose content was attributed to mannose receptor and asialoglycoprotein receptor, which are the endocytotic receptors expressed on hepatocytes and endothelial cells. Depending on the expression system, antibodies with different glycosylation patterns demonstrate different binding affinity to various Fcγ Rs resulting in altered ADCC activities or CDC activity, which has implications for both efficacy and safety.44 The tools to evaluate the mAb PK based on its physiochemical properties are furthered discussed in the “How to derisk mAbs with undesirable PK properties in early lead selection?” section.
ADA‐mediated clearance and immunogenicity
mAb‐based therapeutics may induce humoral immune responses when administered. Immunogenicity reflects the capacity of a given therapeutics to induce such a response and is characterized by the generation of antidrug antibodies (ADA). ADA response may result in the formation of immune complexes with the mAbs. Varying in size and structure, immune complexes are more likely cleared via FcγR‐mediated endocytosis at a different rate than mAb alone (faster or sometimes slower) and accelerate (or less frequently delay) the overall mAb clearance.45 ADA response can reduce efficacy and induce adverse events through diverse immunological mechanisms and through PK/PD interactions. For example, studies have shown that ADA response in rheumatoid arthritis patients treated with anti‐TNF immunobiologicals leads to loss of therapeutic benefit after an initial positive response.46 From a survey of recently approved mAbs, immunogenicity incidence rates varied from 0% to as high as 42% (Table 3).
In general, minimizing or eliminating murine sequences reduces the frequency of mAb‐induced immune responses and hypersensitivity reactions. However, sequence alone is not a reliable predictor of immunogenicity. Determinants of immunogenicity include product characteristics such as oxidation and deamidation sites, as well as target biology, patient population, dosing routes, and regimen.47 Immunogenicity in preclinical species cannot predict the incidence of human ADAs but may have utility in characterizing the consequences of potential ADAs.48 Despite these challenges, there are efforts aimed at predicting immunogenicity using in silico and in vitro tools.49
CONSIDERATIONS FOR CHARACTERIZING mAb PK IN DRUG DEVELOPMENT
PK characterization serves distinct roles at different stages of discovery and development. Early initiation of PK/PD assessment facilitates understanding of the biology by validating and confirming target engagement.50 Once at the lead selection and optimization stage, PK screening should be included as one of the lead “developability” criteria along with efficacy and toxicity evaluation to minimize the risk of off‐target binding.51 At the investigational new drug (IND) enabling stage, integrated PK/PD knowledge along with safety studies serve a critical role in translating preclinical findings to first‐in‐human dose and regimen selection in the clinic.52, 53 Once in clinical development, clinical pharmacology studies can identify the covariates and potential sources of intersubject variability; establish exposure–response relationships in patients, and aid in development of a dosing regimen.40 The alignment of major PK/PD‐related studies with the decision points of mAbs development are shown in Figure 3.
Figure 3.
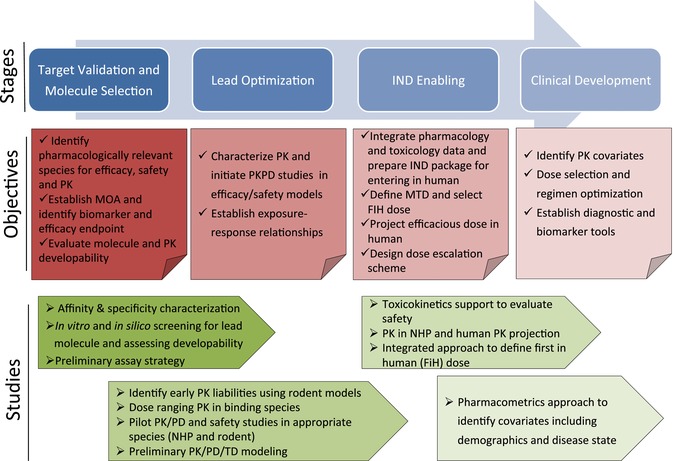
The alignment of major PK/PD‐related studies with the decision points of mAb development in both objectives and recommended studies.
FREQUENTLY ASKED QUESTIONS ON PK RELATED ACTIVITIES DURING EARLY STAGE DRUG DEVELOPMENT
What are the most appropriate assays to quantify mAbs?
Currently, the majority of bioanalytical methods used to determine mAb concentrations are ligand‐based assays (e.g., ELISA).54 In addition to ligand‐based assays, LC‐MS technologies have also been utilized for high‐throughput quantification of mAbs.55 Depending on the specific reagent used, ELISAs may detect one of the multiple forms of mAbs in circulation, including free mAb, antigen‐bound mAb, total antibody, and ADA‐mAb immunocomplex. Free mAb is often the most desirable analyte since it is assumed to be the driver for biological activities. In practical terms, the assay strategy is often evolving with the information required for decision‐making and the availability of reagents. For example, in the early stages of lead selection, a generic assay for total antibody that uses anti‐human IgG as a capture reagent might be sufficient in rodent PK studies, while a specific assay that utilizes antigen‐specific reagents or anti‐idiotype (anti‐ID) antibody is required for human studies. When the target antigen is soluble, free and total antigen concentrations may also be required for appropriate PK/PD assessment. For example, an anti‐ID antibody was used to measure unbound anti‐PCSK9 antibody (evolocumab), while unbound PCSK9 was measured using evolocumab for capture and a second biotin‐conjugated anti‐PCSK9 antibody for detection.56 PK/PD analysis using unbound evolocumab, unbound PCSK9, and LDL‐C data has enabled dose and regimen selection in clinical trials37 (see “Target‐mediated or specific clearance” for further details).
