

ABSTRACT
Ultrafine anaphase bridges (UFBs) are a potential source of genome instability that is a hallmark of cancer. UFBs can arise from DNA catenanes at centromeres/rDNA loci, late replication intermediates induced by replication stress, and DNA linkages at telomeres. Recently, it was reported that DNA intertwinements generated by homologous recombination give rise to a new class of UFBs, which have been termed homologous recombination ultrafine bridges (HR-UFBs). HR-UFBs are decorated with PICH and BLM in anaphase, and are subsequently converted to RPA-coated, single-stranded DNA bridges. Breakage of these sister chromatid entanglements leads to DNA damage that can be repaired by non-homologous end joining in the next cell cycle, but the potential consequences include DNA rearrangements, chromosome translocations and fusions. Visualisation of these HR-UFBs, and knowledge of how they arise, provides a molecular basis to explain how upregulation of homologous recombination or failure to resolve recombination intermediates leads to the development of chromosomal instability observed in certain cancers.
KEYWORDS: Recombination intermediate, Holliday junction, chromosome segregation, chromosomal instability, 53BP1, MUS81
Introduction
Sister chromatid non-disjunction occurs when sister chromatids remain physically connected at the onset of anaphase, leading to chromosome mis-segregation events that are commonly observed in cancer cells [1,2]. UFBs, visualized as long fine DNA threads that cannot be detected by conventional DNA staining and are devoid of histones, were previously identified as a special class of mitotic DNA structures that interlink two separating sister chromatids [3,4]. Their discovery came from the immunofluorescent staining of proteins that bind them, including BLM (Bloom’s syndrome helicase), PICH (PLK1-interacting checkpoint helicase) and RPA (replication protein A) [5–8].
UFBs can be classified by both the genomic loci from which they originate and their underlying structures. Four major types of UFB have previously been described (Figure 1). First, the most common UFBs are centromeric UFBs (C-UFBs) that arise from double-stranded DNA (dsDNA) catenanes at centromeres, and are characterized by the association of centromeric markers (e.g. CENP-A, HEC1) at the bridges’ termini. C-UFBs exist in every mitosis and their numbers are increased by treatment with the topoisomerase IIα inhibitor ICRF-193, indicating that they are readily removed by this topoisomerase [5,6,9,10]. Second, DNA catenanes that persist at ribosomal DNA (rDNA) loci give rise to R-UFBs that colocalise with the ribosomal RNA transcription factor UBF, a marker for rDNA [11]. Third, fragile site UFBs (FS-UFBs) arise from late replication intermediates at common fragile sites (CFSs) where replication is often delayed, especially under conditions that induce replication stress (e.g. treatment with the DNA polymerase inhibitor aphidicolin). The Fanconi anemia proteins, FANCI and FANCD2 associate with CFSs after replication stress and localize to the termini of FS-UFBs [8,12–14]. Finally, telomeric UFBs (T-UFBs) can be induced by interfering with the replication of telomeres or by overexpression of the shelterin component TRF2 that induces chromosome end-to-end fusions [15–17]. Inhibition of topoisomerase IIα also induces T-UFBs [18], indicating that T-UFBs consist of DNA replication intermediates and catenanes.
Figure 1.

Schematic diagram indicating the five types of anaphase UFBs. (1) Centromeric UFBs (C-UFBs) emerge from centromeres, possess double-stranded catenanes, and can be induced by inhibition of topoisomerase IIα by ICRF-193. (2) Ribosomal UFBs (R-UFBs) emerge from catenated rDNA and are marked by UBF (green circles). (3) Fragile site UFBs (FS-UFBs) emerge from incompletely replicated DNA at CFSs and are flanked by FANCD2 twin foci (yellow rhombus). They can be induced by DNA polymerase inhibitors (e.g. aphidicolin) that induce replication stress. (4) Telomeric UFBs (T-UFBs) originate from telomeric regions and can be induced by replication stress and/or overexpression of the shelterin protein TRF2 leading to telomere fusions. (5) Homologous recombination UFBs (HR-UFBs) originate from unresolved recombination intermediates. They can be induced by inhibition of GEN1 and MUS81, two nucleases that mediate Holliday junction resolution, or by depletion of 53BP1 which leads to upregulation of homologous recombination.
