

ABSTRACT
Here, we wish to propose a new systematic approach to cancer therapy, based on the targeting of mitochondrial metabolism, especially in cancer stem cells (CSCs). In the future, we envision that anti-mitochondrial therapy would ultimately be practiced as an add-on to more conventional therapy, largely for the prevention of tumor recurrence and cancer metastasis. This mitochondrial based oncology platform would require a panel of FDA-approved therapeutics (e.g. Doxycycline) that can safely be used to inhibit mitochondrial OXPHOS and/or biogenesis in CSCs. In addition, new therapeutics that target mitochondria could also be developed, to optimize their ability to eradicate CSCs. Finally, in this context, mitochondrial-based biomarkers (i.e. “Mito-signatures”) could be utilized as companion diagnostics, to identify high-risk cancer patients at diagnosis, facilitating the early detection of tumor recurrence and the prevention of treatment failure. In summary, we suggest that new clinical trials are warranted to test and possibly implement this emerging treatment strategy, in a variety of human cancer types. This general approach, using FDA-approved antibiotics to target mitochondria, was effective in killing CSCs originating from many different cancer types, including DCIS, breast (ER(+) and ER(-)), prostate, ovarian, lung and pancreatic cancers, as well as melanoma and glioblastoma, among others. Thus, we propose the term MITO-ONC-RX, to describe this anti-mitochondrial platform for targeting CSCs. The use of re-purposed FDA-approved drugs will undoubtedly help to accelerate the clinical evaluation of this approach, as these drugs can move directly into Phase II clinical trials, saving considerable amounts of time (10–15 y) and billions in financial resources.
KEYWORDS: Mitochondria, drug discovery, oncology platform, Mito-therapeutics, Mito-signatures, mito-biomarkers, Mitoriboscins, Mitoketoscins, Mitoflavoscins
1. Mitochondrial function is required for CSC propagation
Tumors and their microenvironment are incredibly heterogeneous structures, which behave like metabolic ecosystems [1–12]. In accordance with this cellular and metabolic heterogeneity, it is now well accepted that more than a single type of cancer cell exists [1–12]. For example, within a given epithelial cancer cell line (such as MCF7), there are “bulk” cancer cells (~ 85–95%; the majority of the population), as well as various types of progenitor cells (< 5%), and cancer stem cells (CSCs; < 1%). CSCs and progenitor cells are thought to be the most dangerous, as they behave as tumor-initiating cells (TICs) in vivo and can undergo metastasis; in contrast, “bulk” cancer cells are largely non-tumorigenic.
Because CSCs are so “rare”, very little is actually known about their metabolic properties. To functionally enrich for CSCs, the entire cell population can be trypsinized and seeded as a single-cell suspension onto low-attachment plates [1,2]. Under these conditions, the majority of “bulk” cancer cells die (> 90%) via apoptosis, while only the CSCs survive and propagate, ultimately resulting after 5 d in the formation of 3D spheroid structures. Under these conditions, each 3D spheroid is clonally formed from a single CSC. For breast CSCs, these 3D spheroids are also known as tumor-spheres or mammo-spheres [1–12]. The generation of these 3D spheroids is thought to mimic the process of tumor formation and/or metastasis, providing an excellent model for drug discovery and functional validation.
Thus, to begin to understand the potential metabolic differences between “bulk” cancer cells and CSCs, we first compared cultured breast cancer cells grown either as monolayers or 3D spheroids. Then, they were subjected to profiling, via unbiased label-free proteomics analysis [13].
As a result of this molecular comparison, it became clear that over 60 nuclear-encoded mitochondrial proteins were specifically up-regulated in 3D spheroid structures, relative to monolayer cells processed in parallel [13]. Virtually identical results were obtained with two distinct ER(+) breast cancer cell lines (MCF7 and T47D; > 40 overlapping mitochondrial proteins). Informatics analysis of the list of up-regulated mitochondrial proteins was consistent with an increase in mitochondrial mass, due either to i) increased mitochondrial biogenesis or ii) a shut down in mitophagy, or both. As such, these results indicated that high mitochondrial mass is a new characteristic feature of the CSC phenotype [13]. These results also suggested the testable hypothesis that CSCs are critically dependent on OXPHOS and/or new mitochondrial biogenesis (protein translation), for their survival and propagation. In accordance with this hypothesis, 3D spheroid formation was effectively blocked using specific mitochondrial inhibitors, such as oligomycin, which targets mitochondrial Complex V and shuts off ATP synthesis [13]. However, oligomycin is quite toxic, so it cannot be used as an anti-cancer therapeutic.
As such, the question remains: How can we safely target mitochondria in CSCs, without inducing serious side effects in normal cells?
MitoTracker is a non-toxic fluorescent probe that can be used to directly measure mitochondrial mass in live cells by flow cytometry [14]. In order to further validate the functional relationship between high mitochondrial mass and “stemness”, we employed staining with MitoTracker to metabolically fractionate the MCF7 cell line into “Mito-high” and “Mito-low” cell sub-populations [14]. For this purpose, we used MitoTracker Deep Red FM, which is a far-red fluorescent dye (abs/em ~ 644/665 nm) that stains mitochondria in live cells. As predicted the “Mito-high” cell population, with increased mitochondrial mass, showed the greatest capacity for i) 3D spheroid formation and ii) tumor initiation in a pre-clinical animal model in vivo [14]. Therefore, mitochondrial mass is a critical determinant of stemness in cancer cells. In addition, elevated telomerase activity (hTERT), a functional marker of proliferation and immortality in CSCs, was also specifically associated with high mitochondrial mass [15].
In light of the above results, a targeted reduction in mitochondrial mass or OXPHOS should provide a specific Achilles’ heel for the eradication of CSCs [13–15].
2. Mitochondrial-based therapeutic strategies for eradicating CSCs: FDA-approved drug repurposing and natural products
Over the last several years, our laboratory has focused on the identification and repurposing of FDA-approved drugs that can be used to inhibit the propagation of CSCs. These different antibiotics include members of the i) Tetracycline family (Doxycycline/Tigecycline) [16] and the ii) Erythromycin family (Azithromycin) [16], as well as iii) anti-parasitic drugs (Pyrvinium pamoate and Atovaquone) [16,17] and iv) anti-microbials targeting drug-resistant mycobacterium (Bedaquiline; TB, tuberculosis) [18], (Figure 1, and Table 1).
Figure 1.
