

Abstract
Cell contractility regulates multiple cell behaviors which contribute to both normal and pathological processes. However, measuring cell contractility remains a technical challenge in complex biological samples. The current state of the art technologies employed to measure cell contractility have inherent limitations that greatly limit the experimental conditions under which they can be used. Here, we use quantitative polarization microscopy to extract information about cell contractility. We show that the optical retardance signal measured from the cell body is proportional to cell contractility in two-dimensional and three-dimensional platforms, and as such can be used as a straightforward, tractable methodology to assess cell contractility in a variety of systems. This label-free optical method provides a novel and flexible way to assess cellular forces of single cells and monolayers in several cell types, fixed or live, in addition to cells present in situ in mouse tumor tissue samples. This easily implementable and experimentally versatile method will significantly contribute to the cell mechanics field.
Keywords: Cell Contractility, Traction Force Microscopy, Optical Retardance, 3D Collagen, Actin
Graphical Abstract
Cell contractility is crucial for numerous cell behaviors which contribute to normal and pathological processes. In this work, we demonstrate that quantitative polarization microscopy (QPOL) provides a readout of cell contractility. By modulating cell contractility, we show in multiple complex biological systems that the birefringence signal directly scales with cell contractility. This imaging method provide an easy and quantitative way to measure cell contractility without exogenous dyes or fluorescent labels.
INTRODUCTION
Cellular contractility influences cell behaviors involved in both normal and pathological processes, including cell adhesion and migration, growth factor signal transduction, and cell differentiation[1–4]. In turn, cell contractility is influenced by the mechanical properties of the extracellular matrix (ECM)[5]. Notably, several pathologies, including cancer, are characterized by increased ECM deposition and crosslinking that results in increased stiffness of the microenvironment[6, 7]. As such, our ability to measure these contractile changes in cells is fundamental to our understanding of numerous disease states. Still, measuring the cell contractile state remains a challenge, particularly in complex 3D biological samples. In this context, a tractable method that can measure cell contractility in 3D could provide a critical tool for the mechanobiology field.
The existing technologies available to measure cell contractility require extensive technical skills, can be quite expensive system upfront, and usually suffer from low throughput and limited 3D application[8–11]. In addition, most biological materials are highly heterogeneous and have non-linear mechanical properties[12–14]. It is therefore challenging to measure and accurately compute cell contractility when using native ECM scaffolds[8, 11, 15]. Given these limitations, there is a need for a technique that allows a more direct measurement of cell contractility. Quantitative polarization microscopy (QPOL) takes advantage of a material’s birefringent properties to image structures without additional exogenous dyes or fluorescent labels. QPOL can measure the changes in the birefringence of a material[16]. Of note, birefringent materials subjected to strain exhibit photoelasticity, which means that their birefringence changes proportionally to the applied strain. Interestingly, actin behaves as a birefringent material[17, 18], which could provide a direct readout of cell contractility that can be measured with QPOL. Thus, we hypothesize QPOL could be used to measure cell contractility in complex, heterogeneous in vitro systems without exogenous dyes or reporters.
In the work reported here, we adapted QPOL to directly measure cell contractility independently of the underlying substrate in both 2D and 3D. The optical retardance of highly invasive MDA-MB-231 tumor cells and non-tumorigenic MCF10A cells was measured using QPOL. The optical retardance signal is dependent on cell contractility based on experiments using the contractility inhibitor, Y27632, actin depolymerization agents, Latrunculin A and Cytochalasin D, and myosin activators, Calyculin A and Rho activator II. The optical retardance signal was found to increase proportionally to ECM stiffness for both cells seeded on polyacrylamide gels with tunable stiffness or cells embedded in type I collagen matrices of increasing density or crosslinking. Moreover, measurements of cell generated force by traction force microscopy (TFM) showed a positive linear correlation with the acquired retardance signal. The retardance signals of cells within confluent MCF10A monolayers, treated with a contractility inhibitor or activator, and tissue sections from low and high stiffness mouse tumors were also measured, demonstrating that QPOL can also provide valuable information about cell contractility in more complex biological systems. Our results highlight the potential of QPOL as a versatile, tractable tool to assess cell contractility.
MATERIALS AND METHODS
Reagents
Primary and secondary antibodies used were as follows: rabbit polyclonal anti-phosphorylated myosin light chain 2 (Thr18/Ser19) (Cell Signaling Technology, #3674); Alexa 568 Fluor donkey anti-rabbit (ThermoFisher Scientific, A10042). Alexa Fluor 488 Phalloidin (A12379) and 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI, D1306) were obtained from ThermoFisher. Y27632, Latrunculin A, Cytochalasin D, Calyculin A were purchased from Calbiochem. Rho Activator II was purchased from Cytoskeleton. Dulbecco’s modified Eagle’s medium (DMEM), DMEM/F12, and PBS were purchased from Gibco (Life Technologies); Fetal bovine serum (FBS) was purchased from Atlanta Biologicals; Epithelial growth factor (EGF) was purchased from Invitrogen; basic fibroblast growth factor (bFGF) was purchased from PeproTech; all other chemicals were from Sigma-Aldrich.
Cell culture
Human breast tumor MDA-MB-231 cells were cultured in DMEM medium with 10% (vol/vol) FBS and 1% (vol/vol) penicillin-streptomycin. Non-tumorigenic breast epithelial MCF10A cells were cultured in DMEM/F12 Media supplemented with 5% horse serum, 20 ng mL−1 EGF, 10 mg mL−1 insulin, 0.5 mg mL−1 hydrocortisone, 100 ng mL−1 cholera toxin, and 1% (vol/vol) penicillin–streptomycin. Both cell lines were maintained at 37°C, humidified atmosphere of 5% (vol/vol) CO2 in air. All cell lines were tested for mycoplasma contamination, and experiments were conducted under mycoplasma-negative conditions. For single cell studies, cells were seeded onto polyacrylamide substrates or collagen-coated bottom cover glass MatTek dishes at 1000 cells mL−1. For monolayer studies, cells were seeded at 50,000 cells mL−1 and allowed to grow for 2 days to reach confluence. In some experiments, cells were pre-treated for 3 hours with Rho activator II (1 μg/mL) or for 30 min with either Y27632 (10 μM), Latrunculin A (2 μM), Cytochalasin D (5 μM), or Calyculin A (1 nM) to modulate cell contractility as described before[19]. Cells were fixed with 3.7% (vol/vol) formaldehyde in PBS for 10 min at room temperature. Alternatively, MCF10A cell monolayers were fixed with 3.7% (vol/vol) formaldehyde in microtubule stabilizing buffer (100 mM PIPES pH 6.8, 2 mM MgCl2 and 5 mM EGTA) at 37°C for 10 min. After fixing, all cells were washed with PBS three times. Cells were kept in PBS during the imaging process.