Conditions that increase the frequently of UFB formation, or interfere with UFB resolution by inhibiting the functions of UFB-binding proteins, can lead to DNA damage and cell division defects, such as cytokinesis failure and micronucleus formation [19,20]. Therefore, it is important to understand how UFBs originate and how they are resolved before cytokinesis.
Unresolved DNA intermediates that arise from homologous recombination (HR) provide a covalent linkage between sister chromatids, and were also proposed to generate DNA bridges/UFBs that interfere with proper chromosome segregation [21]. Recently, two laboratories confirmed this notion and identified a new class of UFBs that are generated by homologous recombination [22,23]. In this article, we summarize and discuss how HR-UFBs are generated, which proteins are recruited, and their roles in bridge processing. Finally, we describe how HR-UFBs can lead to chromosomal instability.
The origin of HR-UFBs
HR-UFBs arise from the recombinational repair of DNA damage, usually double-stranded breaks (DSBs). Breaks are repaired by DNA end resection followed by invasion of the resulting 3ʹ-single-stranded DNA (ssDNA) tail into the homologous sister chromatid to form a D-loop structure, which then serves as an initiation site for subsequent DNA synthesis [24]. HR often leads to the formation of DNA joint molecules in which the two recombining DNAs are covalently connected by a four-way DNA junction or Holliday junction (HJ) [25–27]. These intermediates can result in chromosome segregation defects if they are not processed before anaphase onset [28–32].
Recombination intermediates can be removed by two primary mechanisms. The first involves the BTR complex (BLM–Topoisomerase IIIα–RMI1–RMI2), which mediates the dissolution of double HJs [33–38]. Persistent recombination intermediates that escape the attentions of BTR, or are refractory to dissolution (e.g. single HJs and D-loop structures), are processed by a second mechanism which involves structure-selective endonuclease (SSE)-mediated resolution [31,32,39,40]. There are two genetically distinct resolution pathways: one is mediated by the SLX1–SLX4, MUS81-EME1 and XPF-ERCC1 (SMX) tri-nuclease complex [27,31,41–46], and the other is mediated by GEN1 endonuclease [47–50].
Recently, we analysed the cellular consequences of inactivating these resolution pathways in human cells by targeting GEN1 and MUS81 [22], and observed that resolvase-deficient cells undergo a cell cycle delay and massive cell death. These phenotypes are due to the accumulation of unresolved recombination intermediates that persist until anaphase and give rise to a high frequency of UFBs that are decorated with PICH, BLM and RPA (Figure 2). They were termed homologous recombination ultrafine bridges (HR-UFBs) to distinguish them from centromeric UFBs, replication stress-associated UFBs, and telomeric UFBs (Figure 1).
Figure 2.

HR-UFBs arise in resolvase-deficient cells. U2OS cells were treated with siRNA against MUS81 and GEN1 to inactivate the SMX and GEN1 Holliday junction resolvases. 24 hours after siRNA transfection, the cells were treated with cisplatin (1 μM for 1 h and released into fresh media for 24 h) in order to induce DNA damage. RPA2, BLM and DNA were visualized using anti-RPA2 antibody (green), anti-BLM antibody (red), and DAPI (blue). Images were acquired using a Zeiss AXIO imager M2 microscope. Scale bar, 10 μm. For detailed methods, see Chan et al., 2018.