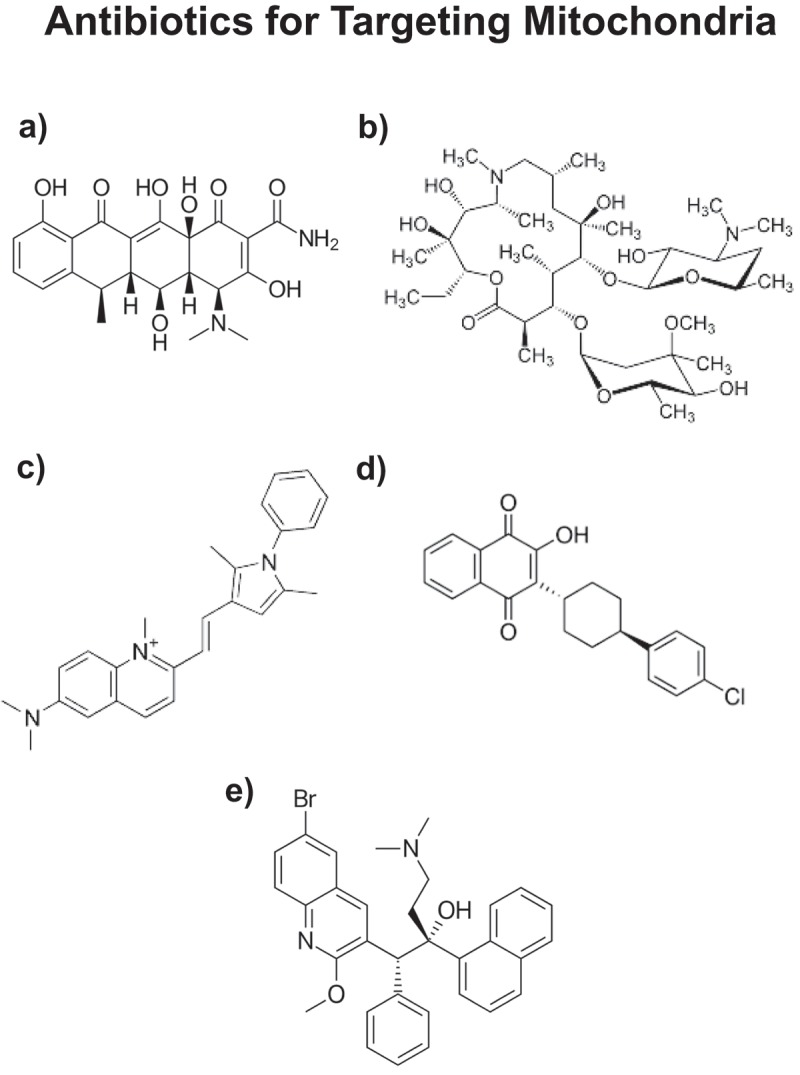
Repurposing FDA-approved antibiotics for targeting mitochondria in CSCs. a) Doxycycline; b) Azithromycin; c) Pyrvinium (pamoate salt; not shown); d) Atovaquone; and e) Bedaquiline. Doxycycline and Azithromycin (a,b) are known to inhibit mitochondrial protein translation as an off-target side effect. They are used clinically as antibiotics to inhibit bacterial protein synthesis. Similarly, Pyrvinium pamoate and Atovaquone (c,d) are known to inhibit OXPHOS (related to mitochondrial complex II/III), as a side effect. Bedaquiline (e) was originally designed to inhibit the bacterial ATP-synthase, which is analogous to mitochondrial complex V. All of these FDA-approved drugs (a-e) have been shown to inhibit the anchorage-independent propagation of CSCs, by targeting mitochondrial function.
Table 1.
FDA approved drugs successfully used to eradicate CSCs.
| Drug Name | Inhibition of | FDA-approved | Refs |
|---|---|---|---|
| Doxycycline | Mito Biogenesis | Yes | [16,19,22] |
| Tigecycline | Mito Biogenesis | Yes | [16] |
| Azithromycin | Mito Biogenesis | Yes | [16] |
| Pyrvinium Pamoate |
OXPHOS/Complex II | Yes | [16] |
| Atovaquone | OXPHOS/Complex III | Yes | [17] |
| Bedaquiline | Complex V | Yes | [18] |
| Palbociclib | CDK4/6 | Yes | [15] |
One aspect that all these antibiotics have in common is that they exert manageable anti-mitochondrial side-effects, which can be repurposed as their therapeutic effects, to eradicate CSCs [19,20]. More specifically, Doxcycyline and Azithromycin inhibit mitochondrial protein translation, effectively blocking new mitochondrial biogenesis [16–20]. Since most tissues already have large numbers of mitochondria (especially skeletal muscle, heart and brain), normal cells remain unaffected. In contrast, CSCs appear to be “addicted” to the generation of new mitochondria, providing a novel basis for the selectivity of these antibiotics in targeting CSCs. New mitochondrial biogenesis may also be required for asymmetric cell division in CSCs.
In addition, Pyrvinium pamoate and Atovaquone behave as mitochondrial OXPHOS inhibitors, affecting Complexes II and III [16,17]. Bedaquiline inhibits the mitochondrial ATP-synthase (Complex V) [18]. Finally, we also identified a series of experimental and natural compounds targeting CSCs, including glycolysis inhibitors (Vitamin C and Silibinin) [20] and mitochondrial inhibitors (Actinonin; CAPE, from Honey bee propolis) [20], as well as inhibitors of protein synthesis (puromycin) and NAD(+) recycling (FK-866) [20,21] (Figure 2 and 3).
Figure 2.
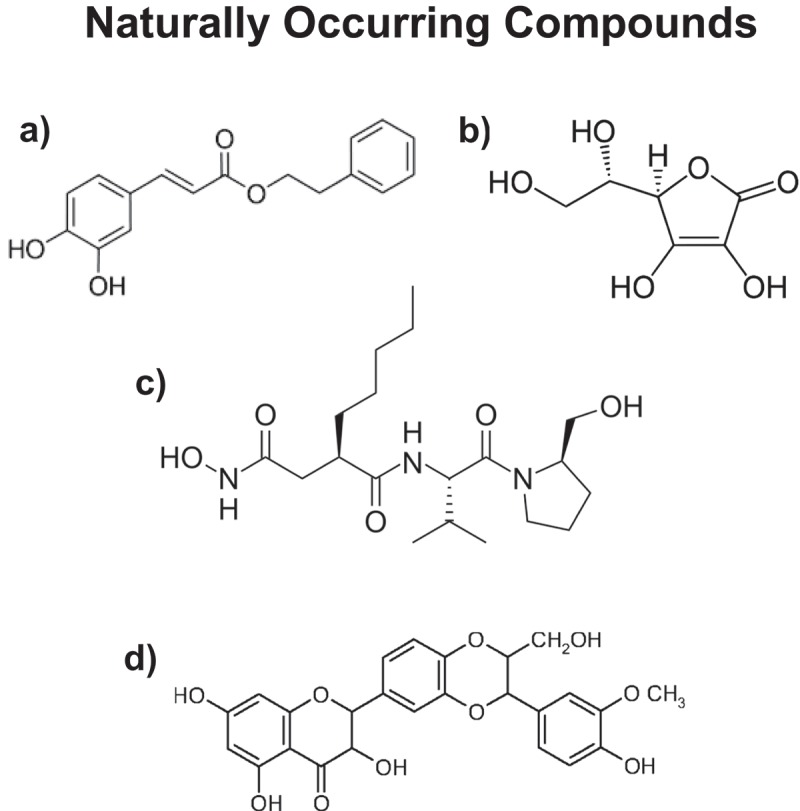
Natural products used for targeting CSCs. a) Caffeic acid phenyl ester (CAPE); b) Vitamin C (Ascorbic acid); c) Actinonin and d) Silibinin. CAPE (from bee propolis) inhibits OXPHOS; Vitamin C blocks GAPDH (a glycolytic enzyme), which is directly upstream of mitochondrial metabolism; Actinonin inhibits the initiation of both bacterial and mitochondrial protein translation; Silibinin (from milk thistle) decreases glucose uptake, by targeting the GLUT family of transporters. These natural products all inhibit CSC expansion.
Figure 3.