How to derisk mAbs with undesirable PK properties in early lead selection?
Marketed antibodies often show relatively narrow ranges of PK parameters (Table 3). In reality, candidate molecules at the early stage of discovery often display a much broader and sometimes undesirable range of PK behavior.57 Given the relative abundance during lead generation and the intense investment needed at later development stages, it is essential to include “PK developability” as one of the lead selection criteria. Recent publications highlighted the feasibility and multiple approaches employed in such efforts. The study by Jarasch et al. presented a set of comprehensive methods to assess “PK developability” to help with lead candidate selection51 and Jain et al. demonstrated the feasibility of using multiple biophysical metrics to characterize mAbs for drug‐like behavior.58
A comprehensive review by Dostalek et al. summarized the various in silico, in vitro, and in vivo tools in predicting PK liability of lead candidates.59 Those tools often focus on two general areas of interest: antibody integrity/stability and nonspecific or off‐target binding. Antibody integrity and stability predictions can be determined using in silico sequence analysis. This type of analysis can identify potential post‐translational modification hotspots (i.e., cysteines, asparagine/aspartate degradation, glycosylation sites, etc.), assess charge and hydrophobicity,42, 60 and model three‐dimensional structure. In vitro assays can be used to analyze mAb hydrophobicity using hydrophobic interaction chromatography, Fv‐FcRn interactions, and overall IgG integrity using FcRn affinity chromatography.61 Nonspecific binding can be assessed using ELISA‐based baculovirus (BV) binding assays,57 and flow cytometric‐based binding assays using soluble HEK293 and CHO membrane proteins.62
How to select preclinical species for PK studies?
In general, we conduct preclinical PK studies to answer two categorical questions: whether the lead candidate mAb exhibit optimal efficacy and safety profiles to justify further development and whether the antibody has the desired PK behavior that will enable dosing regimen selection that is compatible with predefined target candidate profile. Fully integrated PK/PD evaluations in both efficacy and safety studies and/or stand‐alone studies help address these two questions. For both purposes, PK studies in pharmacologically relevant species provide the best information and optimal support for safety and efficacy evaluations.
Selecting the relevant preclinical species for mAb development involves several steps: first, the target protein sequence comparison, particularly the sequence of binding epitopes between human and preclinical species, typically rodents and non‐human primates. A high degree of homology between epitopes often signals the likelihood of crossreactivity for a given mAb. Next, the binding affinity of mAb with targets derived from different species needs to be assessed through surface plasmon resonance (SPR) or other ligand binding assays. Empirically, species with affinity differences within 10‐fold of affinity to a human antigen can be considered for further evaluation. Biological activity of the antibody in cell‐based assays provides another means of assessing species relevance. Ideally, lead candidate mAbs should have a comparable binding affinity to its antigen in at least one preclinical species to facilitate both toxicity and efficacy evaluation.
It is not unusual to have lead candidates that show limited binding to rodent and NHPs are the only relevant species that support toxicity or PD studies. When rodents are not a relevant species for safety assessment, it may still be necessary to characterize mAb PK in rodents if they are used to assess efficacy such as mouse xenograft models with human tumors. In addition, given the limited predictive ability from in vitro and in silico studies, it may be informative to conduct PK studies in rodents to obtain an early read on in vivo PK.59 A cassette dosing strategy along with differentiating assays has been explored in screening multiple antibodies in a single study for maximal efficiency.63
How to project mAb PK in human?
The practice of projecting mAb PK in humans is well established,63, 64, 65, 66, 67 and two general approaches for antibodies with linear PK are commonly used.
The first approach is empirical allometric scaling that is based on a power law relationship between body weight and PK parameters (V or CL) as described by Eq. 1.
| (1) |
where Y is the parameter of interest (V or CL); a and b are the empirical values to scale parameters of interest; BW is the body weight of the species of interest.
Among different variations of allometric scaling methods, the “simplified allometry,” which only uses data from cynomolgus monkey instead of multiple species, is the most effective approach given the anatomical, physiological, and biological similarities between cynomolgus monkey and human, including FcRn binding affinity and antibody binding specificity. Using this method, the volume of distribution scales proportionally with body weight and clearance often follows an exponent between 0.8–0.9 along with body weight scaling.66
The second approach is the species‐invariant time method (or elementary Dendrick plot), which is based on the assumption that the dose‐normalized drug concentrations in preclinical species are equivalent to humans, while “PK‐time” in animals is transformed to human equivalent time based on body weight. This method provides projection of human PK profiles with specific dose and dosing regimens. The species‐invariant time method was reported to have better predictions than simplified allometry for both membrane‐bound and soluble targets.65
Mechanistic approaches are often used when mAbs have a dose‐dependent PK profile. Specifically, by incorporating the Michaelis–Menten equation into a conventional two‐compartment model, target‐mediated drug disposition (TMDD) modeling has been employed successfully to capture the dynamic interactions between antibody and antigen.67 Recently, physiologically based PK (PBPK) modeling has been adapted in mAb scaling despite its complexity.29 By integrating physiological parameters, in vitro and in vivo PK data into anatomically and physiologically meaningful compartments, PBPK modeling can capture both drug‐related and system‐related properties across species and add additional insights in translational projection.