In parallel with these studies, work from another laboratory also described HR-generated UFBs that arose in 53BP1-depleted human cancer cells [23]. 53BP1 is regarded as a “gatekeeper” of DSB repair that plays a crucial role in determining DSB-repair pathway choice. In G1 phase of the cell cycle, 53BP1 rapidly accumulates at DSB sites and antagonizes DNA end resection, thereby favouring non-homologous end joining (NHEJ). In S and G2 phase, however, BRCA1 promotes the removal of 53BP1 to allow resection and DSB repair by HR [51,52]. Recently, 53BP1 was also proposed to suppress excessive DSB resection in S/G2 phase to prevent highly mutagenic form of HR (e.g. repair via single-strand annealing) [53]. Tiwari et al. observed that 53BP1 depletion in cancer cells leads to sister chromatid non-disjunction mediated by UFBs. They proposed that loss of 53BP1 activity leads to a distinct type of replication intermediate which are converted to DNA joint molecules by HR. Alternatively, loss of 53BP1 may favour the initiation of HR at stalled/damaged replication forks.
Several observations support the notion that HR-UFBs are distinct from all types of previously described UFBs [22,23]. First, in contrast to FS-UFBs, HR-UFBs do not associate with FANCD2 foci, a marker of late replication intermediates. Second, inhibition of the HR machinery by depletion of RAD51 or BRCA2 suppressed the formation of these FANCD2-negative UFBs. Finally, expression of the bacterial HJ resolvase RusA reduced the formation of HR-UFBs in resolvase (GEN1 and MUS81)-deficient cells. Together, these studies concluded that HR-UFBs are induced either when recombination intermediates fail to be resolved or when HR-mediated repair is upregulated.
Interestingly, regions of sister chromatid bridging induced by 53BP1 depletion mapped close to a well-known fragile site (FRA16D in the WWOX locus) and to centromeres [23]. These results indicate that genomic loci that show high fragility and spontaneous breakage are likely to be repaired by HR in the absence of the anti-recombinogenic activity of 53BP1, leading to increased HR-mediated sister DNA intertwinements that are visualized as UFBs in anaphase. On the other hand, HR-UFBs observed in resolvase-deficient cells are not associated with centromeres [22], indicating that HR rarely occurs between sister centromeres in the presence of 53BP1. In future, it would be interesting to identify the genomic loci that are prone to give rise to HR-UFBs in undamaged resolvase-deficient cells as they may represent novel hotspots of chromosome breakage and recombination.
Proteins that recognize and process HR-UFBs
PICH appears to be the first protein recruited to all known types of UFBs. It is recruited to HR-UFBs in early anaphase when the two sister chromatids are just separating [22,23]. PICH is a member of the SNF2 family of DNA-dependent ATPases that possesses dsDNA translocase activity [5]. A key property of PICH is that it displays a high affinity for stretched dsDNA [54]. Therefore, PICH may act as a “tension sensor” to decorate UFBs as they are put under tension generated by the mitotic spindle. It has been shown that the timely resolution of C-UFBs depends on the ATPase activity of PICH as replacement of wild-type PICH with an ATPase-dead mutant of PICH prolongs C-UFB persistence [20]. Whether the ATPase activity of PICH is involved in resolving HR-UFBs remains to be determined.
PICH colocalizes with topoisomerase IIα on C-UFBs and stimulates topoisomerase IIα-mediated decatenation activity in vitro [20]. It also serves as the main recruitment factor for a variety of proteins to UFBs. One of the most important and well-studied UFB-binding proteins recruited by PICH is the Bloom’s syndrome helicase BLM [6,19]. BLM is a RecQ family helicase that can efficiently unwind a variety of DNA structures [55]. BLM interacts with topoisomerase IIIα, RMI1 and RMI2 to form the BTR complex that mediates the dissolution of double HJs. RMI1 and topoisomerase IIIα were shown to colocalise with BLM on C-UFBs, indicating that the whole BTR complex is recruited by PICH to UFBs [6]. Also, as observed with PICH, BLM is recruited to all known types of UFB and depletion of BLM increases the level of PICH-coated UFBs [6,8,19,22], indicating that BLM plays an essential role in UFB resolution.