Mouse studies
All mice were maintained following a protocol approved by the Cornell University Institutional Animal Care and Use Committee. MMTV-PyMT transgenic and WT control mice, both on the FVB strain background, were obtained from the Jackson Laboratory. Female mice between 10 and 12 weeks old were euthanized by CO2 asphyxiation and necropsied. Mammary tumors were collected and snap frozen in liquid nitrogen. Snap frozen mouse tumor samples were embedded within OCT compound and processed in a cryostat to obtain 8 μm-thick sections. The tumor samples were immediately fixed after sectioning in 4% (wt/vol) paraformaldehyde for 10 min at room temperature. The sections were then washed two times in PBS. Slides were mounted in Vectashield mounting media.
Alternatively, tumor organoids were obtained from freshly harvested tumors as described[20]. Briefly, freshly extracted tumors were kept in DMEM on ice. The tumors were then minced and digested for 40 min at 37°C in a DMEM/F12 solution supplemented with 0.5 μg mL−1collagenase IV, 0.2% (vol/vol) trypsin, 5% (vol/vol) FBS and 1 nM insulin. The tumors were washed 3 times in DMEM/F12 and then treated with DNase for 4 min at room temperature. The organoids and single cells were then separated using differential centrifugation at 1000 rpm at least four times. Isolated organoids were then embedded within a 3D collagen gel. After 1 hour, a DMEM/F12 organoid branching media supplemented with 1% (vov/vol) ITS growth supplement and 2.5 nM bFGF and allowed to invade for 24 hours.
Polyacrylamide substrate synthesis
Polyacrylamide (PA) gel substrates of different stiffnesses (0.5 kPa-30 kPa) were prepared and characterized as described previously[21]. Gels were functionalized with N-6-((acryloyl)amino)hexanoic acid to allow covalent attachment of type I rat-tail collagen. Gels were polymerized and covalently bound to activated glass coverslips and were then coated with a 100 μg mL−1 collagen solution.
Collagen 3D scaffolds
Type I collagen was isolated from rat-tail tendons and extracted in 0.1% sterile acetic acid at 4°C as described previously[22, 23]. Stiffness of the collagen scaffolds was controlled either by changing the collagen density or by increasing collagen crosslinking through non-enzymatic glycation. Glycated collagen solutions were prepared by mixing 10 mg mL−1 collagen stock solutions with 0.5 M ribose to form solutions containing 0, 50, or 100 mM ribose in 0.1% (vol/vol) sterile acetic acid on ice and then incubated for 5 days at 4°C. To prepare the 3D scaffolds, the collagen solutions were neutralized with 1 N sodium hydroxide and mixed with 1 M HEPES and sodium bicarbonate in 10x PBS, to form 1.5, 3 or 5 mg mL−1 collagen gels with final concentrations of 1x PBS, 25 mM HEPES and 44 mM sodium bicarbonate.
Immunofluorescence
Fixed cells were first permeabilized with 0.02% (vol/vol) triton in PBS for 20 min. The cells were then immunostained for phosphorylated Myosin Light Chain 2 (p-MLC) (1:100 dilution) in PBS overnight at 4 °C followed by Alexa Fluor 568 donkey anti-rabbit (1:200) for 1 hour at room temperature. Actin was stained with Alexa Fluor 488 Phalloidin (1:100). The cell nuclei were stained with DAPI (1:1000). Fluorescent images were acquired with an inverted Axiovert microscope (Zeiss).
Quantitative polarization microscopy
The polarization microscope is built on an inverted Axiovert microscope equipped with a Zeiss Axiocam 506 color camera. Briefly, a linear polarizer actuated by a motorized rotation stage (Thorlabs, max speed of 20 degrees sec−1) was positioned in the illumination plane above the condenser, and a circular polarizer (analyzer) was positioned in the imaging plane. Images were acquired using a 40× 0.9 NA polarization objective (Zeiss). Image sequences were acquired with 5 degree intervals of the rotating polarizer over a range of 0 to 180 degrees using Zen software. Acquisition time for each individual picture was set at 20 ms. A timed delay of 2 s was used between each acquisition to allow rotation of the polarizing element. The polarized image sequences were then processed with a custom Matlab code to obtain a pixel-by-pixel retardance map. The resulting retardance images were then background subtracted and analyzed with ImageJ. The retardance of the whole cell was quantified as the average value of the background-subtracted retardance of the cell area.
Traction force microscopy
For traction force microscopy assays, 5 kPa PA gels embedded with 0.5 μm diameter fluorescent beads (Life Technologies) were used. Cells were seeded on the PA gels and allowed to adhere for 24 hour. Transmission images of single cells were then taken immediately followed by fluorescent images of the bead field at the gel surface. A polarization image sequence of the same cell was immediately acquired. Since the QPOL is not equipped with an environmental chamber, a maximum of 5 cells per PA gels were imaged over a time span of 5 min. Cells were then removed with 0.25% (vol/vol) trypsin/EDTA (Life Technologies) and a second fluorescent image of the bead field was captured. Bead displacements between the stressed and null state were calculated using the LIBTRC analysis library developed by M. Dembo (Dept. of Biomedical Engineering, Boston University). The overall cellular force the total magnitude of the force, |F|, is the integral of the traction vector magnitudes over the total cell area[24]. Generated data points are from 3 independent experiments for both MCF10A (n=8 cells) and MDA-MB-231 (n=12 cells).
Statistical test
Prism program was used for data analysis. Parametric one-way or two-way ANOVAs with post hoc Tukey’s honest significance test were performed where appropriate. P < 0.05 was considered statistically significant.