What are the considerations in first‐in‐human (FIH) dose selection for mAbs?
The framework of an FIH dose selection for mAbs is based on regulatory guidance.68, 69 The specific choice for FIH dose depends on several factors: species cross‐reactivity, relevance of animal models, steepness of dose response, PK/PD relationship, nature of toxicity such as predictability, reversibility, tolerability, manageability, and the precedence of molecules in similar pathways.70 Surveys on FIH dose selection showed that guidance based on no observed adverse effects levels (NOAEL) was the most frequently used method, while the PK/PD model‐based approach was gaining acceptance.71
Relevance of preclinical species deserves special consideration for both pharmacology and safety evaluation. NHPs are the most frequently used species for selecting FIH dose due to their highly similar physiology to humans, similar FcRn binding affinity to human IgG, and overall similar antibody disposition. Based on literature, approximately 58% of studies used NHPs only and nearly 80% used both NHPs and rodents to determine FiH dose.71
Recent surveys on the strategies and outcomes of mAbs tested in FIH trials between 2000 and 2013 found that severe mAb‐related toxicities were infrequent, and the usefulness of toxicity as the criterion to guide dose escalation was limited.71, 72 However, in the wake of the TGN1412 incident,73 which led to the introduction of the minimal anticipated biological effect level (MABEL)‐ or minimum effective dose (MED)‐based approach,74 additional precautions have been taken for molecules of novel pathways or for immune modulators. The MABEL approach was used more frequently in recent years, reflecting the increasing repertoire of immune modulators and the recognition of translational insufficiency for this family of molecules.75 While the MABEL‐based approach is the most conservative approach leading to a lower starting dose, it could achieve the desired objectives of FIH clinical trials effectively with a more aggressive dose escalation.72
PK/PD modeling incorporating both in vitro and in vivo data is an important tool in FIH dose selection. The mechanistic insights it provides is particularly relevant in applying a MABEL approach.76 For example, a TMDD/PKPD model was developed for TAM‐163, an agonist antibody targeting tyrosine receptor kinase‐B (TrkB). The integration of exposure, target coverage, and pharmacological activity identified an efficacy threshold to achieve significant weight gain in monkeys.71 The subsequent scaling to humans led to the projection of minimal and maximal pharmacologically active doses. The recommended FIH dose for TAM‐163 based on the MABEL approach was projected to achieve <10% target coverage at Cmax.
How to project efficacious dose in human and the utility of the projection?
Projecting human efficacious dose is based on two key assumptions: exposure–response relationships translate across species and mAb PK is predictable in humans at a relevant dose range. It includes the following steps: 1) define the efficacy end points and establish an exposure–response relationships in the efficacy model; 2) project human PK profiles from cynomolgus monkey using a simplified allometric scaling or time‐invariant time method (TMDD modeling if expecting nonlinear PK); and 3) integrate efficacy PK/PD models with human PK projections and estimate human efficacious dose.77
The translatability of human efficacious dose projection depends on confidence in selecting the most relevant PD/efficacy end points and identifying key PK drivers (Cmax, Ctrough, or AUC, etc.) for efficacy. PD or efficacy endpoint selection is critical in translation between preclinical studies and clinical development since the underlying assumption is that exposure–response relationships are translatable across species. It remains an empirical and case‐by‐case practice for mAbs, especially when clinical outcomes are different from preclinical efficacy end points. For example, in a retrospective population PK/PD analysis on eight targeted and cytotoxic agents with known clinical outcome, Wong and colleagues showed that greater than 60% tumor growth inhibition in preclinical models at clinically relevant exposures was more likely to lead to responses in the clinic.78 A similar report on reverse translatability by Spilker et al. demonstrated the same key concept of a PK‐based strategy to select nonclinical doses/regimen in mice for approved drugs. By identifying “clinically relevant dose” instead of “maximally tolerated dose,” this approach can maximize the translatability of efficacy results, which is particularly relevant in selecting candidates for combination with approved therapeutics.80 Alternatively, a translatable mechanistic PK/PD model can be set up as a more advanced method if the mode of action is known.
Dosing regimen in clinic selection poses another translational challenge. One compelling example is the regimen finding strategy employed by Jumbe et al. to determine the appropriate dose and schedule to achieve the desired tumor suppression for trastuzumab‐DM1 (T‐DM1).79 The authors developed a population‐based semi‐mechanistic PK/PD model to describe the T‐DM1 antitumor activity by consolidating the exposure–effect relationship determined across multiple animal models. Subsequently, the treatment regimen, targeted exposure parameters (AUC, Cmax, or time above minimal concentration threshold), and tumoristatic concentration were derived from the model to support early clinical trial design.
Frequently, the recommended phase II dose (RP2D) is determined in the absence of a maximum tolerated dose (MTD) for mAbs. In these cases, clinical PK data and projected efficacious dose based on preclinical studies were the main criteria for choosing an RP2D.80 The ability of PK/PD modeling to predict the correct dose and dosing regimen takes on additional urgency for novel checkpoint inhibitors in cancer immunotherapy since the majority of these therapeutics exhibit heterogeneous tumor responses and lack predictive biomarkers.81 Translational PK/PD modeling of pembrolizumab, a humanized mAb against PD‐1, exemplified the power of this approach. Lindauer et al. utilized physiologically based PK and systems pharmacology approaches combining in vitro binding data, mechanistic tumor growth inhibition models, and drug exposure‐time‐to‐event relationships to successfully identify the lowest effective dose for evaluation.82
What are the key objectives of mAb early clinical development?