RIF1 (Rapl-interacting factor 1) is another protein that is recruited by PICH to C-UFBs [56]. RIF1 plays multiple functions in different phases of the cell cycle. In G1 phase, 53BP1 recruits RIF1 to DSB sites and they cooperate to prevent resection and promote NHEJ [57–59]. RIF1 also plays important roles in DNA replication. RIF1 colocalizes with replication forks mostly at pericentromeric heterochromatin in mid-S phase and is required for the regulation of replication timing and the assembly of newly replicated heterochromatin [60,61]. Although RIF1 interacts directly with BLM [62], the localization of RIF1 on C-UFBs does not depend on BLM, and vice versa [56]. Depletion of RIF1 increases the formation of micronuclei and G1-phase 53BP1 nuclear bodies in response to ICRF-193 treatment, suggesting that RIF1 is required for the timely resolution of C-UFBs. We find that RIF1 is also recruited to HR-UFBs in resolvase-deficient cells in anaphase, but not in telophase when the bridges are predominantly coated with RPA (Figure 3(a)). Importantly, depletion of BLM abolishes RPA binding to the HR-UFBs but has no impact on RIF1 localization (Figure 3(b)). These results indicate that RIF1 mainly localizes on double-stranded UFBs before they are converted to ssDNA by BLM. This is consistent with biochemical studies of RIF1 showing that its C-terminal region preferentially binds DNA forks and HJs compared with ssDNA [62]. However, the exact role of RIF1 in processing UFBs remains unclear. Other factors, such as TOPBP1 [63,64] and FANCM [65] also localize to certain types of UFBs (TOPBP1 on C-UFBs and FANCM on FS-UFBs).
Figure 3.
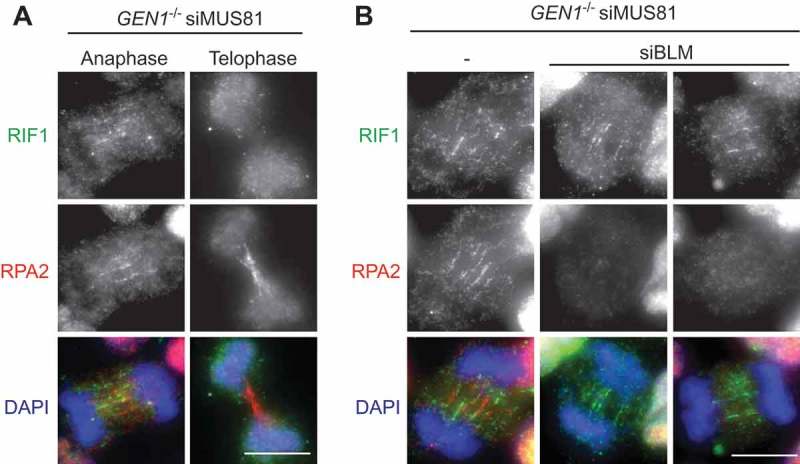
Localisation of RIF1 on double-stranded HR-UFBs.(a) GEN1–/– 293 cells generated by CRISPR-Cas9 editing were treated with siRNA against MUS81. 24 hours after siRNA transfection, the cells were treated with cisplatin (1 μM for 1 h and released into fresh media for 24 h). RPA2, RIF1 and DNA were visualized using anti-RPA2 antibody (red), anti-RIF1 antibody (green), and DAPI (blue) as indicated. Examples of anaphase and telophase cells are shown. (b) GEN1–/– 293 cells were treated with siRNA against MUS81 alone or together with siRNA against BLM. 24 hours after siRNA transfection, the cells were treated with cisplatin (1 μM for 1 h and released into fresh media for 24 h). RPA2, RIF1 and DNA were visualized as indicated. Scale bars, 10 μm.