RESULTS
Quantitative Polarization Microscopy can measure the polarization signal from live and fixed samples
The QPOL system was built from a conventional inverted microscope by inserting a linear polarizer between the monochromatic light source and the condenser, with a circular polarization analyzer positioned after the objective (Fig. 1A). Using the equations derived for such a polarizer configuration[25], one can obtain the following function describing the pixel-by-pixel intensity, I:
| (1) |
Where I0 is the intensity of initial input, ϕ(x, y) is the orientation of the main birefringent axis of each component within the sample, χ is the orientation of the linear polarizer relative to the sample and δ(x, y) is the retardance of the light due to the sample birefringence. The transmitted light intensity of each image taken as the polarizer was rotated incrementally was documented. Each pixel of the resulting image sequence can then be best described as a periodic signal modulated by the nth orientation χ of the polarizer. The signal of the resulting pixel-by-pixel modulation can be obtained using the following coefficients:
| (2) |
Which are the Fourier series coefficients that describe the signal average a0 and the fundamental frequencies a1 and a2. Then, the relationship between the amplitude of the measured signal swing and the retardance δ can be described as:
| (3) |
Using the QPOL system, we proceeded to measure the retardance of MDA-MB-231 cells seeded on collagen-coated glass slides. Superposition of the QPOL signal with phalloidin stained cells showed regions where visible actin fibers clearly overlapped with the retardance signal (Fig. 1B). We then measured the retardance of both live and fixed MDA-MB-231 cells. Both the live and fixed cells showed similar retardance distribution (Fig. 1C), and the fixation did not affect the measured average retardance (Fig. 1D). In addition, we also measured the retardance in live cells by collecting a series of time-lapse images of MDA-MB-231 cells (Fig. 1E). Overall, our results indicate that retardance signal can be obtained from both fixed and unfixed cells.
Figure 1 |.
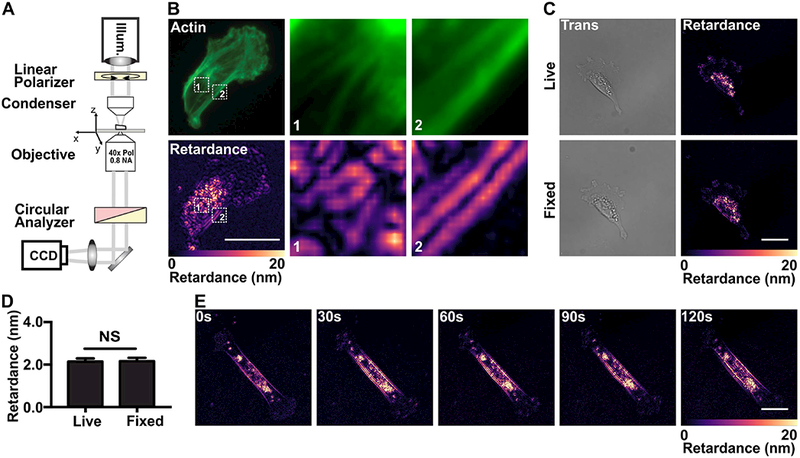
Setup of the QPOL system and effects of fixation on cellular retardance.
(A) Optical diagram of QPOL showing the position of optical component including the illumination (Illum.) and the acquisition camera (CCD). (B) Fluorescent image of a MDA-MB-231 cell showing fibrillar actin along with the corresponding retardance (Retardance) signal. Magnified views of regions 1 and 2 (numbered dashed boxes) from each image set are provided. (C) Images showing the comparison between the transmitted light (Trans) and retardance (right) immediately before (Live) and after fixation (Fixed) of a MDA-MD-231 cell seeded on collagen-coated glass slides. (D) Corresponding quantification of the average retardance of live (n=39) and fixed cells (n=39). (E) Image sequences showing the retardance of a live MDA-MB-231 cell over the course of 120s. (Data presented as mean ± SEM, Scale bar = 25 μm, NS indicates p>0.05).
Cell contractility is correlated to optical retardance
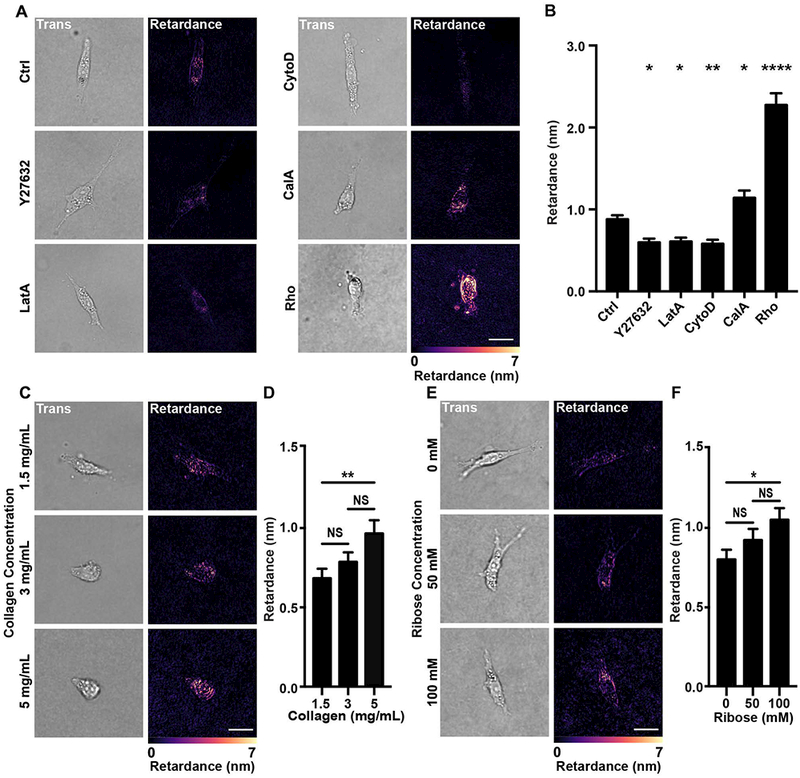
Cell contractility is mainly dependent on acto-myosin mediated forces[5, 26]. In this context, we proceeded to assess how cellular optical retardance was related to actomyosin-mediated cell contractility. To this end, we measured the retardance of MDA-MB-231 cells treated with either actin depolymerization agents, Latrunculin A and Cytochalasin D, the cell contractility inhibitor, Y27632, or the contractility activators, Calyculin A and Rho activator II. The actin depolymerizing agents Latrunculin A and Cytochalasin D are known inhibitors of cell contractility [27, 28]. Use of these agents resulted in loss of myosin activation (Y27632, Latrunculin A, and Cytochalasin D), actin depolymerization (Latrunculin A and Cytochalasin D), or increased myosin activation (Calyculin A, Rho activator II) compared to untreated samples (Fig. 2A). Concurrently, addition of these drugs led to significant changes in the measured retardance that were positively correlated with either the associated decrease or increase of cell contractility induced by these drugs (Fig. 2B and C).