Clinical pharmacology along with pharmacometric approaches help identify patient‐specific covariates, assess the source of variability, explore dose/exposure response relationships, dissect the role of predictive and prognostic biomarkers, and ultimately, establish the dose and dosing regimen for a given treatment.83
NEXT‐GENERATION ANTIBODY‐BASED THERAPEUTICS AND THEIR UNIQUE PK CHALLENGES
Driven by biological insights and technical advances in antibody engineering, antibody therapeutics have blossomed into a plethora of diverse platforms.2 Among them, bispecific or multispecific antibodies, antibody–drug conjugates, glyco‐engineered antibodies, and various novel antibody scaffolds present new challenges in applying quantitative pharmacology and translational PK/PD.
Bispecific antibodies
Bispecific antibodies (bsAbs) target multiple antigens from distinct signaling pathways or redundant antigens in the same pathway. Currently, there are over 60 different bsAbs formats with variations in modular structure, size, affinity/avidity, PK behavior, and intended clinical application.84
One of the most active areas of bsAbs application is in the field of cancer immunotherapy.85, 86, 87 Cancer immunotherapy works by unleashing the body's own immune system to eliminate or control cancer. The basic tenet for bsAbs in cancer immunotherapy has been the simultaneous binding of bsAbs to cytotoxic T cells and antigen‐expressing tumors. This is best exemplified by the first approved bsAbs, catumaxomab (a bsAb binds both CD3 on cytotoxic T cells and EpCAM on human adenocarcinomas,88) and blinatumomab (a CD19 × CD3 targeting bispecific scFv antibody fragment.89) A comprehensive panel of studies is required to assess whether bsAbs are able to preserve antigen‐binding affinity, biological activities of their parental monospecific antibodies, and PK properties. For example, the PK of the anti‐Her2/CD3T cell‐dependent bispecific (TDB) antibody in rats demonstrated similar clearance and half‐life as trastuzumab.92 Novel approaches in reducing production complexity requires comprehensive PK evaluation, as demonstrated by Dillon et al., who showed that bsAbs produced from a single host cell through multiple engineering strategies have comparable PK profiles in mice as the in vitro‐assembled bispecific IgG.90
Target‐mediated drug disposition along with efficacy and toxicity needs to be carefully addressed for bsAbs. In an effort to maximize the therapeutic window for the anti‐HER2/CD3 T‐cell‐dependent bispecific (TDB) antibody, Mandikian et al. showed that the relative HER2/CD3 affinity is a critical driver for TDB distribution and catabolism in transgenic mouse models. They also showed that there was a strong correlation between CD3 affinity and distribution to T‐cell‐rich tissues, with higher CD3 affinity reducing systemic exposure and shifting TDB distribution away from tumor to T cell‐containing tissues91 (Mandikian et al., 2018).
While development of bsAbs is intrinsically more complex, it is also an area where PK/PD and systems pharmacology modeling can make an immediate impact. Another important application of bsAbs is to deliver mAbs to areas generally thought to be restricted to antibodies. For example, mAbs exhibit limited distribution to the brain, and mAb concentrations was determined to be 0.1–0.2 % and 0.02 % of the steady‐state circulating mAb concentration in rodents and cynomolgus monkey, respectively.92, 93 To overcome this limitation, a transferrin receptor (TfR) and β‐secretase 1 (BACE1) bsAbs was generated. By targeting TfR, a transcytosis efficient membrane protein expressed on endothelial cells in brain, these bsAb constructs drastically improved CNS delivery in both rodents and NHPs.94 As expected, altering the affinity to TfR impacted systemic exposure and safety profiles. Gadkar et al. built a PK/PD model using data from bsAbs with a range of affinities that captured the dependence of both systemic and brain exposure on TfR affinity and the subsequent impact on efficacy and safety.87
Antibodies with modified effector functions and FcRn modifications
Antibodies with enhanced effector functions: antibody‐dependent cell‐mediated cytotoxicity (ADCC), antibody‐dependent cellular phagocytosis (ADCP), complement‐dependent cytotoxicity (CDC), can improve potency. The anti‐CD20 antibodies, obinutuzumab and ocrelizumab, with their enhanced FcγR binding affinity showed greater potency compared to a first‐generation antibody, rituximab.95 On the other hand, for certain immuno‐oncology indications, reduced effector functions are desirable for greater safety. Atezolizumab, an human IgG1 mAb targeting PD‐L1, was engineered with a mutation in the Fc domain (N298A) to reduce ADCC and CDC.96 Modulation of FcγR binding usually does not change PK profiles.97
Engineered antibodies with modified FcRn binding affinity can increase or decrease the mAb half‐life. As expected, increasing the affinity of the IgG‐FcRn interaction at pH 6.0 results in longer serum half‐life. For example, M252Y/S254T/T256E (YTE) mutations on motavizumab led to a 10‐fold increase in affinity for human FcRn at pH 6.0 with no change in affinity at pH 7.4 and significantly increased its half‐life in monkeys and in healthy adult humans.32 A couple of recent reviews summarize additional applications of FcRn modulations.98, 99
CONCLUSION
In the 40 years since the introduction of hybridoma technology, antibody‐based therapeutics have advanced rapidly and become an integral part of medical intervention. The modular architecture of antibodies has been exploited to create different antibody formats with the most desirable mechanism of action, PK profiles, and ultimately optimal clinical outcome. Along with the considerable success in antibody‐based therapies in recent years, we have witnessed the increased application of PK/PD approaches in drug development. A systematic implementation of PK/PD strategy with appropriate study design and gating criteria helps the selection and the optimization of antibody candidates prior to entering into safety studies in human, and continuing translational PK/PD and clinical pharmacology pave a successful an ultimate path to approval.
