

Abstract
Glycoconjugates are directly or indirectly involved in many biological processes. Due to their complex structures, the structural elucidation of glycans and the exploration of their role in biological systems have been challenging. Glycan pools generated through release from glycoprotein, or glycolipid mixtures can often be very complex. For the sake of procedural simplicity, many glycan profiling studies choose to concentrate on a single class of glycoconjugates. In this paper, we demonstrate it feasible to cover glycosphingolipids, N-glycans and O-glycans isolated from the same sample. Small volumes of human blood serum and ascites fluid as well as small mouse brain tissue samples are sufficient to profile sequentially glycans from all three classes of glycoconjugates and even positively identify some mixture components through MALDI-MS and LC-ESI-MS. The results show that comprehensive glycan profiles can be obtained from the equivalent of 500 μg protein starting material or possibly less. These methodological improvements can help accelerating future glycomic comprehensive studies, especially for precious clinical samples.
Keywords: integrated glycomics scheme, N- and O-glycans, glycosphingolipid (GSL)-glycans
Introduction
Glycosylation, in its many forms and structural details, appears to play a major role in most, if not all, biological processes. It is now generally appreciated that more than 50 % of all proteins are glycosylated [1] by the glycan structures assembled and modified through the action of glycosyltransferases in the endoplasmic reticulum and Golgi apparatus and subsequently linked to their respective conjugating counterparts. It is further estimated that some 250-500 genes in the human genome are directly involved in the overall glycosylation process, giving rise to a huge variety of possible glycan structures [2, 3]. Since glycosylation appears distinct for different cell types and their biological status, it is imperative that glycomic measurements address the extent of such structural variations over an extensive concentration range. Glycosylation complexity in glycolipids can also be substantial. Depending on a cell type, up to 10 % of glycosphingolipids can be encountered in the lipid fractions of different cellular materials [4].
The general biological importance of glycosylation has made it an increasingly popular target to access biomarkers in stem cells[5–7] and human diseases [8–12], all typically requiring to handle small biological specimens. The high sensitivity and specificity of the corresponding glycomic profiling measurements are currently best assured through the use of modern mass spectrometry (MS) and/or its combination with capillary separation techniques [13–18]. During the recent years, there has also been a remarkable progress made in combining these measurement techniques with advances in sample preparation and microderivatization methodologies [19–24]. However, there are only a few methods reported for isolating different classes of glycoconjugates from the same sample. Notably, Fujitani et al.[25] developed a method to isolate most major glycans from human stem cell samples through the use of glycoblotting, however, two separate extraction procedures still had to be performed for free oligosaccharide/N-glycans/O-glycans and glycosphingolipids/glycosaminoglycans. Some other methods [26, 27] opted to combine an isolation procedure to determine both N- and O-glycans through MS analyses. To the best of our knowledge, no attempt has been reported toward a sequential isolation of glycosphingolipids, N-glycans and O-glycans from the same biological material performed at microscale.
The remarkable detection performance of MS measurements now permits determinations of glycoconjugates from extremely small biological samples, as exemplified by profiling N-glycans from low-microliter volumes of blood sera[8] and O-glycans from miligrams of mucosal biopsies [28]. However, the bottleneck of highly sensitive measurements is often in the effective microscale cleavage of glycoconjugates and their extraction and purification prior to analysis. For the sake of enhanced MS detection, sensitivity and information content, the glycoconjugates can further be derivatized through the use of quantitative permethylation [29, 30]. To enable the sequential acquisition of the glycan profiles from glycosphingolipids, N-oligosaccharides and O-oligosaccharides from the same samples, we examined here the possibility of combining the acetone precipitation[31], which extracts nonpolar glycolipids from the glycosylated proteins, with other effective procedures pursued by our laboratory. A further use of centrifugal filtration [32] in concert with the previously established glycan cleavage procedures, enables us to obtain three complementary glycan profiles from minute volumes of physiological fluids or small tissue samples. The utility of this “combined approach” is demonstrated here with the examples of human blood serum, ascites fluids, and a small tissue sample of the mouse cortex.