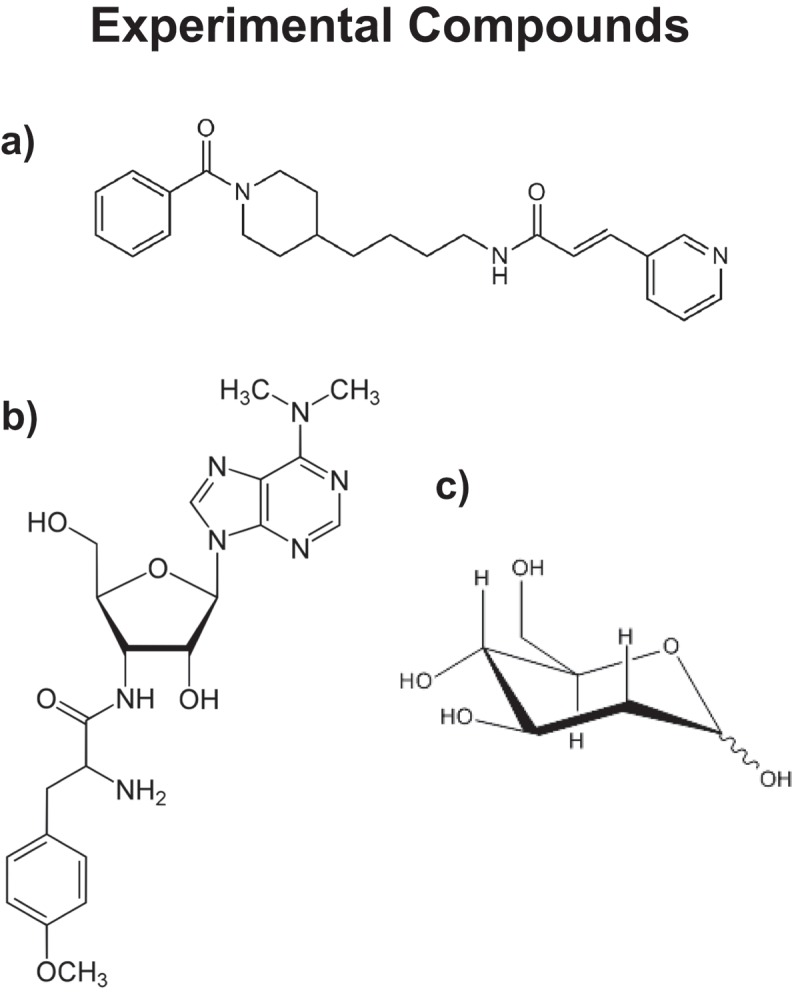
Experimental compounds used for targeting CSCs. a) FK-866 (NAMPT inhibitor); b) Puromycin; and c) 2-Deoxy-glucose (2-DG). FK-866 potently inhibits the NAD(+) salvage pathway; Puromycin inhibits protein synthesis (both cellular and mitochondrial), as it resembles the structure of a tRNA; 2-DG is a modified form of glucose, in which the OH-group at the 2 position has been removed and replaced with a hydrogen atom. As a consequence, it cannot undergo complete glycolysis and competitively inhibits the pathway, blocking the formation of glucose-6-phosphate from glucose. These three experimental compounds all block CSC propagation, by interfering with energy metabolism.
As CSCs appear to be highly proliferative, due to their over-expression of telomerase (hTERT), they are quite sensitive to Palbociclib, an FDA-approved CDK4/6 inhibitor, with an IC-50 of ~ 100 nM [15] (Table 1). Therefore, inhibition of CSC proliferation is an alternative or could be used in conjunction with anti-mitochondrial therapy [15].
Regarding therapy with Doxycycline, one possible concern that arises is the potential for the development of drug-resistance in CSCs [22]. To address this issue directly, we developed and characterized the phenotypic behavior of Doxy-resistant (DoxyR)-MCF7 cells [22]. Remarkably, what we observed is that DoxyR-CSCs show a dramatic shift towards aerobic glycolysis, due to a loss of mitochondrial function, ultimately resulting in metabolic inflexibility [22]. As expected, they showed an up to 35-fold loss of mitochondrial-DNA encoded proteins (mt-DNA) that are absolutely required for OXPHOS activity, such as MT-ND3, MT-CO2, MT-ATP6 and MT-ATP8 [22]. Also, DoxyR-CSCs appeared to be more “quiescent”, with > 50% reductions in proliferation and cell migration, as well as a significantly impaired ability to form 3D spheroids. As a consequence, our results showed that DoxyR-CSCs are especially sensitive to other metabolic therapies, including inhibitors of i) OXPHOS (Atovaquone, Irinotecan, Sorafenib, Niclosamide), ii) glycolysis (Vitamin C and Stiripentol) and iii) autophagy (Chloroquine).
Importantly, many of these drugs are already clinically-approved (Atovaquone, Irinotecan, Sorafenib, Niclosamide, Stiripentol, Chloroquine) or they are natural products (Vitamin C and Berberine), making them readily available for clinical trials (Table 2). Therefore, the efficacy of Doxycycline could be potentially improved, by developing combination therapies with these other metabolic inhibitors, based on the concepts of metabolic inflexibility and synthetic lethality in cancer cells [22].
Table 2.
Nine drugs, or nutraceuticals, that were successfully used in conjunction with Doxycycline, to eradicate CSCs.
| Drug Name | Target | FDA-approved |
|---|---|---|
| Atovaquone | OXPHOS | Yes |
| Irinotecan | OXPHOS | Yes |
| Sorafenib | OXPHOS | Yes |
| Niclosamide | OXPHOS | Yes |
| Berberine | OXPHOS | Natural supplement |
| 2-DG* | Glycolysis | Experimental |
| Vitamin C | Glycolysis | Natural supplement |
| Stiripentol | Glycolysis | Clinically-approved (Europe/Canada/Japan) |
| Chloroquine | Autophagy | Yes |
*2-deoxy-glucose; See reference [22], for additional information.
3. Mitochondrial-based therapeutic strategies for eradicating CSCs: developing new mitochondrial inhibitors
Because of the success of our drug repurposing efforts, we also decided to develop a series of new therapeutics, by focusing on key mitochondrial targets [23–26]. These results are briefly summarized in Table 3.
Table 3.
Novel anti-mitochondrial drug design: Four new therapeutic classes.
| Drug Name | Target | Metabolic Process | Refs |
|---|---|---|---|
| Mitoriboscins | Mito-ribosome | Mito Protein Synthesis | [23] |
| Mitoketoscins | OXCT1/ACAT1 | Ketone Metabolism | [24] |
| Mitoflavoscins | Mito Complex I/II | Flavin-containing proteins | [25] |
| TPP* | Mitochondria | Mito-targeting-signal(s) | [26] |
*Tri-phenyl-phosphonium
As a consequence, based on in silico drug design (i.e. computational chemistry) and phenotypic drug screening, we identified several new families of mito-ribosome inhibitors, termed Mitoriboscins [23], (Figure 4). They were specifically designed to target the mitochondrial ribosome, to inhibit mitochondrial protein synthesis. By targeting the mitochondrial enzymes OXCT1 and ACAT1, we also developed new mitochondrial inhibitors that interfere with ketone metabolism, by mimicking the structure of CoA [24], (Figures 5 and 6). In addition, we identified a novel approach to acutely induce a Vitamin B2 (riboflavin) deficiency, that potently inhibits CSC propagation, with an IC-50 of ~ 3 nM [25], (Figure 7, Upper). Thus, this drug is approximately 30 times more potent than Palbociclib for targeting CSCs.
Figure 4.