While development of classical mAbs against novel targets are evolving at a fast pace, development of next‐generation biotherapeutics such as bispecific or multispecific antibodies, antibody–drug conjugates, glycol‐engineered antibodies, and various novel antibody scaffolds are gaining momentum. These formats tend to have complex moieties with unpredictable interactions among them, which necessitate careful selection of lead candidates based on their biochemical, biophysical, in vivo efficacy, PK, and safety properties. Our understanding in mAb PK and experience with translational PK/PD will facilitate new modalities to reach patients with unmet needs.
Conflict of Interest
The authors declare no competing interests for this work.
Funding
No funding was received for this work.
References
- 1. Reichert, J.M. Antibodies to watch in 2017. MAbs. 9, 167–181 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weiner, G.J. Building better monoclonal antibody‐based therapeutics. Nat. Rev. Cancer. 15, 361–370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. FDA . Purple Book: Lists of Licensed Biological Products with Reference Product Exclusivity and Biosimilarity or Interchangeability Evaluations 2016 [Available from: https://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm411418.htm.
- 4. Cruse, J.M. & Lewis, R. Atlas of Immunology, Third Edition, (CRC Press Taylor & Francis Group; 2010). [Google Scholar]
- 5. Kohler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 256, 495–497 (1975). [DOI] [PubMed] [Google Scholar]
- 6. Liu, J.K. The history of monoclonal antibody development—Progress, remaining challenges and future innovations. Ann. Med. Surg. (Lond). 3, 113–116 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Almagro, J.C. & Fransson, J. Humanization of antibodies. Front. Biosci. 13, 1619–1633 (2008). [DOI] [PubMed] [Google Scholar]
- 8. Mondon, P. , Dubreuil, O. , Bouayadi, K. & Kharrat, H. Human antibody libraries: a race to engineer and explore a larger diversity. Front. Biosci. 13, 1117–1129 (2008). [DOI] [PubMed] [Google Scholar]
- 9. Ayyar, B.V. , Arora, S. & O'Kennedy, R. Coming‐of‐Age of Antibodies in Cancer Therapeutics. Trends Pharmacol. Sci. 37, 1009–1028 (2016). [DOI] [PubMed] [Google Scholar]
- 10. Janeway, C. Immunobiology, 5th Edition Garland Publishing; 2001.
- 11. Li, P. , Zheng, Y. & Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti‐TNF Biologics. Front. Pharmacol. 8, 460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pardoll, D.M. Immunology beats cancer: a blueprint for successful translation. Nat. Immunol. 13, 1129–1132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chan, A.C. & Carter, P.J. Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 10, 301–316 (2010). [DOI] [PubMed] [Google Scholar]
- 14. Lim, S.H. , Beers, S.A. , French, R.R. , Johnson, P.W. , Glennie, M.J. & Cragg, M.S. Anti‐CD20 monoclonal antibodies: historical and future perspectives. Haematologica. 95, 135–143 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Renukuntla, J. , Vadlapudi, A.D. , Patel, A. , Boddu, S.H. & Mitra, A.K. Approaches for enhancing oral bioavailability of peptides and proteins. Int. J. Pharm. 447, 75–93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morishita, M. & Peppas, N.A. Is the oral route possible for peptide and protein drug delivery? Drug Discov. Today. 11, 905–910 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Singh, R. , Singh, S. & Lillard, J.W., Jr. Past, present, and future technologies for oral delivery of therapeutic proteins. J. Pharm. Sci. 97, 2497–2523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jackisch, C. , Muller, V. , Maintz, C. , Hell, S. & Ataseven, B. Subcutaneous Administration of Monoclonal Antibodies in Oncology. Geburtshilfe Frauenheilkd. 74, 343–349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Glassman, P.M. , Abuqayyas, L. & Balthasar, J.P. Assessments of antibody biodistribution. J. Clin. Pharmacol. 55 (Suppl 3), S29–S38, (2015). [DOI] [PubMed] [Google Scholar]
- 20. Richter, W.F. , Bhansali, S.G. & Morris, M.E. Mechanistic determinants of biotherapeutics absorption following SC administration. AAPS J. 14, 559–570 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Datta‐Mannan, A. , Witcher, D.R. , Lu, J. & Wroblewski, V.J. Influence of improved FcRn binding on the subcutaneous bioavailability of monoclonal antibodies in cynomolgus monkeys. MAbs. 4, 267–273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng, Y. et al Minipig as a potential translatable model for monoclonal antibody pharmacokinetics after intravenous and subcutaneous administration. MAbs. 4, 243–255 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lobo, E.D. , Hansen, R.J. & Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 93, 2645–2668 (2004). [DOI] [PubMed] [Google Scholar]