Experimental
Biological samples
Human serum was received from Bioreclamation LLC (Westbury, NY) and the ascites fluids from Indiana University School of Medicine, Indianapolis (courtesy of Dr. Daniela Matei, Indiana University School of Medicine, Indianapolis, IN). Mouse tissues were obtained from Dr. Sachiko Koyama (Indiana University School of Medicine, Medical Sciences Program, Bloomington, IN).
Chemicals
Trifluoroacetic acid and HPLC grade water, acetonitrile and methanol were purchased from EMD Chemicals (Gibbstown, NJ). Chloroform, acetic acid and acetone was obtained from Macron Fine Chemicals (Center Valley, PA). Sodium hydroxide beads, 20-40 mesh size, and methyl iodide was acquired from Sigma-Aldrich (St. Louis, MO). Dimethylformamide and sodium chloride were purchased from Mallinckrodt (St Louis, MO). The following enzymes with their sources are: N-glycanase/PNGase F (Prozyme, Hayward, CA), Pronase (Roche, Indianapolis, IN), porcine trypsin (Sigma-Aldrich, St. Louis, MO), endoglycoceramidase II (rEGCase II) enzyme (Takara Bio USA, Mountain View, CA). 2,5-dihydroxybenzoic acid (2,5-DHB) was received from Alfa Aesar (Ward Hill, MA).
Empty reaction vessels and micro-spin columns with different matrixes (active charcoal, C8, C18, amine) were purchased from Harvard Apparatus (Holliston, MA). Amicon Ultra-0.5 Centrifugal Filter Unit with Ultracel-10 membrane was from EMD Millipore (a part of Millipore Sigma, Billerica, MA).
Sample Preparation
Brain cortex cuts from three mice were weighted and suspended in 10 mM phosphate buffer (pH 7.5) to a final concentration of 50 mg/mL. The tissues were homogenized by using a TissueRuptor (Qiagen, Germantown, MD), for 2 times x 1 min at MAX speed. The aliquots were incubated in 1% sodium dodecylsulfate (SDS) at 60 °C for 30 min to ensure that the proteins were contained in the solution prior to protein concentration measurement with bicinchoninic acid assay (BCA assay kit, ThermoFisher, Rockford, IL). Three mouse cortex extracts, three human ascites fluids and three human blood serum samples were used to extract sequentially glycosphingolipids, N-glycans and O-glycans. Five hundred μg protein aliquots samples (from cortex tissues or ascites fluid) and 50 μL human blood serum were used for the glycan analysis, respectively. An overview of the complete sample preparation can be seen in Figure 1.
Figure 1.
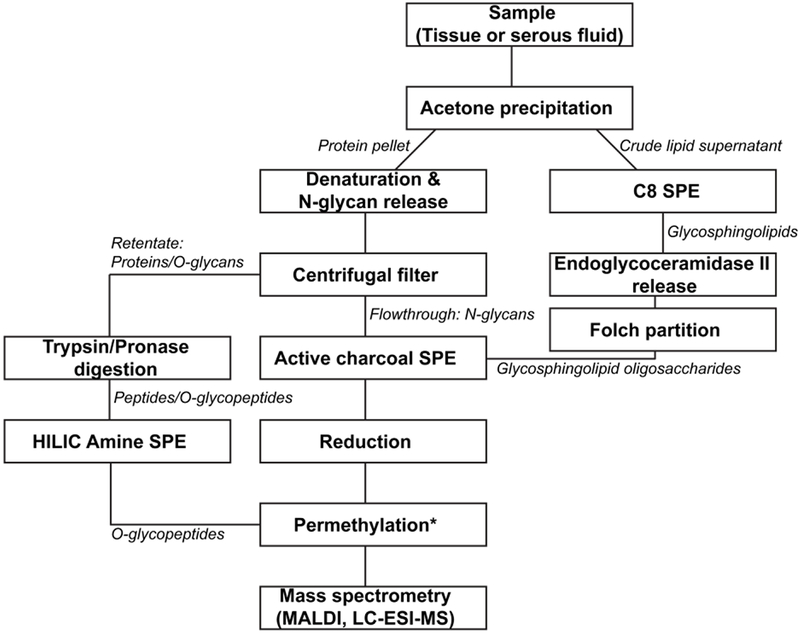
Schematic representation of glycomic procedures. For O-glycopeptides, the permethylation step simultaneously releases and permethylates O-glycans. Abreviations: SPE, Solid-Phase Extraction; HILIC, Hydrophilic Interaction Liquid Chromatography; MALDI, Matrix-Assisted Laser Desorption/Ionization; LC-ESI-MS, Liquid Chromatography Electrospray Ionization Mass Spectrometry.