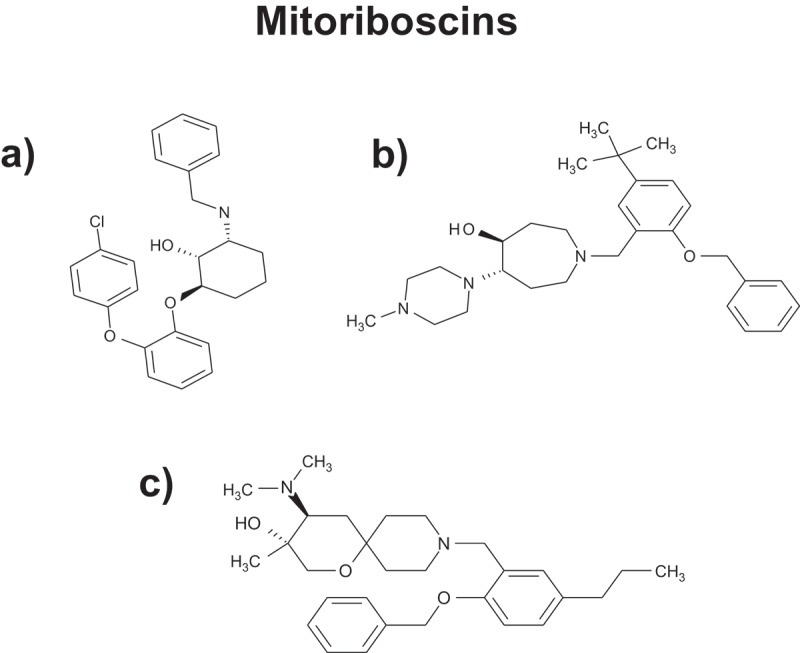
Mitoriboscins: Novel inhibitors for targeting the mitochondrial ribosome. Several examples of Mitoriboscins are illustrated. These compounds were identified by combining computational chemistry (in silico drug design), together with phenotypic library screening, to detect ATP depletion. The target used was the 3D structure of the large mitochondrial ribosome, as determined by cryo-EM (electron microscopy). For further details on these compounds, please see reference [23].
Figure 5.
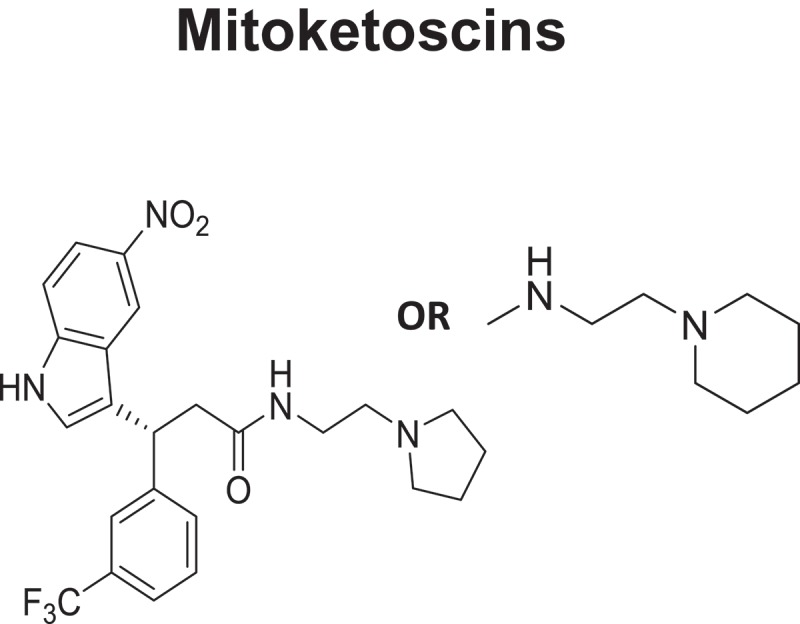
Mitoketoscins: Therapeutics for targeting mitochondrial ketone metabolism. The pharmacaphore for Mitoketoscins is shown. These compounds were identified by combining computational chemistry (in silico drug design), together with phenotypic library screening, to detect ATP depletion. The targets used were the crystal structures of OXCT1 and ACAT1, mitochondrial enzymes involved in the conversion of serum ketone bodies back into Acetyl-CoA, for use in the TCA cycle.
Figure 6.
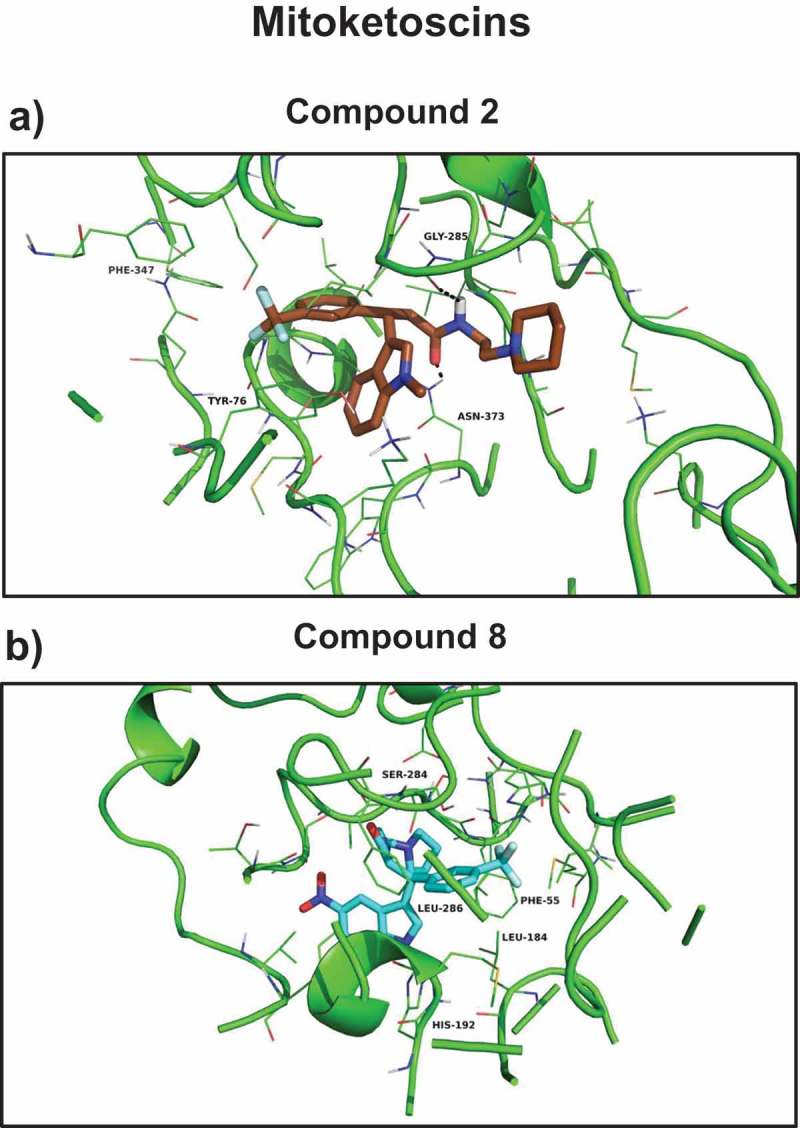
Mitoketoscins: Docking images with OXCT1 and ACAT1. (a) Compound 2 docking at the succinyl-CoA binding site of 3-oxoacid CoA-transferase 1 (OXCT1); (b) compound 8 docking at the CoA binding site of human acetyl-CoA acetyltransferase (ACAT1). Modified and reproduced from [24].
Figure 7.