A unified view for HR-UFB, FS-UFBs and C-UFB processing
Although the underlying DNA structures of different types of UFBs are likely to be different (Figure 1), the same set of proteins (PICH, BLM and RPA) are recruited to them, suggesting that a common mechanism may be employed for their processing. HR-UFBs are first decorated mainly with PICH and BLM in early anaphase. In telophase, most of the HR-UFBs are exclusively coated with RPA, indicating that duplex DNA bridges are converted to ssDNA. Furthermore, RPA binding to HR-UFBs and C-UFBs are dependent on PICH/BLM [22,56]. These results lead us to propose a common mechanism for the processing of HR-UFBs, FS-UFBs and C-UFBs in late anaphase/telophase: PICH recruits BLM to unwind dsDNA present in the UFBs to generate single-stranded bridges that are coated with RPA [22]. Whether ssDNA formation is actually induced to facilitate further processing, or spindle-force driven breakage, or it represents intermediates of a failed resolution attempt is presently unclear. In contrast to C-UFBs, FS-UFBs and HR-UFBs, chromatin bridges generated by telomere fusions are processed by the cytoplasmic exonuclease TREX1 to generate RPA-coated single-stranded DNA [16]. These results suggest that DNA bridges that arise from chromosome end-to-end fusions undergo a different mechanism of processing compared with other types of UFBs.
A role of LEM-3/ANKLE1 in the resolution of DNA bridges?
Recently, it was proposed that the LEM-3 nuclease might play a role in resolving DNA bridges [66,67]. Using Caenorhabditis elegans embryos, it was found that LEM-3 accumulates at the midbody when chromatin bridges are trapped at the cleavage plane, and that it is required for the resolution of chromatin bridges formed by incomplete DNA replication and recombination. Moreover, LEM-3 was shown to be synthetic lethal in combination with SLX-4 and MUS-81 (the worm orthologs of SLX4 and MUS81), indicating that it may function as a backup to resolve persistent DNA intermediates that arise during mitotic and meiotic division in C. elegans.
The mammalian ortholog of LEM-3 is known as ANKLE1 [68,69]. LEM-3/ANKLE1 contains N-terminal Ankyrin repeats, a LEM domain and a C-terminal GIY-YIG nuclease motif that is similar to that found in SLX1 nuclease. Although there is presently little known about the specificity of the LEM-3/ANKLE1 nuclease activity, it has been shown to cut both single-stranded and duplex DNA [68,69]. Based on these studies, it is conceivable that UFBs arising from unresolved replication/recombination intermediates might also be acted upon by ANKLE1. However, given that UFBs have not been observed in C. elegans embryos and a PICH ortholog has yet to be described for C. elegans [5,66], any involvement of ANKLE1 in the processing of UFBs in higher organisms remains to be determined.
Unresolved HR-UFBs lead to chromosomal instability
More than 70 years ago, Barbara McClintock proposed that anaphase bridges can drive chromosome fusions and rearrangements via a so-called breakage-fusion-bridge cycle, where chromatin bridges break apart during cytokinesis and the broken ends subsequently rejoin or rearrange with other broken chromosomes [70,71]. It is conceivable that UFBs, which are thought to be fragile, are readily broken during telophase/cytokinesis. Indeed, we have shown that breakage of HR-UFBs induces DNA damage in the following G1 phase of the cell cycle and that most of the G1 DNA damage in resolvase-deficient cells is dependent on cell division [22]. One possibility is that the UFBs are trapped within the cleavage plane during ingression of the cleavage furrow and become broken. Previously, it was shown that lagging chromosomes can be damaged during cytokinesis, leading to chromosome translocations via NHEJ in the following cell cycle [72]. Similarly, breakage of HR-UFBs leads to an increased frequency of chromosome fusions generated by NHEJ [22]. Chromosomal instability is further exaggerated as chromosome fusions lead to elevated levels of lagging chromosomes and chromatin bridges in the next round of mitosis.
Besides the conventional bridge-breakage model, it has been suggested that there is a distinct sister chromatid damage mechanism that is termed “sister-chromatid rupture-bridging” [23]. The proposal is that breakage or rupture of the sister-chromatid axes occurs at the UFB sites after the onset of anaphase. The breakage events are independent of spindle pulling and cytokinesis, and appears to require APC/C activation. Importantly, it was suggested that rupture occurs at or near centromeres and drives the formation of some signature chromosome rearrangements such as whole-arm (Robertsonian-like) deletions/translocations and isochromosome formation, similar to events that are observed in certain cancer cells. These two mechanisms of chromosome breakage are not mutually exclusive and can operate in parallel to promote gross chromosome abnormalities and genome instability. In future studies, it will be important to determine the genetic alterations that occur in genomic regions prone to form HR-UFBs, as this could reveal the precise mechanisms of genome instability caused by UFB breakage.