Figure 2 |.
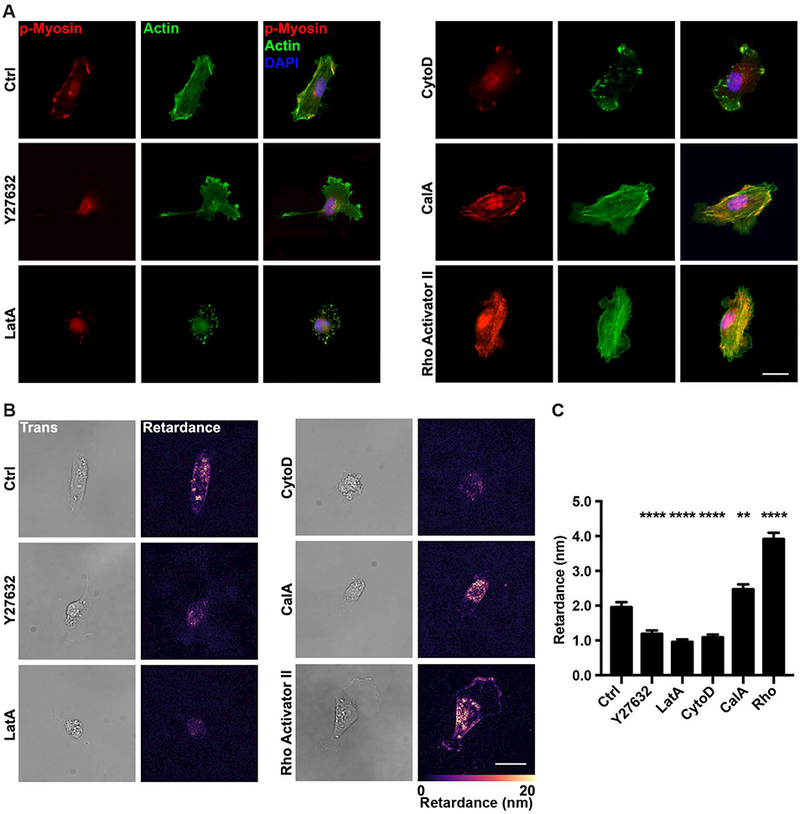
Contractility modulating drugs alter the retardance of MDA-MB-231 cells.
(A) Representative fluorescence images of MDA-MB-231 cells stained for actin and p-MLC (p-Myosin) seeded on collagen-coated glass slides and treated with vehicle (Ctrl), Y27632, Cytochalasin D (CytoD), Latrunculin A (LatA), Calyculin A (CalA) or Rho activator II (Rho). The merged image includes the DAPI stained nucleus in blue. (B) Transmitted light (left) and computed retardance (right) images of the cells treated with Y27632, CytoD, LatA, CalA or Rho along with (C) the corresponding average retardance (Ctrl n=83, Y27632 n=84, LatA n=89, CytoD n=114, CalA n=85). (Data presented as mean ± SEM, Scale bar = 25 μm. ** indicates P<0.005, **** indicates P<0.0001 vs Ctrl).
Cell contractility of both tumorigenic and non-tumorigenic cells has been reported to increase along with the increasing of substrate stiffness[21]. In addition, tumorigenic cells are typically more contractile than their non-tumorigenic counterparts[21, 29]. Therefore, we measured the retardance of tumorigenic MDA-MB-231 cells and non-tumorigenic MCF10A cells seeded on 2D collagen-coated polyacrylamide substrates with stiffnesses ranging from 0.5 kPa to 30 kPa to mimic tumor stiffness as reported previously[30, 31]. Interestingly, the retardance signal of both cell lines increased as a function of substrate stiffness (Fig. 3A and B). Moreover, the average retardance of the MCF10A cells was significantly lower than for the MDA-MB-231 cells at all stiffness conditions (Fig. 3C). Both of these findings are consistent with our previous traction force measurements[21]. We performed TFM and QPOL measurements at the same time to further ascertain if the retardance signal was proportional to cell contractility. Notably, there was a strong positive linear correlation of the force measurements with measures of the cell retardance in both MDA-MB-231 and MCF10A cells (Fig. 3D). Together, our data indicate that cell optical retardance signal is a function of its contractility.
Figure 3 |.
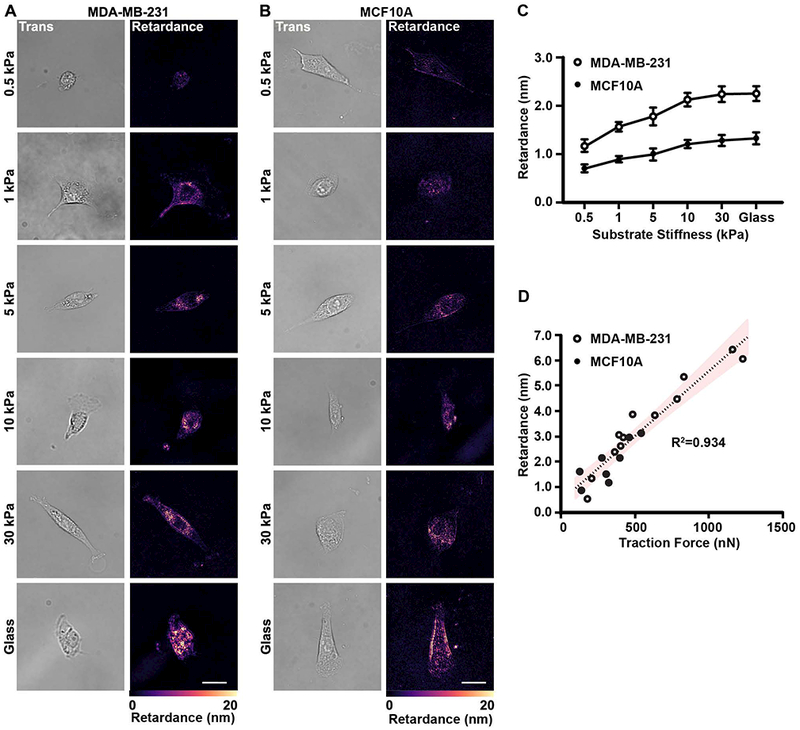
Retardance correlates with substrate stiffness-mediated cell contractility.