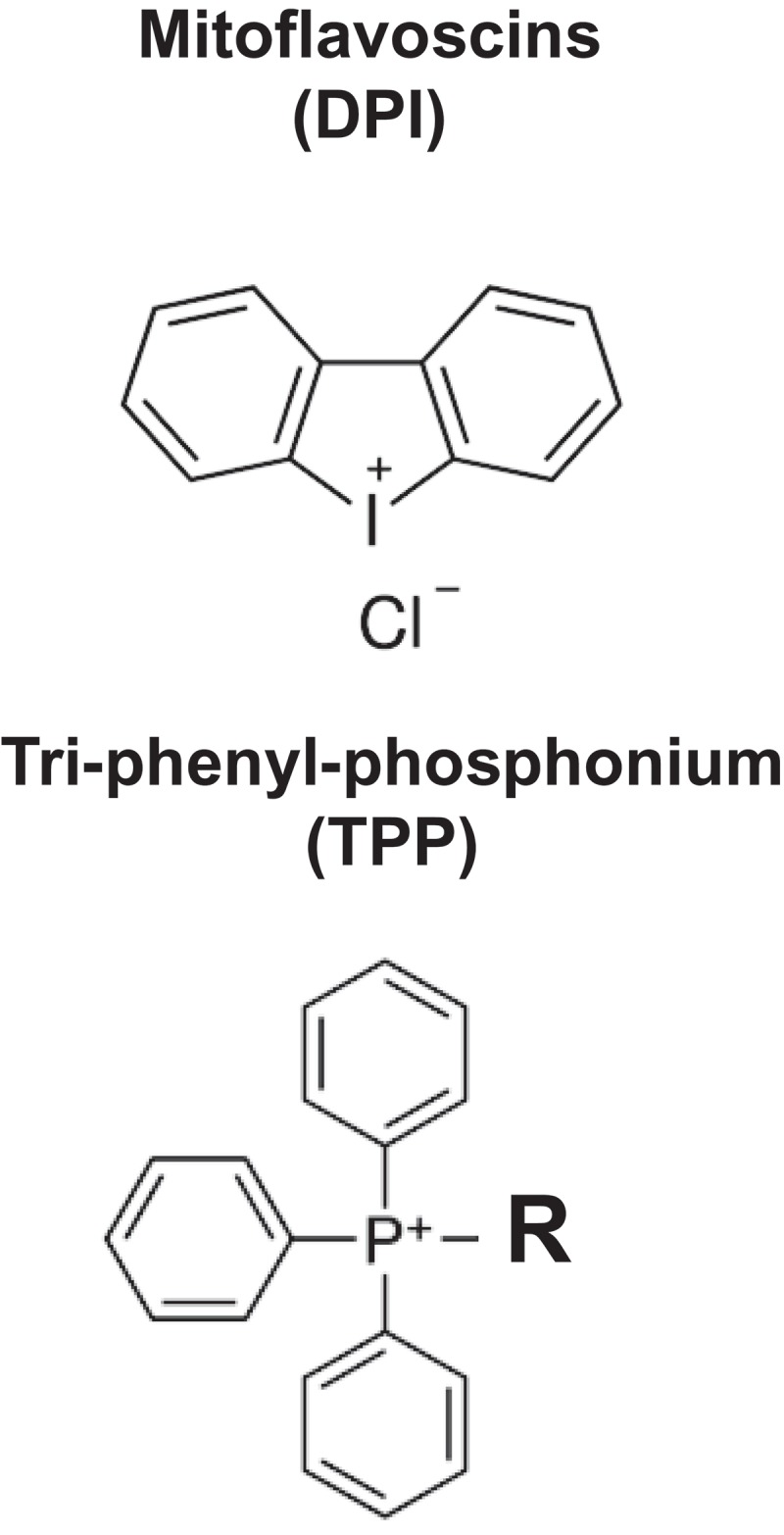
Mitoflavoscins and Tri-phenyl-phosphonium (TPP). Upper, The structure of DPI (Diphenyleneiodonium chloride), a Mitoflavoscin, is shown. Lower, The structure of a representative TPP compoundis is shown.
The use of tri-phenyl-phosphonium (TPP) also appears to be a very promising approach, as it behaves as a chemical mitochondrial targeting signal (MTS) (Figure 7, Lower) [26]. Interestingly, a series of TPP compounds we tested were non-toxic in normal fibroblasts, but were selectively toxic in “bulk” cancer cells and CSCs [26]. Therefore, these TPP compounds appear to be able to metabolically distinguish between “normal cell” mitochondria and “malignant” mitochondria [26]. Since “bulk” cancer cells and CSCs likely have mitochondria with higher membrane potentials, this could explain the selective targeting of cancer cell mitochondria with TPP compounds.
Finally, we have begun to search for new naturally-occurring mitochondrial inhibitors, that can be used to more effectively target CSCs (Table 4). In this context, we recently showed that two molecules that are novel components of Bergamot, namely Brutieridin and Melitidin, act as specific Statin-like drugs and inhibit mevolonate metabolism, as well as CSC propagation (Figure 8) [27]. Interestingly, Bergamot is currently used as a fragrance in many health and well-being products (soaps and perfumes), and is a significant component of Earl Grey English breakfast tea.
Table 4.
Other experimental mitochondrial inhibitors for targeting CSCs.
| Drug Name | Target | Metabolic Process Inhibited | Refs |
|---|---|---|---|
| mDIVI1 | DRP1* | Mitochondrial Fission/Fusion | [28] |
| Brutieridin | HMGR** | Mevalonate Metabolism; Statin-like | [27] |
| Melitidin | HMGR** | Mevalonate Metabolism; Statin-like | [27] |
*Dynamin-related protein 1
**3-hydroxy-3-methylglutaryl-CoA-reductase.
Figure 8.
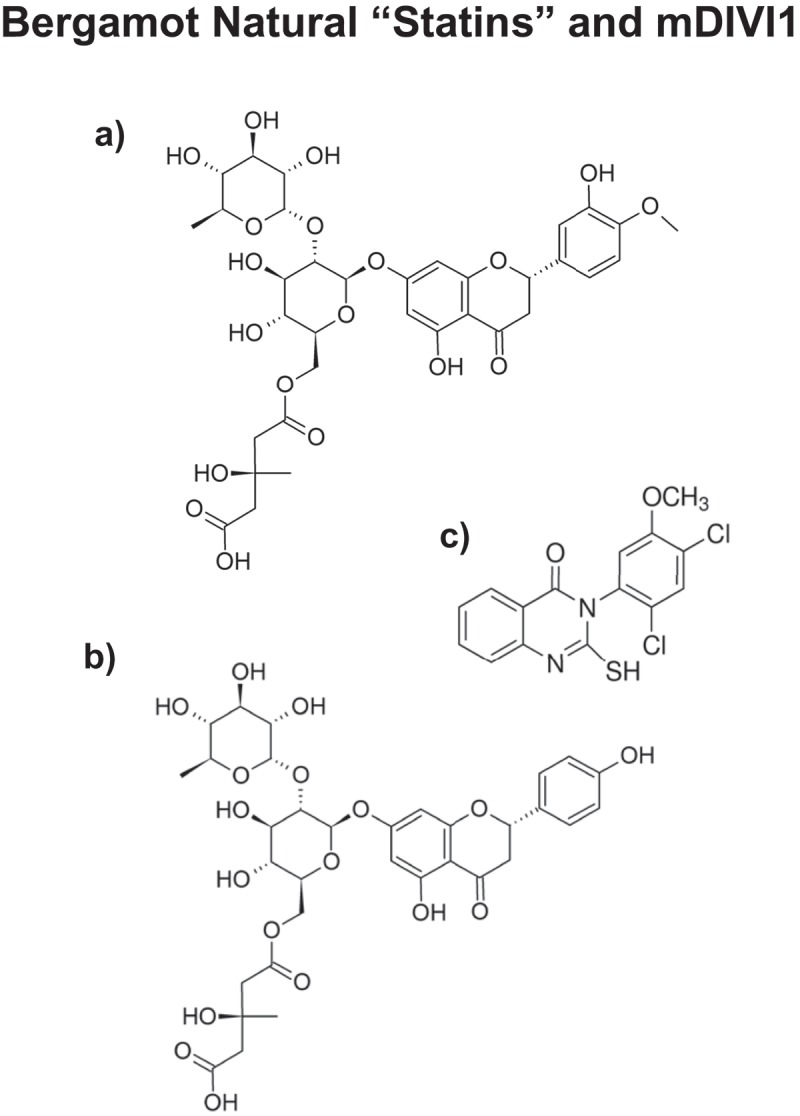
Components of Bergamot function as natural “statin-like” molecules. The chemical structures of a) Brutieridin and b) Melitidin are shown. The structure of c) mDIVI1 is also shown for comparison.