Concluding remarks
Sister entanglements, such as late replication/recombination intermediates, often escape cell cycle checkpoint surveillance as they do not contain DNA ends or significant amounts of ssDNA to the trigger DNA damage response. In addition, they do not interfere with microtubule-kinetochore attachment which would otherwise trigger activation of the spindle assembly checkpoint. They therefore persist to anaphase and lead to segregation defects. The discovery of UFBs provides an explanation of how genomic aberrations can accumulate in cancer cells that are both checkpoint and repair proficient. In this review, we have summarized two recent studies, conducted using resolvase-deficient and 53BP1-deficient model systems, that provide a detailed model of how HR can lead to the formation of persistent recombination intermediates that give rise to HR-UFBs [22,23]. Future research should focus on both the molecular pathways involved in UFB processing and the genetic alterations that occur in regions prone to form UFBs to reveal the precise mechanisms of genome instability caused by UFBs.
Funding Statement
Work in the author’s laboratory is supported by the Francis Crick Institute, The European Research Council and the Louis Jeantet Foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].McGranahan N, Burrell RA, Endesfelder D, et al. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jallepalli PV, Lengauer C.. Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer. 2001;1:109–117. [DOI] [PubMed] [Google Scholar]
- [3].Liu Y, Nielsen CF, Yao Q, et al. The origins and processing of ultra fine anaphase DNA bridges. Curr Opin Genet Dev. 2014;26:1–5. [DOI] [PubMed] [Google Scholar]
- [4].Sarlos K, Biebricher A, Petermann EJG, et al. Knotty problems during mitosis: mechanistic insight into the processing of ultrafine DNA bridges in anaphase. Cold Spring Harb Symp Quant Biol. 2017;82:187–195. [DOI] [PubMed] [Google Scholar]
- [5].Baumann C, Korner R, Hofmann K, et al. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell. 2007;128:101–114. [DOI] [PubMed] [Google Scholar]
- [6].Chan KL, North PS, Hickson ID.. BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J. 2007;26:3397–3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chan KL, Hickson ID. On the origins of ultra-fine anaphase bridges. Cell Cycle. 2009;8:3065–3066. [DOI] [PubMed] [Google Scholar]
- [8].Chan KL, Palmai-Pallag T, Ying S, et al. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–760. [DOI] [PubMed] [Google Scholar]
- [9].Wang LH, Schwarzbraun T, Speicher MR, et al. Persistence of DNA threads in human anaphase cells suggests late completion of sister chromatid decatenation. Chromosoma. 2008;117:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang LH, Mayer B, Stemmann O, et al. Centromere DNA decatenation depends on cohesin removal and is required for mammalian cell division. J Cell Sci. 2010;123:806–813. [DOI] [PubMed] [Google Scholar]
- [11].Nielsen CF, Hickson ID. PICH promotes mitotic chromosome segregation: identification of a novel role in rDNA disjunction. Cell Cycle. 2016;15:2704–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Naim V, Rosselli F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat Cell Biol. 2009;11:761–768. [DOI] [PubMed] [Google Scholar]
- [13].Ying S, Minocherhomji S, Chan KL, et al. MUS81 promotes common fragile site expression. Nat Cell Biol. 2013;15:1001–1007. [DOI] [PubMed] [Google Scholar]
- [14].Naim V, Wilhelm T, Debatisse M, et al. ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol. 2013;15:1008–1015. [DOI] [PubMed] [Google Scholar]
- [15].Barefield C, Karlseder J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 2012;40:7358–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Maciejowski J, Li Y, Bosco N, et al. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nera B, Huang HS, Lai T, et al. Elevated levels of TRF2 induce telomeric ultrafine anaphase bridges and rapid telomere deletions. Nat Commun. 2015;6:10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].d’Alcontres MS, Palacios JA, Mejias D, et al. TopoIIalpha prevents telomere fragility and formation of ultra thin DNA bridges during mitosis through TRF1-dependent binding to telomeres. Cell Cycle. 2014;13:1463–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ke Y, Huh JW, Warrington R, et al. PICH and BLM limit histone association with anaphase centromeric DNA threads and promote their resolution. EMBO J. 2011;30:3309–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nielsen CF, Huttner D, Bizard AH, et al. PICH promotes sister chromatid disjunction and co-operates with topoisomerase II in mitosis. Nat Commun. 2015;6:8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sarbajna S, West SC. Holliday junction processing enzymes as guardians of genome stability. Trends Biochem Sci. 2014;39:409–419. [DOI] [PubMed] [Google Scholar]