(A) Transmitted light (Trans) and retardance (Retardance) images of MDA-MB-231 cells seeded on polyacrylamide substrates with a range of stiffness (0.5, 1, 5, 10 and 30 kPa) and on glass. (B) Transmitted light and retardance images of MCF10A cells seeded on polyacrylamide substrates with the same stiffness values as in (3a). (C) Corresponding quantification of the average retardance as a function of increasing stiffness for both MDA-MB-231 (0.5 kPa n=77, 1 kPa n=80, 5 kPa n=94, 10 kPa n=90, 30 kPa n=88, glass n=87) and MCF10A (0.5 kPa n=77, 1 kPa n=90, 5 kPa n=110, 10 kPa n=96, 30 kPa n=105, glass n=93). Data presented as mean ± SEM. (D) Scatter plot of the retardance of single cells as a function of the total traction forces for MDA-MB-231 (n=12) and MCF10A (n=7) cells. The resulting data was fit with the linear regression (dashed line) and confidence interval (95%). The R2 value is also provided. (Scale bar = 25 μm)
To determine whether the QPOL system can measure cell contractility in 3D culture conditions, we embedded cells within a 3D type I collagen matrix. We first confirmed that we were indeed measuring actomyosin-mediated contractility using MDA-MB-231 cells embedded in 1.5 mg mL−1 type I collagen gels. Notably, the MDA-MB-231 cell retardance signal was significantly decreased compared to the control when either myosin was inhibited with Y27632 or when actin was depolymerized using Cytochalasin D or Latrunculin A, while the retardance signal was increased when cell contractility was increased with Calyculin A or Rho activator II (Fig. 4A and B). In addition, 3D collagen scaffolds were tuned through manipulation of collagen density or crosslinking. Collagen was crosslinked through a ribose-mediated glycation process using 0, 50 and 100 mM ribose to create collagen gels of stiffness of 175, 300 and 500 Pa respectively[32]. Under these conditions, the MDA-MB-231 cells retardance increased proportionally as a function of either collagen concentration or ribose concentration (Fig. 4C–F). Interestingly, the average optical signal from cell embedded within 500 Pa crosslinked collagen gels was comparable to the signal of cells seeded on 500 Pa polyacrylamide gels. Overall, the results above indicate that QPOL microscopy also provides a reliable assessment of cell contractility in 3D systems.
Figure 4 |.
Retardance of MDA-MB-231 cells embedded in 3D type I collagen matrix.
(A) Transmitted light (Trans) and retardance (Retardance) images of MDA-MB-231 cells treated with either vehicle (Ctrl), Y27632, Cytochalasin D (CytoD), Latrunculin A (LatA), Calyculin A (CalA) and Rho activator II (Rho) along with (B) the corresponding average retardance (Ctrl n=163, Y27632 n=71, LatA n=67, CytoD n=89, CalA n=73, Rho n=70). (C) Transmitted light and retardance images of MDA-MB-231 cells embedded in 1.5, 3, and 5 mg mL−1 type I collagen matrix along with (D) the corresponding average retardance (1.5 mg mL−1 n=74, 3 mg mL−1 n=71, 5 mg mL−1 n=75). (E) Images showing the transmitted light and retardance images of MDA-MB-231 cells embedded in ribose-crosslinked collagen type I matrix along with (F) the corresponding average retardance (0 mM n=71, 50 mM n=73, 100 mM n=73). (Data presented as mean ± SEM, Scale bar = 25 μm, * indicates P< 0.05 and ** indicates P<0.0050, **** indicates P<0.0001 vs Ctrl).
Measurement of cell contractility in complex biological samples using Quantitative Polarization Microscopy
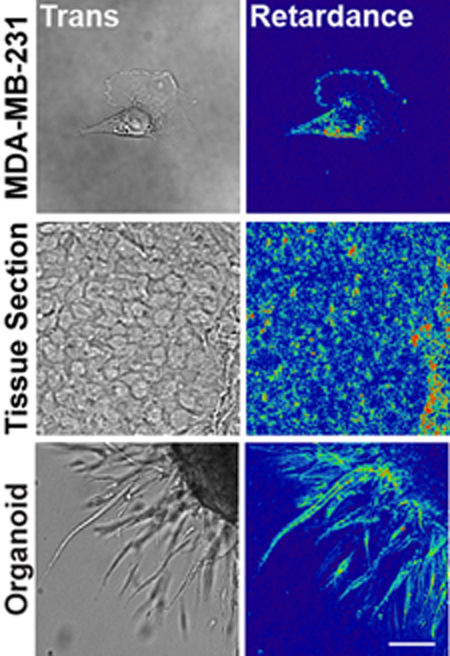
We expanded our QPOL analysis to complex biological samples, including cell monolayers, ex vivo organoids and tumor tissues to further evaluate the capabilities of our technique. Measuring cellular contractility is difficult in these complex systems and in many cases has not been attempted[8, 10, 11]. In the case of epithelial cell monolayers, which are known to exert contractile forces through their cell-cell junctions[33], the retardance signal was localized around the cell edge and showed good overlap with the cobblestone architecture of the epithelial monolayer (Fig. 5A). Similar to what we had observed in single cells, use of the cell contractility inhibitor Y275632 significantly lowered the retardance signal compared to the control, while the use of Rho activator II increased the signal (Fig. 5A and B). To further evaluate the imaging capability of QPOL, we also investigated whether we could apply our technique to study cell invasion using an ex vivo 3D tumor organoid model. Interestingly, the cells invading in the 3D collagen scaffold showed retardance signal, indicating that QPOL could be potentially used to measure cell contractility of cells in the ex vivo 3D tumor organoid model (Fig. 5C). It has recently been reported that QPOL could be used to obtain relevant information from tumor samples stroma[34]. In this context, we used histological tumor sections from PyMT mice treated with or without β-amino proprionitrile (BAPN) treatment. BAPN inhibits collagen crosslinking, which results in mice having more compliant tumors and lower activation of cell contractility markers[35–37]. Several evidences indicate that cell contractility is expected to be lower in more compliant tumors[38]. Indeed, p-MLC staining of tumor sections confirmed that cells from mice treated with BAPN had lower cell contractility compared with those from the control group (Fig. 5D). Accordingly, we observed a lower QPOL signal in tumor sections from mice treated with BAPN compared to the untreated control (Fig. 5E). These results are consistent with the reduced stiffness and contractility of tumor tissue observed in BAPN treated mice[22]. Of particular relevance, the quantification of the retardance revealed that we could distinguish between soft and stiff tumors (Fig. 5F). Together, our data indicate that QPOL is a potent approach to measure cell contractility under many conditions, including complex multi-cellular biological systems.