Interference with the normal process of mitochondrial fission-fusion cycles, by targeting the DRP1 protein, may also represent a new viable strategy for eradicating CSCs (See Figure 8 and Table 4) [28].
4. Mitochondrial inhibitors also show broad-spectrum antibiotic activity
The identification and design of new mitochondrial inhibitors has other interesting medical applications and benefits, such as the development of new anti-bacterial and anti-fungal agents, to combat antibiotic-resistance. According to the “Endo-symbiotic Theory of Mitochondrial Evolution”, mitochondria first originated historically from the engulfment of aerobic bacteria, an event that occurred ~ 1.45 billion years ago [29–32]. As a result, mitochondria still share strong structural and functional similarities with bacteria, explaining the off-target effects of antibiotics, which often show manageable mitochondrial side-effects [29–32]. Conversely, it would then be predicted that mitochondrial inhibitors may also show some moderate anti-bacterial and anti-fungal side effects [29–32].
To directly test this hypothesis, we evaluated the anti-bacterial and anti-fungal activity of the Mitoriboscins [23]. Interestingly, several Mitoriboscins showed anti-bacterial activity towards both gram-positive and gram-negative organism(s), as well as pathogenic yeast (Candida albicans) and even Methicillin-resistant Staphylococcus aureus (MRSA) [23]. Therefore, this systematic approach, using cancer cells for initial drug screening, may also be useful for developing new antibiotics, to combat drug-resistant micro-organisms [23].
5. Mitochondrial-based companion diagnostics: treatment stratification and predicting the response to therapy
Over one thousand mitochondrial proteins are encoded by the nuclear genome. Therefore, we have begun to assess their potential prognostic value as novel biomarkers and companion diagnostics. Our hypothesis was that the over-expression of a given mitochondrial protein in cancer cells and CSCs may be associated with tumor recurrence and metastasis, due to the emergence of drug resistance, ultimately resulting in treatment failure (Figure 9).
Figure 9.
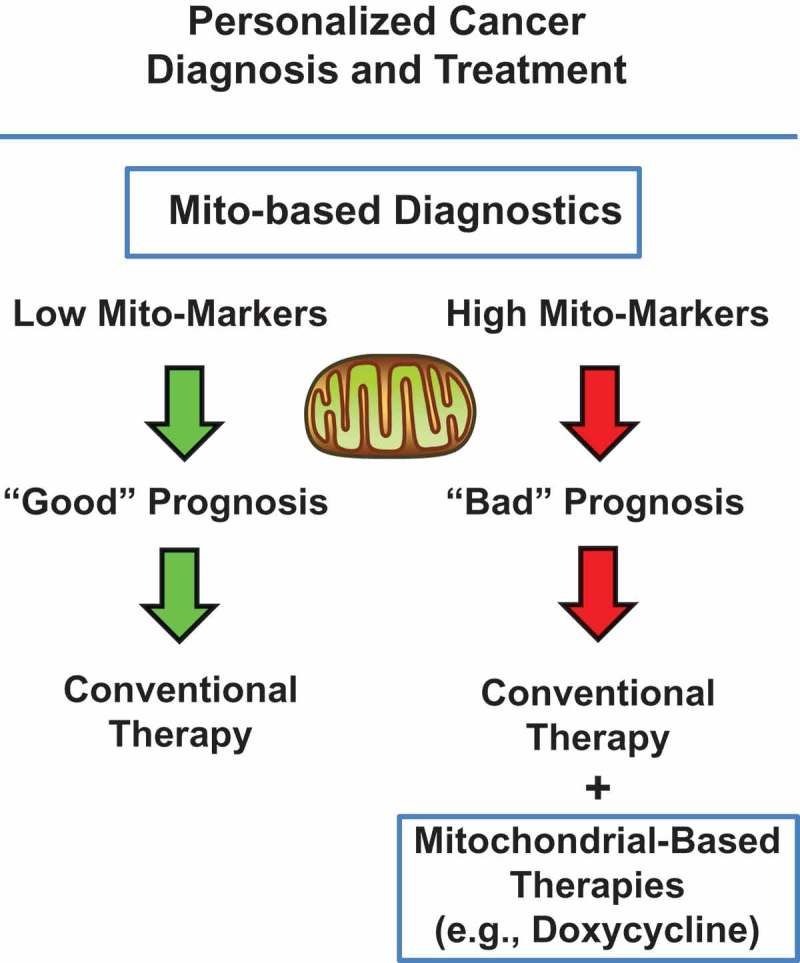
Identifying mitochondrial-based companion diagnostics for more personalized cancer therapy. Briefly, mitochondrial markers could be used to stratify cancer patients into high-risk and low-risk groups, at diagnosis. Patients with high levels of mitochondrial markers (“bad prognosis”) could be treated with mitochondrial-based therapies, as an add-on to the standard of care, in order to effectively prevent tumor recurrence, metastasis and drug-resistance. Modified from [32,33].
To test this hypothesis, we used an online survival-analysis tool (known as the KM-plotter; http://kmplot.com/analysis/) to perform Kaplan-Meier (K-M) studies on > 400 nuclear mitochondrial gene transcripts, to interrogate publically available microarray data from patients with four distinct epithelial cancer types: i) breast, ii) ovarian, iii) lung and iv) gastric [32,33]. This approach allowed us to directly perform in silico validation of these potential mitochondrial biomarkers [33,34].
Remarkably, in all four anatomic cancer types, we observed that the over-expression of mitochondrial gene transcripts was specifically associated with poor clinical outcome. Importantly, this approach effectively predicted tamoxifen-resistance in ER(+) breast cancer patients (Figure 10), as well as Taxol- and Platin-resistance in ovarian cancer patients (Figure 11) [32,33]. These results are functionally supported by further experimental observations demonstrating that Tamoxifen-resistant MCF7 cells (TAMR) show a significant increase in mitochondrial oxygen consumption and ATP production [35].
Figure 10.
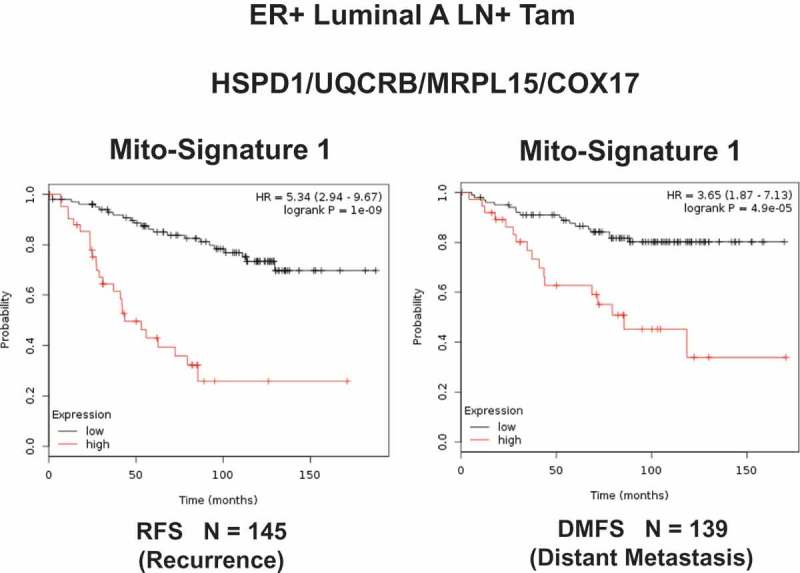
A short mitochondrial gene signature predicts tumor recurrence and distant metastasis, in high-risk breast cancer patients, receiving endocrine therapy. Note that high expression of this Mito-Signature predicts treatment failure on Tamoxifen (Tam). These represent ER-positive patients, with the luminal A sub-type of breast cancer, who showed local lymph node metastasis at diagnosis, with 10–15 y of follow-up data. RFS, recurrence-free survival; DMFS, distant metastasis-free survival. Reproduced with permission from [32].