- [22].Chan YW, Fugger K, West SC. Unresolved recombination intermediates lead to ultra-fine anaphase bridges, chromosome breaks and aberrations. Nat Cell Biol. 2018;20:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tiwari A, Addis Jones O, Chan KL. 53BP1 can limit sister-chromatid rupture and rearrangements driven by a distinct ultrafine DNA bridging-breakage process. Nat Commun. 2018;9:677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Holliday R. A mechanism for gene conversion in fungi. Genet Res (Camb). 1964;5:282–304. [DOI] [PubMed] [Google Scholar]
- [26].West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–445. [DOI] [PubMed] [Google Scholar]
- [27].Wyatt HD, West SC. Holliday junction resolvases. Cold Spring Harb Perspect Biol. 2014;6:a023192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sarbajna S, Davies D, West SC. Roles of SLX1-SLX4, MUS81-EME1, and GEN1 in avoiding genome instability and mitotic catastrophe. Genes Dev. 2014;28:1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wechsler T, Newman S, West SC. Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature. 2011;471:642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Garner E, Kim Y, Lach FP, et al. Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 are essential for the resolution of replication-induced Holliday junctions. Cell Rep. 2013;5:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wyatt HD, Sarbajna S, Matos J, et al. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for Holliday junction resolution in human cells. Mol Cell. 2013;52:234–247. [DOI] [PubMed] [Google Scholar]
- [32].Garcia-Luis J, Machin F. Mus81-Mms4 and Yen1 resolve a novel anaphase bridge formed by noncanonical Holliday junctions. Nat Commun. 2014;5:5652. [DOI] [PubMed] [Google Scholar]
- [33].Wu L, Hickson ID. The Bloom’s syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. [DOI] [PubMed] [Google Scholar]
- [34].Raynard S, Bussen W, Sung P. A double Holliday junction dissolvasome comprising BLM, topoisomerase IIIα, and BLAP75. J Biol Chem. 2006;281:13861–13864. [DOI] [PubMed] [Google Scholar]
- [35].Wu L, Bachrati CZ, Ou J, et al. BLAP75/RMI1 promotes the BLM-dependent dissolution of homologous recombination intermediates. Proc Natl Acad Sci USA. 2006;103:4068–4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Singh TR, Ali AM, Busygina V, et al. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 2008;22:2856–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Xu D, Guo R, Sobeck A, et al. RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev. 2008;22:2843–2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cejka P, Plank JL, Bachrati CZ, et al. Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nat Struct Mol Biol. 2010;17:1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Matos J, Blanco MG, Maslen S, et al. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011;147:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chan YW, West SC. Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nat Commun. 2014;5:4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Andersen SL, Bergstralh DT, Kohl KP, et al. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol Cell. 2009;35:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fekairi S, Scaglione S, Chahwan C, et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009;138:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Munoz IM, Hain K, Declais AC, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell. 2009;35:116–127. [DOI] [PubMed] [Google Scholar]
- [44].Svendsen JM, Smogorzewska A, Sowa ME, et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Castor D, Nair N, Declais AC, et al. Cooperative control of Holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol Cell. 2013;52:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wyatt HD, Laister RC, Martin SR, et al. The SMX DNA repair tri-nuclease. Mol Cell. 2017;65:848–860 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ip SC, Rass U, Blanco MG, et al. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456:357–361. [DOI] [PubMed] [Google Scholar]