- 24. Rudnick, S.I. & Adams, G.P. Affinity and avidity in antibody‐based tumor targeting. Cancer Biother. Radiopharm. 24, 155–161 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boswell, C.A. , Tesar, D.B. , Mukhyala, K. , Theil, F.P. , Fielder, P.J. & Khawli, L.A. Effects of charge on antibody tissue distribution and pharmacokinetics. Bioconjug. Chem. 21, 2153–2163 (2010). [DOI] [PubMed] [Google Scholar]
- 26. Deng, R. , Jin, F. , Prabhu, S. & Iyer, S. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opin. Drug Metab. Toxicol. 8, 141–160 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Rudnick, S.I. et al Influence of affinity and antigen internalization on the uptake and penetration of Anti‐HER2 antibodies in solid tumors. Cancer Res. 71, 2250–2259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shah, D.K. & Betts, A.M. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. MAbs. 5, 297–305 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ferl, G.Z. , Theil, F.P. & Wong, H. Physiologically based pharmacokinetic models of small molecules and therapeutic antibodies: a mini‐review on fundamental concepts and applications. Biopharm. Drug Dispos. 37, 75–92 (2016). [DOI] [PubMed] [Google Scholar]
- 30. Roopenian, D.C. & Akilesh, S. FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. 7, 715–725 (2007). [DOI] [PubMed] [Google Scholar]
- 31. Dirks, N.L. & Meibohm, B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 49, 633–659 (2010). [DOI] [PubMed] [Google Scholar]
- 32. Robbie, G.J. et al A novel investigational Fc‐modified humanized monoclonal antibody, motavizumab‐YTE, has an extended half‐life in healthy adults. Antimicrob. Agents Chemother. 57, 6147–6153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang, W. , Wang, E.Q. & Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 84, 548–558 (2008). [DOI] [PubMed] [Google Scholar]
- 34. Golay, J. et al Lessons for the clinic from rituximab pharmacokinetics and pharmacodynamics. MAbs. 5, 826–837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mellman, I. & Plutner, H. Internalization and degradation of macrophage Fc receptors bound to polyvalent immune complexes. J. Cell Biol. 98, 1170–1177 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mould, D.R. et al Population pharmacokinetics‐pharmacodynamics of alemtuzumab (Campath) in patients with chronic lymphocytic leukaemia and its link to treatment response. Br. J. Clin. Pharmacol. 64, 278–291 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gibbs, J.P. et al Impact of Target‐Mediated Elimination on the Dose and Regimen of Evolocumab, a Human Monoclonal Antibody Against Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9). J. Clin. Pharmacol. 57, 616–626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schoch, A. et al Charge‐mediated influence of the antibody variable domain on FcRn‐dependent pharmacokinetics. Proc. Natl. Acad. Sci. USA 112, 5997–6002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bumbaca, D. , Boswell, C.A. , Fielder, P.J. & Khawli, L.A. Physiochemical and biochemical factors influencing the pharmacokinetics of antibody therapeutics. AAPS J. 14, 554–558 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gill, K.L. , Machavaram, K.K. , Rose, R.H. & Chetty, M. Potential Sources of Inter‐Subject Variability in Monoclonal Antibody Pharmacokinetics. Clin. Pharmacokinet. 55, 789–805 (2016). [DOI] [PubMed] [Google Scholar]
- 41. Igawa, T. et al Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng. Des. Sel. 23, 385–392 (2010). [DOI] [PubMed] [Google Scholar]
- 42. Bumbaca Yadav, D. et al Evaluating the Use of Antibody Variable Region (Fv) Charge as a Risk Assessment Tool for Predicting Typical Cynomolgus Monkey Pharmacokinetics. J. Biol. Chem. 290, 29732–29741 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goetze, A.M. et al High‐mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 21, 949–959 (2011). [DOI] [PubMed] [Google Scholar]
- 44. Kanda, Y. et al Comparison of biological activity among nonfucosylated therapeutic IgG1 antibodies with three different N‐linked Fc oligosaccharides: the high‐mannose, hybrid, and complex types. Glycobiology. 17, 104–118 (2007). [DOI] [PubMed] [Google Scholar]
- 45. Bendtzen, K. Immunogenicity of Anti‐TNF‐alpha Biotherapies: II. Clinical Relevance of Methods Used for Anti‐Drug Antibody Detection. Front Immunol. 6, 109 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Prado, M.S. , Bendtzen, K. & Andrade, L.E.C. Biological anti‐TNF drugs: Immunogenicity underlying treatment failure and adverse events. Expert Opin. Drug Metab. Toxicol. 2017. [DOI] [PubMed] [Google Scholar]
- 47. Singh, S.K. Impact of product‐related factors on immunogenicity of biotherapeutics. J. Pharm. Sci. 100, 354–387 (2011). [DOI] [PubMed] [Google Scholar]
- 48. FDA . Guidance for Industry ‐ Immunogenicity Assessment for Therapeutic Protein Products 2014 [Available from: https://www.fda.gov/downloads/drugs/guidances/ucm338856.pdf.