Figure 11.
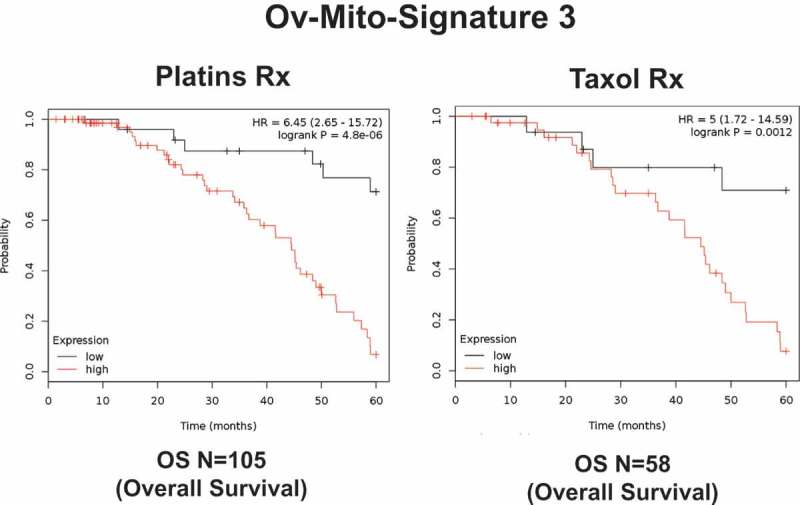
A short mitochondrial gene signature predicts drug-resistance and treatment failure in ovarian cancer patients. Note that high expression of this Mito-Signature predicts treatment failure on Platins and Taxol, in ovarian cancer patients. These patients with serous ovarian cancer had at least 5-years of follow-up data. Reproduced with permission from [33].
Therefore, in the future, we propose that high-risk patients could be first identified at diagnosis, by the high expression of mitochondrial markers or “Mito-signatures” in their primary tumors [33,34]. Here, we define “Mito-signatures” as combinations of mitochondrial markers (mRNA levels) that can be used to more accurately predict clinical outcome in patient populations.
Then, these patients could be also treated with existing FDA-approved antibiotics or novel mitochondrial therapeutics, as an add-on to conventional anti-cancer therapy [33,34].
6. Other advantages of anti-mitochondrial therapy with Doxycycline
Doxycycline shows many other interesting anti-cancer properties that should be further explored. For example, Doxycycline behaves as a radio-sensitizer, making CSCs approximately 3 to 5 times more sensitive to radiation treatment [36]. In addition, Doxycycline effectively targets hypoxic CSCs and overcomes Paclitaxel-resistance, under conditions of hypoxia; this may have important implications for achieving more effective anti-angiogenic therapy [37]. Finally, Doxycycline appears to be effective as a mutation-independent approach for targeting CSCs, as it inhibits both activated H-Ras (G12V) and c-Myc oncogenes, as well as other environmental oncogenic stimuli (mitochondrial oxidative stress/ROS), via the specific targeting of mitochondrial biogenesis [38].
7. Conclusions
In conclusion, we propose a new, integrated, multi-disciplinary, medical oncology platform to target cancer stem cells (CSCs), in most cancer types. This new mitochondrial oncology platform, known as MITO-ONC-RX, consists of both therapeutic and diagnostic modalities (Figure 12). As increased mitochondrial biogenesis and OXPHOS are key characteristics of CSCs, we propose to target metabolism in CSCs, using FDA drug repurposing and/or new first-in-class therapeutics, such as the i) Mitoriboscins, the ii) Mitoketoscins and the iii) Mitoflavoscins, as well as iv) TPP-based compounds. In this context, we have also discovered a series of mitochondrial markers (companion diagnostics) for identifying and selecting the high-risk cancer patient population that is most likely to benefit from anti-mitochondrial therapy.
Figure 12.
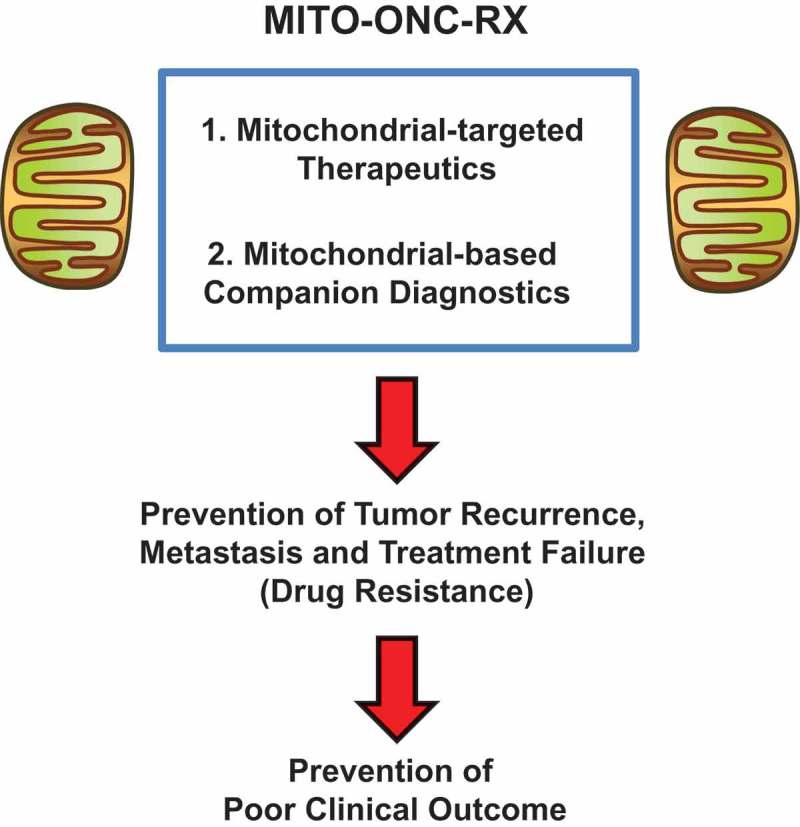
A mitochondrial-based oncology platform for cancer therapy: MITO-ONC-RX. This oncology platform would consist of two basic components: i) Mito-therapeutics and ii) Mito-diagnostics. These components could then be tailored and optimized, for a given cancer type.
The essential role of mitochondria in the propagation of CSCs has now been confirmed and extended by other laboratories, in London and at MD Anderson Cancer Center, as well as other institutions (See references #2 and #39, for specific citations). In further support of this notion, please see the following articles for further reading on the subject [39–45].
Funding Statement
In recent years, our overall research goals have been supported by the University of Manchester, the University of Salford, British Schools and Universities Foundation, the Healthy Life Foundation and the Foxpoint Foundation, as well as Lunella Biotech, Inc.
Acknowledgments
We are grateful to the University of Manchester, which allocated start-up funds and administered a donation, to provide all the necessary resources required to start and complete these drug discovery projects. At the University of Salford, the Sotgia and Lisanti Laboratories were supported by the British Schools and Universities Foundation, the Healthy Life Foundation and the Foxpoint Foundation. New ongoing studies are also supported by grant funding provided by Lunella Biotech, Inc.
Author contributions
Professors Lisanti and Sotgia conceived and prepared the initial draft of this review article, including the figures and tables. This draft manuscript was then further edited, by Drs. Bela Ozsvari, Marco Fiorillo, Ernestina Marianna De Francesco and Gloria Bonuccelli.
Disclosure statement
MPL and FS hold a minority interest in Lunella Biotech, Inc.