- [48].Rass U, Compton SA, Matos J, et al. Mechanism of Holliday junction resolution by the human GEN1 protein. Genes Dev. 2010;24:1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Chan YW, West S. GEN1 promotes Holliday junction resolution by a coordinated nick and counter-nick mechanism. Nucleic Acids Res. 2015;43:10882–10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shah Punatar R, Martin MJ, Wyatt HD, et al. Resolution of single and double Holliday junction recombination intermediates by GEN1. Proc Natl Acad Sci USA. 2017;114:443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol. 2014;34:1380–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7–18. [DOI] [PubMed] [Google Scholar]
- [53].Ochs F, Somyajit K, Altmeyer M, et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 2016;23:714–721. [DOI] [PubMed] [Google Scholar]
- [54].Biebricher A, Hirano S, Enzlin JH, et al. PICH: a DNA translocase specially adapted for processing anaphase bridge DNA. Mol Cell. 2013;51:691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Croteau DL, Popuri V, Opresko PL, et al. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Hengeveld RC, De Boer HR, Schoonen PM, et al. RIF1 is required for resolution of ultrafine DNA bridges in anaphase to ensure genomic stability. Dev Cell. 2015;34:466–474. [DOI] [PubMed] [Google Scholar]
- [57].Chapman JR, Barral P, Vannier JB, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Di Virgilio M, Callen E, Yamane A, et al. RIF1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–883. [DOI] [PubMed] [Google Scholar]
- [60].Buonomo SB, Wu Y, Ferguson D, et al. Mammalian RIF1 contributes to replication stress survival and homology-directed repair. J Cell Biol. 2009;187:385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cornacchia D, Dileep V, Quivy JP, et al. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J. 2012;31:3678–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Xu D, Muniandy P, Leo E, et al. RIF1 provides a new DNA-binding interface for the Bloom syndrome complex to maintain normal replication. EMBO J. 2010;29:3140–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Germann SM, Schramke V, Pedersen RT, et al. TOPBP1/DPB11 binds DNA anaphase bridges to prevent genome instability. J Cell Biol. 2014;204:45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Broderick R, Nieminuszczy J, Blackford AN, et al. TOPBP1 recruits TOP2A to ultra-fine anaphase bridges to aid in their resolution. Nat Commun. 2015;6:6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vinciguerra P, Godinho SA, Parmar K, et al. Cytokinesis failure occurs in Fanconi anemia pathway-deficient murine and human bone marrow hematopoietic cells. J Clin Invest. 2010;120:3834–3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hong Y, Sonneville R, Wang B, et al. LEM-3 is a midbody-tethered DNA nuclease that resolves chromatin bridges during late mitosis. Nat Commun. 2018;9:728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hong Y, Velkova M, Silva N, et al. The conserved LEM-3/Ankle1 nuclease is involved in the combinatorial regulation of meiotic recombination repair and chromosome segregation in Caenorhabditis elegans. PLoS Genet. 2018;14:e1007453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Brachner A, Braun J, Ghodgaonkar M, et al. The endonuclease Ankle1 requires its LEM and GIY-YIG motifs for DNA cleavage in vivo. J Cell Sci. 2012;125:1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Dittrich CM, Kratz K, Sendoel A, et al. LEM-3 - A LEM domain containing nuclease involved in the DNA damage response in C. elegans. PLoS One. 2012;7:e24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].McClintock B. The fusion of broken ends of chromosomes following nuclear fusion. Proc Natl Acad Sci USA. 1942;28:458–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gisselsson D, Pettersson L, Hoglund M, et al. Chromosomal breakage-fusion-bridge events cause genetic intratumor heterogeneity. Proc Natl Acad Sci USA. 2000;97:5357–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Janssen A, van der Burg M, Szuhai K, et al. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–1898. [DOI] [PubMed] [Google Scholar]