Figure 5 |.
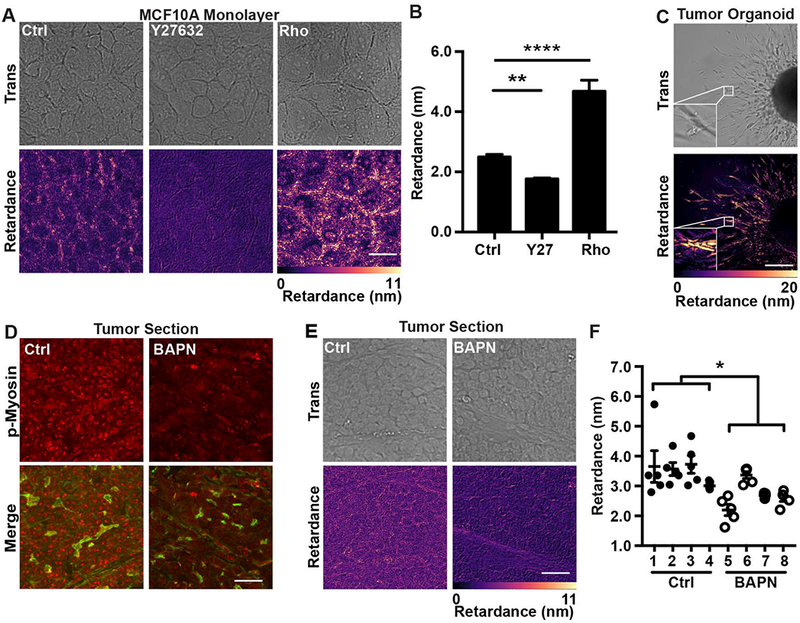
Cell contractility influences the retardance of complex biological samples in vitro, ex vivo and in vivo.
(A) Transmitted light (Trans) and retardance (Retardance) images of MCF10A cell monolayers treated with the vehicle (Ctrl), Y27632 or with the Rho activator II (Rho) along with (B) the corresponding average retardance (n=45). (C) Retardance of ex vivo tumor organoid isolated from a MMtV-PyMT mouse and embedded within a 3D collagen gel. The inset image shows the increased signal intensity in the invading cells. (D) Representative fluorescence images of tumor tissue sections from PyMT mice stained for p-MLC (p-Myosin; red) and actin (Merge; green) and treated with vehicle (Ctrl) or BAPN (BAPN). (E) Transmitted light and retardance images of mammary tumor sections with (BAPN) or without (Ctrl) BAPN treatment along with (F) the corresponding quantification of the retardance of Ctrl (n=4 mice) and BAPN (n=4 mice) treated tumor sections from individual MMTV-PyMT. Each tumor section was imaged in five different locations. (Data presented as mean ± SEM, Scale bar = 25 μm, * indicates P< 0.05, ** indicates P<0.0050, **** indicates P<0.0001 vs. Ctrl)
DISCUSSION
In this work, we show that it is possible to measure cell contractility using QPOL microscopy in both 2D and 3D. Notably, we demonstrate that the retardance signal is linearly proportional to the cell contractility state in both 2D and 3D using contractility modulating drugs. The retardance signal scales with matrix stiffness-mediated cell contractility increases in both polyacrylamide and collagen gel systems. Using QPOL, we are able to distinguish between the differential contractility of highly invasive MDA-MB-231 cells and non-tumorigenic epithelial MCF10A cells based on the retardance signal. We then demonstrate that QPOL can be used to assess cell contractility in complex biological systems, including cell monolayers, tumor sections, and ex vivo tumor organoids embedded in 3D matrix.
While QPOL can be used to access cell contractility in 2D directly, the most significant advantage of QPOL is that it can be used to measure contractility of cells in complex biological samples with quantifiable results comparable to 3D TFM. Measuring cell contractility in 3D systems has proven to be a difficult challenge, most notably considering that most native ECM components are mechanically heterogeneous and non-linear[14, 39, 40]. Indeed, ECM mechanical properties must be known to correctly assess a stress-strain field propagation and thus the forces generated in a system. In addition, the currently available methods are computationally intensive and require technical expertise. Notably, our QPOL technique is based solely on the inherent birefringent properties of the cell and does not require prior knowledge of the ECM mechanical properties. This allows measurement of cell contractility in highly heterogeneous biomaterials with non-linear mechanical properties such as collagen gels. Of note, the collagen used in our experiments also exhibited birefringent properties[34], which could be a source of noise and account for some of the background signal. However, the resolution of our system did not allow us to image individual collagen fibers. It remains possible that, with the correct sensitivity to change in retardance, QPOL could be used to measure the strain in the ECM scaffold.
While we have not examined the ability of this system to perform time lapse in depth, the signal similarity between alive and fixed cells does suggest QPOL could be a powerful tool to gain more insight in the role of cell contractility in several time-dependent processes. One important possible limitation is the speed of acquisition between successive images, which is limited by the rotation speed of the polarizer. Acquisition of a full dataset takes approximately 72 s with our current setup when rotating the polarizing element in 5 degree steps. A slow acquisition speed presents an issue since cells may have the time to change their morphology over the course of the multi-images acquisition, thus shifting the spatial position of the birefringent components between each frame. Notably, the rate of membrane protrusion and the speed at which a cell can migrate can be as high as 10 μm min−1 [41, 42]. Any slight frame-to-frame shift can introduce artifacts from the computational step that provides false retardance signals. In addition, the traction forces exerted by a cell in some individual focal adhesions can fluctuate by as much as 50% over 10 seconds[43], potentially changing the overall mechanical stress in a cell and resulting in a perturbed QPOL signal. The use of polarization components with quicker response time, such as a spatial light modulator or fast rotation stage, to keep the acquisition time below a few seconds would help limit the apparition of such artifacts. Alternatively, the use of freshly fixed samples avoids this problem altogether.