References
- [1].Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, et al. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017. January;14(1):11–31. [DOI] [PubMed] [Google Scholar]
- [2].Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, et al. Cancer stem cell metabolism. Breast Cancer Res. 2016. May 24;18(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Aguilar-Gallardo C, Simón C.. Cells, stem cells, and cancer stem cells. Semin Reprod Med. 2013. January;31(1):5–13. [DOI] [PubMed] [Google Scholar]
- [4].Apostoli AJ, Ailles L. Clonal evolution and tumor-initiating cells: new dimensions in cancer patient treatment. Crit Rev Clin Lab Sci. 2016;53(1):40–51. [DOI] [PubMed] [Google Scholar]
- [5].Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the Reverse Warburg Effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017. June;44(3):198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Reina-Campos M, Moscat J, Diaz-Meco M. Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol. 2017. October;48:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Nowell PC. The clonal evolution of tumor cell populations. Science. 1976. October 1;194(4260):23–28. [DOI] [PubMed] [Google Scholar]
- [8].Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012. January 18;481(7381):306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Visvader JE. Cells of origin in cancer. Nature. 2011. January 20;469(7330):314–322. [DOI] [PubMed] [Google Scholar]
- [10].Dando I, Dalla Pozza E, Biondani G, et al. The metabolic landscape of cancer stem cells. IUBMB Life. 2015. September;67(9):687–693. [DOI] [PubMed] [Google Scholar]
- [11].Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013. September 19;501(7467):328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017. February 16;16(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lamb R, Harrison H, Hulit J, et al. Mitochondria as new therapeutic targets for eradicating cancer stem cells: quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014. November 30;5(22):11029–11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Farnie G, Sotgia F, Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget. 2015. October 13;6(31):30472–30486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bonuccelli G, Peiris-Pages M, Ozsvari B, et al. Targeting cancer stem cell propagation with palbociclib, a CDK4/6 inhibitor: telomerase drives tumor cell heterogeneity. Oncotarget. 2017. February 7;8(6):9868–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015. March 10;6(7):4569–4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fiorillo M, Lamb R, Tanowitz HB, et al. Repurposing atovaquone: targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget. 2016. June 7;7(23):34084–34099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fiorillo M, Lamb R, Tanowitz HB, et al. Bedaquiline, an FDA-approved antibiotic, inhibits mitochondrial function and potently blocks the proliferative expansion of stem-like cancer cells (CSCs). Aging (Albany NY). 2016. August 8;8(8):1593–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].De Luca A, Fiorillo M, Peiris-Pagès M, et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget. 2015. June 20;6(17):14777–14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bonuccelli G, De Francesco EM, de Boer R, et al. NADH autofluorescence, a new metabolic biomarker for cancer stem cells: identification of Vitamin C and CAPE as natural products targeting “stemness”. Oncotarget. 2017. March 28;8(13):20667–20678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lamb R, Harrison H, Smith DL, et al. Targeting tumor-initiating cells: eliminating anabolic cancer stem cells with inhibitors of protein synthesis or by mimicking caloric restriction. Oncotarget. 2015. March 10;6(7):4585–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].De Francesco EM, Bonuccelli G, Maggiolini M, et al. Vitamin C and Doxycycline: A synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget. 2017. June 9;8(40):67269–67286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ozsvari B, Fiorillo M, Bonuccelli G, et al. Mitoriboscins: mitochondrial-based therapeutics targeting cancer stem cells (CSCs), bacteria and pathogenic yeast. Oncotarget. 2017. July 7;8(40):67457–67472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ozsvari B, Sotgia F, Simmons K, et al. Mitoketoscins: novel mitochondrial inhibitors for targeting ketone metabolism in cancer stem cells (CSCs). Oncotarget. 2017. September 24;8(45):78340–78350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ozsvari B, Bonuccelli G, Sanchez-Alvarez R, et al. Targeting flavin-containing enzymes eliminates cancer stem cells (CSCs), by inhibiting mitochondrial respiration: vitamin B2 (Riboflavin) in cancer therapy. Aging (Albany NY). 2017. December 16;9(12):2610–2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ozsvari B, Sotgia F, Lisanti MP. Exploiting mitochondrial targeting signal(s), TPP and bis-TPP, for eradicating cancer stem cells (CSCs). Aging (Albany NY). 2018. February 19;10(2):229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fiorillo M, Peiris-Pagès M, Sanchez-Alvarez R, et al. Bergamot natural products eradicate cancer stem cells (CSCs) by targeting mevalonate, Rho-GDI-signalling and mitochondrial metabolism. BBA Bioenergetics. 2018;1859:984–996. In Press. [DOI] [PubMed] [Google Scholar]
- [28].Peiris-Pagès M, Bonuccelli G, Sotgia F, et al. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget. 2018. January 19;9(17):13254–13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Martin W, Mentel M. The origin of mitochondria. Nat Educ. 2010;3(9):58. [Google Scholar]
- [30].MW Gray. Mitochondrial Evolution. Cold Spring Harb Perspect Biol. 2012. September;4(9):a011403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Martin WF, Garg S, Zimorski V. Endosymbiotic theories for eukaryote origin. Philos Trans R Soc Lond B Biol Sci. 2015. September 26;370(1678):20140330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zimorski V, Ku C, Martin WF, et al. Endosymbiotic theory for organelle origins. Curr Opin Microbiol. 2014. December;22:38–48. [DOI] [PubMed] [Google Scholar]
- [33].Sotgia F, Fiorillo M, Lisanti MP. Mitochondrial markers predict recurrence, metastasis and tamoxifen-resistance in breast cancer patients: early detection of treatment failure with companion diagnostics. Oncotarget. 2017. July 27;8(40):68730–68745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sotgia F, Lisanti MP. Mitochondrial mRNA transcripts predict overall survival, tumor recurrence and progression in serous ovarian cancer: companion diagnostics for cancer therapy. Oncotarget. 2017. August 6;8(40):66925–66939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fiorillo M, Sotgia F, Sisci D, et al. Mitochondrial “power” drives tamoxifen resistance: NQO1 and GCLC are new therapeutic targets in breast cancer. Oncotarget. 2017. March 2;8(12):20309–20327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lamb R, Fiorillo M, Chadwick A, et al. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: implications for more effective radiation therapy. Oncotarget. 2015. June 10;6(16):14005–14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].De Francesco EM, Maggiolini M, Tanowitz HB, et al. Targeting hypoxic cancer stem cells (CSCs) with Doxycycline: implications for optimizing anti-angiogenic therapy. Oncotarget. 2017. June 12;8(34):56126–56142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ozsvari B, Sotgia F, Lisanti MP. A new mutation-independent approach to cancer therapy: inhibiting oncogenic RAS and MYC, by targeting mitochondrial biogenesis. Aging (Albany NY). 2017. October 27;9(10):2098–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].De Francesco EM, Sotgia F, Lisanti MP. Cancer stem cells (CSCs): metabolic strategies for their identification and eradication. Biochem J. 2018. May 9;475(9):1611–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kalghatgi S, Spina CS, Costello JC, et al. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci Transl Med. 2013. July 3;5(192):192ra85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Song IS, Jeong JY, Jeong SH, et al. Mitochondria as therapeutic targets for cancer stem cells. World J Stem Cells. 2015. March 26;7(2):418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Cuyàs E, Martin-Castillo B, Corominas-Faja B, et al. Anti-protozoal and anti-bacterial antibiotics that inhibit protein synthesis kill cancer subtypes enriched for stem cell-like properties. Cell Cycle. 2015;14(22):3527–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Shen YA, Wang CY, Hsieh YT, et al. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle. 2015;14(1):86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016. June 14;114(12):1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Loureiro R, Mesquita KA, Magalhães-Novais S, et al. Mitochondrial biology in cancer stem cells. Semin Cancer Biol. 2017. December;47:18–28. [DOI] [PubMed] [Google Scholar]