- 49. De Groot, A.S. , McMurry, J. & Moise, L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr. Opin. Pharmacol. 8, 620–626 (2008). [DOI] [PubMed] [Google Scholar]
- 50. Cook, D. et al Lessons learned from the fate of AstraZeneca's drug pipeline: a five‐dimensional framework. Nat. Rev. Drug Discov. 13, 419–431 (2014). [DOI] [PubMed] [Google Scholar]
- 51. Jarasch, A. , Koll, H. , Regula, J.T. , Bader, M. , Papadimitriou, A. & Kettenberger, H. Developability assessment during the selection of novel therapeutic antibodies. J. Pharm. Sci. 104, 1885–1898 (2015). [DOI] [PubMed] [Google Scholar]
- 52. Tuntland, T. et al Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research. Front. Pharmacol. 5, 174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang, Y. , Booth, B. , Rahman, A. , Kim, G. , Huang, S.M. & Zineh, I. Toward greater insights on pharmacokinetics and exposure‐response relationships for therapeutic biologics in oncology drug development. Clin. Pharmacol. Ther. 101, 582–584 (2017). [DOI] [PubMed] [Google Scholar]
- 54. Patton, A. , Mullenix, M.C. , Swanson, S.J. & Koren, E. An acid dissociation bridging ELISA for detection of antibodies directed against therapeutic proteins in the presence of antigen. J. Immunol. Methods. 304, 189–195 (2005). [DOI] [PubMed] [Google Scholar]
- 55. Pai, R. et al Therapeutic Antibody‐Induced Vascular Toxicity Due to Off‐Target Activation of Nitric Oxide in Cynomolgus Monkeys. Toxicol. Sci. 151, 245–260 (2016). [DOI] [PubMed] [Google Scholar]
- 56. Colbert, A. , Umble‐Romero, A. , Prokop, S. , Xu, R. , Gibbs, J. & Pederson, S. Characterization of a quantitative method to measure free proprotein convertase subtilisin/kexin type 9 in human serum. MAbs. 6, 1103–1113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hotzel, I. et al A strategy for risk mitigation of antibodies with fast clearance. MAbs. 4, 753–760 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jain, T. et al Biophysical properties of the clinical‐stage antibody landscape. Proc. Natl. Acad. Sci. USA 114, 944–949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dostalek, M. , Prueksaritanont, T. & Kelley, R.F. Pharmacokinetic de‐risking tools for selection of monoclonal antibody lead candidates. MAbs. 9, 756–766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sharma, V.K. et al In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc. Natl. Acad. Sci. USA 111, 18601–18606 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schlothauer, T. et al Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies. MAbs. 5, 576–586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhou, L. et al Stratification of antibody-positive subjects by antibody level reveals an impact of immunogenicity on pharmacokinetics. AAPS J. 15, 30–40 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang, J. , Iyer, S. , Fielder, P.J. , Davis, J.D. & Deng, R. Projecting human pharmacokinetics of monoclonal antibodies from nonclinical data: comparative evaluation of prediction approaches in early drug development. Biopharm. Drug Dispos. 37, 51–65 (2016). [DOI] [PubMed] [Google Scholar]
- 64. Ling, J. , Zhou, H. , Jiao, Q. & Davis, H.M. Interspecies scaling of therapeutic monoclonal antibodies: initial look. J. Clin. Pharmacol. 49, 1382–1402 (2009). [DOI] [PubMed] [Google Scholar]
- 65. Oitate, M. , Nakayama, S. , Ito, T. , Kurihara, A. , Okudaira, N. & Izumi, T. Prediction of human plasma concentration‐time profiles of monoclonal antibodies from monkey data by a species‐invariant time method. Drug Metab. Pharmacokinet. 27, 354–359 (2012). [DOI] [PubMed] [Google Scholar]
- 66. Deng, R. , Iyer, S. , Theil, F.P. , Mortensen, D.L. , Fielder, P.J. & Prabhu, S. Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data: what have we learned? MAbs. 3, 61–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dong, J.Q. et al Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first‐in‐human prediction. Clin. Pharmacokinet. 50, 131–142 (2011). [DOI] [PubMed] [Google Scholar]
- 68. FDA . Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers 2005 [Available from: https://www.fda.gov/downloads/drugs/guidances/ucm078932.pdf.