QPOL is a powerful and versatile technique to measure cell contractility under many conditions, including single cells and cells inside complex biological samples, by taking advantage of the cells’ inherent birefringent properties and the concept of photoelasticity. However, actin is not the only optically birefringent material in the cell. Other cytoskeleton components such as microtubules also exhibit a high degree of birefringence [17, 44]. However, normal expression of cytoskeletal components do not appear to be significantly affected by changes in matrix stiffness or contractility [45], indicating that variation of the QPOL signal we observe here is mainly due to changes in cell contractility. However, different cell types are known to express varying amounts of actin, intermediate filaments family members, or microtubules [46–48]. In addition, several actin accessory proteins effectively act as cytoskeletal crosslinkers that can alter cell mechanical properties [49, 50]. For example, overexpression of α-actinin alone can increase the apparent stiffness of the cell [51]. Of note, the number of crosslinks present in a polymer network can influence its photoelastic response [52]. Therefore, it is quite possible that the cell lineage could influence the baseline birefringent signal as well as the mechanical load distribution within the cytoskeleton. If such is the case, one would have to be careful before using QPOL to compare between cell types of widely different lineages without performing exhaustive calibration. Importantly, though, it remains that based on our results in different experimental conditions, including from in vivo tissue sections and ex vivo tumors, QPOL can easily distinguish between the contractility states of similar cells. Recent work by Eldridge et al. utilized quantitative phase imaging, an approach that also relies on changes in the optical properties of cells, to measures cellular stiffness [53]. Quantitative phase imaging measures the overall phase retardance due to change in refraction index. Interestingly, local changes of the intracellular birefringence would also result in a proportional modulation of the phase, suggesting that the two metrics may be related. Taken together, these observations highlight how cellular optical properties are modulated by the underlaying mechanical properties of the cell.
At the subcellular level, previous work has shown that actin stress fibers are mainly under tensile load due to the action of myosins [54, 55]. Interestingly, the elastic energy stored in actin fibers is highly heterogeneous in a cell and depends on the underlying cytoskeleton architecture and connectivity [55]. In addition, the strain measured in individual actin stress fibers fluctuates along its length [56], highlighting the anisotropic nature of cytoskeletal mechanics. Furthermore, work done with intracellular stress tomography indicates that mechanical stress can focalize far away from the point of force application [57]. Numerical simulations suggest that stress concentration could occur in the perinuclear region of a spreading cell in numerous conditions [58, 59]. In this context, the distribution of the QPOL signal we observe could be representative of the heterogeneous nature of the intracellular strain field. However, further work will be required to fully determine how the QPOL signal is related to cytoskeleton mechanics at the subcellular level.
Overall, our results show that QPOL microscopy can be used to obtain information on cell contractility under different conditions, including single cells as well as complex biological samples such as cell monolayers, ex vivo tumor organoids, and tissue sections. Furthermore, we were able to distinguish between cells from soft and stiff tumors samples using our method. An obvious advantage is that QPOL can be used to measure a signal that is proportional to cell contractility in 2D and 3D systems without any exogenous labels, independent of the substrate mechanical properties, and with minimal computational need. Overall, QPOL provides a powerful platform to study cell contractility in physiologically relevant settings, such as tumor progression.
Acknowledgement
This work was supported in part by the New York Brain Tumor Project of Weill Cornell Medical College, the Kavli Institute of Nanotechnology at Cornell Instrumentation Projects award, a Sloan Foundation and a NSF Graduate Research Fellowship to J.P.M. and the Scholarship for the Next Generation of Scientists from the Cancer Research Society and NIH K99 through Award number CA212270 to F.B.
Footnotes
Author contributions.
W.W., J.P.M. and F.B. performed the experiments. W.W. and F.B. performed the data analysis. J.P.M. and F.B. designed the microscope and the acquisition and analysis software. F.B., W.W. and C.R.K. conceived the experiments. F.B., C.R.K. and S.C.P. supervised the work. All authors contributed to writing the manuscript.
Competing financial interests
The authors declare no competing financial interests.
REFERENCE
- [1].Wozniak MA, Desai R, Solski PA, Der CJ, Keely PJ, J Cell Biol 2003. 163 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006. 126 677. [DOI] [PubMed] [Google Scholar]
- [3].Joshi B, Strugnell SS, Goetz JG, Kojic LD, Cox ME, Griffith OL, Chan SK, Jones SJ, Leung SP, Masoudi H, Leung S, Wiseman SM, Nabi IR, Cancer Res 2008. 68 8210. [DOI] [PubMed] [Google Scholar]
- [4].Zanotelli MR, Bordeleau F, Reinhart-King CA, Current Opinion in Biomedical Engineering 2017. 1 8. [Google Scholar]
- [5].Discher DE, Janmey P, Wang Y.-l., Science 2005. 310 1139. [DOI] [PubMed] [Google Scholar]
- [6].Huang S, Ingber DE, Cancer Cell 2005. 8 175. [DOI] [PubMed] [Google Scholar]
- [7].Paszek MJ, Weaver VM, Journal of mammary gland biology and neoplasia 2004. 9 325. [DOI] [PubMed] [Google Scholar]
- [8].Polacheck WJ, Chen CS, Nat Methods 2016. 13 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tymchenko N, Wallentin J, Petronis S, Bjursten LM, Kasemo B, Gold J, Biophysical journal 2007. 93 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mulligan JA, Bordeleau F, Reinhart-King CA, Adie SG, Biomed Opt Express 2017. 8 1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Koch TM, Munster S, Bonakdar N, Butler JP, Fabry B, PLoS One 2012. 7 e33476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kokini K, Sturgis JE, Robinson JP, Voytik-Harbin SL, J Biomech Eng 2002. 124 214. [DOI] [PubMed] [Google Scholar]