- 69. EMA . Guideline on Strategies to Identify and Mitigate Risks for First‐in‐Human Clinical Trials with Investigational Medicinal Products 2007 [Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf. [DOI] [PMC free article] [PubMed]
- 70. Chapman, K. , Pullen, N. , Graham, M. & Ragan, I. Preclinical safety testing of monoclonal antibodies: the significance of species relevance. Nat. Rev. Drug Discov. 6, 120–126 (2007). [DOI] [PubMed] [Google Scholar]
- 71. Tosi, D. et al Clinical Development Strategies and Outcomes in First‐in‐Human Trials of Monoclonal Antibodies. J. Clin. Oncol. 33, 2158–2165 (2015). [DOI] [PubMed] [Google Scholar]
- 72. Suh, H.Y. , Peck, C.C. , Yu, K.S. & Lee, H. Determination of the starting dose in the first‐in‐human clinical trials with monoclonal antibodies: a systematic review of papers published between 1990 and 2013. Drug Des Devel Ther. 10, 4005–4016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Suntharalingam, G. et al Cytokine storm in a phase 1 trial of the anti‐CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 355, 1018–1028 (2006). [DOI] [PubMed] [Google Scholar]
- 74. EMA . Guideline on Strategies to Identify and Mitigate Risks for First‐in‐Human Clinical Trials with Investigational Medicinal Products 2007. [DOI] [PMC free article] [PubMed]
- 75. Trivedi, A. et al Clinical Pharmacology and Translational Aspects of Bispecific Antibodies. Clin. Transl. Sci. 10, 147–162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. EMA . Guideline on strategies to identify and mitigate risks for first‐in‐human and early clinical trials with investigational medicinal products. 2017. [DOI] [PMC free article] [PubMed]
- 77. Gupta, P. et al Preclinical pharmacokinetics of MHAA4549A, a human monoclonal antibody to influenza A virus, and the prediction of its efficacious clinical dose for the treatment of patients hospitalized with influenza A. MAbs. 8, 991–997 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kamath, A.V. et al Preclinical pharmacokinetics of MEHD7945A, a novel EGFR/HER3 dual‐action antibody, and prediction of its human pharmacokinetics and efficacious clinical dose. Cancer Chemother. Pharmacol. 69, 1063–1069 (2012). [DOI] [PubMed] [Google Scholar]
- 79. Jumbe, N.L. et al Modeling the efficacy of trastuzumab‐DM1, an antibody drug conjugate, in mice. J. Pharmacokinet. Pharmacodyn. 37, 221–242 (2010). [DOI] [PubMed] [Google Scholar]
- 80. Stroh, M. et al Challenges and Opportunities for Quantitative Clinical Pharmacology in Cancer Immunotherapy: Something Old, Something New, Something Borrowed, and Something Blue. CPT Pharmacometrics Syst Pharmacol. 4, 495–497 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garrido, M.J. , Berraondo, P. & Troconiz, I.F. Commentary on Pharmacometrics for Immunotherapy. CPT Pharmacometrics Syst. Pharmacol. 6, 8–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lindauer, A. et al Translational Pharmacokinetic/Pharmacodynamic Modeling of Tumor Growth Inhibition Supports Dose‐Range Selection of the Anti‐PD‐1 Antibody Pembrolizumab. CPT Pharmacometrics Syst. Pharmacol. 6, 11–20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Vicini, P. & Roskos, L.K. Clinical Pharmacology: a Discipline at the Nexus Between Translational Science and Precision Medicine: Commentary on “Enhancing Value of Clinical Pharmacodynamics in Oncology Drug Development: An Alliance Between Quantitative Pharmacology and Translational Science”. Clin. Pharmacol. Ther. 2016. [DOI] [PubMed] [Google Scholar]
- 84. Spiess, C. , Zhai, Q. & Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 67 (2 Pt A), 95–106 (2015). [DOI] [PubMed] [Google Scholar]
- 85. Chames, P. & Baty, D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel? MAbs. 1, 539–547 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Muller, D. & Kontermann, R.E. Bispecific antibodies for cancer immunotherapy: Current perspectives. BioDrugs. 24, 89–98 (2010). [DOI] [PubMed] [Google Scholar]
- 87. Gadkar, K. et al Mathematical PKPD and safety model of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur. J. Pharm. Biopharm. 101, 53–61 (2016). [DOI] [PubMed] [Google Scholar]
- 88. Linke, R. , Klein, A. & Seimetz, D. Catumaxomab: clinical development and future directions. MAbs. 2, 129–136 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bargou, R. et al Tumor regression in cancer patients by very low doses of a T cell‐engaging antibody. Science. 321, 974–977 (2008). [DOI] [PubMed] [Google Scholar]
- 90. Dillon, M. et al Efficient production of bispecific IgG of different isotypes and species of origin in single mammalian cells. MAbs. 9, 213–230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mandikian, D. et al Relative Target Affinities of T‐Cell‐Dependent Bispecific Antibodies Determine Biodistribution in a Solid Tumor Mouse Model. Mol. Cancer Ther. 17, 776–785 (2018). [DOI] [PubMed] [Google Scholar]
- 92. Poduslo, J.F. , Curran, G.L. & Berg, C.T. Macromolecular permeability across the blood‐nerve and blood‐brain barriers. Proc. Natl. Acad. Sci. USA 91, 5705–5709 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yadav, D.B. et al Widespread brain distribution and activity following i.c.v. infusion of anti‐beta‐secretase (BACE1) in nonhuman primates. Br. J. Pharmacol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Yu, Y.J. et al Therapeutic bispecific antibodies cross the blood‐brain barrier in nonhuman primates. Sci. Transl. Med. 6, 261ra154 (2014). [DOI] [PubMed] [Google Scholar]
- 95. Polito, L. , Djemil, A. & Bortolotti, M. Plant Toxin‐Based Immunotoxins for Cancer Therapy: A Short Overview. Biomedicines. 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Deng, R. et al Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti‐PD‐L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs. 8, 593–603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Leabman, M.K. , Meng, Y.G. , Kelley, R.F. , DeForge, L.E. , Cowan, K.J. & Iyer, S. Effects of altered FcgammaR binding on antibody pharmacokinetics in cynomolgus monkeys. MAbs. 5, 896–903 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kuo, T.T. & Aveson, V.G. Neonatal Fc receptor and IgG‐based therapeutics. MAbs. 3, 422–430 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sockolosky, J.T. & Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug. Deliv. Rev. 91, 109–124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kenny, J.R. et al Therapeutic protein drug-drug interactions: navigating the knowledge gaps-highlights from the 2012 AAPS NBC Roundtable and IQ Consortium/FDA workshop. AAPS J. 15, 933–940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]