- [13].Badylak SF, Freytes DO, Gilbert TW, Acta biomaterialia 2009. 5 1. [DOI] [PubMed] [Google Scholar]
- [14].Storm C, Pastore JJ, MacKintosh FC, Lubensky TC, Janmey PA, Nature 2005. 435 191. [DOI] [PubMed] [Google Scholar]
- [15].Steinwachs J, Metzner C, Skodzek K, Lang N, Thievessen I, Mark C, Munster S, Aifantis KE, Fabry B, Nat Methods 2016. 13 171. [DOI] [PubMed] [Google Scholar]
- [16].Glazer AM, Lewis JG, Kaminsky W, Proceedings of the Royal Society a-Mathematical Physical and Engineering Sciences 1996. 452 2751. [Google Scholar]
- [17].Kuhn JR, Wu Z, Poenie M, Biophysical journal 2001. 80 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Helfer E, Panine P, Carlier MF, Davidson P, Biophys J 2005. 89 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kraning-Rush CM, Carey SP, Califano JP, Smith BN, Reinhart-King CA, Physical biology 2011. 8 015009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nguyen-Ngoc K-V, Shamir ER, Huebner RJ, Beck JN, Cheung KJ, Ewald AJ, Methods in molecular biology (Clifton, NJ) 2015. 1189 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kraning-Rush CM, Califano JP, Reinhart-King CA, PLoS One 2012. 7 e32572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, LaValley DJ, Huynh J, Proceedings of the National Academy of Sciences 2016. 201613855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mason BN, Reinhart-King CA, Organogenesis 2013. 9 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Reinhart-King CA, Dembo M, Hammer DA, Biophys J 2005. 89 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Glazer A, Lewis J, Kaminsky W, An automatic optical imaging system for birefringent media, Proceedings of the Royal Society of London A: Mathematical, Physical and Engineering Sciences, The Royal Society, 1996, pp. 2751. [Google Scholar]
- [26].Even-Ram S, Doyle AD, Conti MA, Matsumoto K, Adelstein RS, Yamada KM, Nature cell biology 2007. 9 299. [DOI] [PubMed] [Google Scholar]
- [27].Wakatsuki T, Schwab B, Thompson NC, Elson EL, Journal of cell science 2001. 114 1025. [DOI] [PubMed] [Google Scholar]
- [28].Hui KL, Balagopalan L, Samelson LE, Upadhyaya A, Molecular biology of the cell 2015. 26 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kumar S, Weaver VM, Cancer and Metastasis Reviews 2009. 28 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huynh J, Bordeleau F, Kraning-Rush CM, Reinhart-King CA, Cellular and Molecular Bioengineering 2013. 6 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Cancer Cell 2005. 8 241. [DOI] [PubMed] [Google Scholar]
- [32].Mason BN, Starchenko A, Williams RM, Bonassar LJ, Reinhart-King CA, Acta biomaterialia 2013. 9 4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ward PD, Tippin TK, Thakker DR, Pharmaceutical science & technology today 2000. 3 346. [DOI] [PubMed] [Google Scholar]
- [34].Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen Y, Liphardt J, Hwang E, Weaver V, Integrative Biology 2015. 7 1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Cell 2009. 139 891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kagan HM, Li W, Journal of cellular biochemistry 2003. 88 660. [DOI] [PubMed] [Google Scholar]
- [37].Bordeleau F, Califano JP, Abril YLN, Mason BN, LaValley DJ, Shin SJ, Weiss RS, Reinhart-King CA, Proceedings of the National Academy of Sciences 2015. 112 8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, LeBleu VS, Roy N, Novitskiy SV, Johansen JS, Poli V, Kalluri R, Iacobuzio-Donahue CA, Wood LD, Hebrok M, Hansen K, Moses HL, Weaver VM, Nat Med 2016. 22 497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Trabbic-Carlson K, Setton LA, Chilkoti A, Biomacromolecules 2003. 4 572. [DOI] [PubMed] [Google Scholar]
- [40].Wang N, Ingber DE, Biophysical journal 1994. 66 2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Han S, Huang J, Liu B, Xing B, Bordeleau F, Reinhart-King CA, Li W, Zhang JJ, Huang XY, Mol Oncol 2016. 10 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang Y, Jeong Y, Jhiang SM, Yu L, Menq CH, PLoS One 2014. 9 e98762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Plotnikov SV, Pasapera AM, Sabass B, Waterman CM, Cell 2012. 151 1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Huang XR, Knighton RW, Invest Ophthalmol Vis Sci 2005. 46 4588. [DOI] [PubMed] [Google Scholar]
- [45].LaValley DJ, Zanotelli MR, Bordeleau F, Wang W, Schwager SC, Reinhart-King CA, Convergent Science Physical Oncology 2017. 3 044001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Small J, Gimona M, Acta physiologica scandinavica 1998. 164 341. [DOI] [PubMed] [Google Scholar]
- [47].Eriksson JE, Dechat T, Grin B, Helfand B, Mendez M, Pallari HM, Goldman RD, J Clin Invest 2009. 119 1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Flitney EW, Kuczmarski ER, Adam SA, Goldman RD, The FASEB Journal 2009. 23 2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Esue O, Tseng Y, Wirtz D, PLoS One 2009. 4 e4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bonakdar N, Schilling A, Spörrer M, Lennert P, Mainka A, Winter L, Walko G, Wiche G, Fabry B, Goldmann WH, Experimental cell research 2015. 331 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jackson WM, Jaasma MJ, Baik AD, Keaveny TM, Annals of biomedical engineering 2008. 36 1605. [DOI] [PubMed] [Google Scholar]
- [52].Nanavati H, Desai P, Abhiraman A, Computational and Theoretical Polymer Science 1999. 9 165. [Google Scholar]
- [53].Eldridge WJ, Steelman ZA, Loomis B, Wax A, Biophys J 2017. 112 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kumar S, Maxwell IZ, Heisterkamp A, Polte TR, Lele TP, Salanga M, Mazur E, Ingber DE, Biophysical journal 2006. 90 3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kassianidou E, Brand CA, Schwarz US, Kumar S, Proceedings of the National Academy of Sciences 2017. 201606649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Guolla L, Bertrand M, Haase K, Pelling AE, J Cell Sci 2012. 125 603. [DOI] [PubMed] [Google Scholar]
- [57].Hu S, Chen J, Fabry B, Numaguchi Y, Gouldstone A, Ingber DE, Fredberg JJ, Butler JP, Wang N, American Journal of Physiology-Cell Physiology 2003. 285 C1082. [DOI] [PubMed] [Google Scholar]
- [58].Ronan W, Deshpande VS, McMeeking RM, McGarry JP, Journal of the Mechanical Behavior of Biomedical Materials 2012. 14 143. [DOI] [PubMed] [Google Scholar]
- [59].Milner JS, Grol MW, Beaucage KL, Dixon SJ, Holdsworth DW, Journal of functional biomaterials 2012. 3 209. [DOI] [PMC free article] [PubMed] [Google Scholar]