Acetone precipitation
The acetone precipitation procedure followed the protocol of Gizaw et al. [31] with some modifications. Acetone precipitation was performed in order to extract and separate glycosphingolipids and other nonpolar molecules from the proteins. Some less polar proteins could potentially be at risk of ending up in the supernatant, but glycoproteins are generally quite polar due to the presence of sugar moieties,
Ice-cold acetone was added to the samples in 4:1 ratio while being vortexed. After 1 min of sonication in a water bath, the samples were placed at −80°C for 2 hrs. After the following centrifugation at 14,000 × g for 15 min (4°C), the supernatants , containing nonpolar components, including glycosphingolipids and other lipids, were collected. The pellets of precipitated proteins were washed with 400 μL ice-cold acetonitrile, vortexed and centrifuged at 14,000 × g for 15 min (4°C). The supernatants were collected and pooled with the acetone supernatants to be dried in a CentriVap concentrator (Labconco, Kansas City, MO). The pellets were air-dried for 5 min and then stored in a freezer prior to N- and O-glycan analysis.
Glycosphingolipid recovery
The dried supernatants from the acetone precipitation, containing glycospingolipids, were purified by C8 Micro SpinColumns as previously described by Garcia et al [33], with some modifications. This purification was performed to separate other lipid constituents from the more polar glycosphingolipids. There is some risk of losing the more nonpolar glycosphingolipids such as the monoglycosylceramides, however, their glycans are too small to be detected by the current state-of-the-art MS and can be momentarily ignored within the scope of our studies.
The columns were washed with 3× 400 μL methanol and centrifuged at 400 × g for 30 s. Subsequently, the columns were conditioned with 2× 400 μL 6:4 methanol:water, and centrifuged at 200 g for 2 min. The dried supernatants were diluted in 200 μL 6:4 methanol:water, added to the columns and centrifuged at 200 × g for 2 min. The eluates were then added to the columns again and centrifuged. The samples were washed with 3× 200 μL 6:4 methanol:water, centrifuged at 200 × g for 2 min and then eluted by adding 200 μL methanol to the columns and centrifuged for ~10 s at 200 × g before letting the column equilibrate for 10 min. Then, the columns were centrifuged at 200 × g for 2 min, another 200 μL methanol was added and centrifuged at 200 g for 2 min. The eluates were dried by a CentriVap concentrator.
The enzymatic cleavage of the glycosphingolipids was done with the recombinant endoglycoceramidase II (rEGCase II) enzyme, similarly to the previously described procedure[34], with some modifications to the desalting procedure. To each sample, 70 μL water, 10 μL 50 mM sodium acetate (pH 5) and 10 μL 2 mg/mL sodium deoxycholate were added. The mixtures were vortexed prior to addition of 10 μL rEGCase II preparation diluted 1:4 in 50 mM sodium acetate buffer (2mU) and left at 37°C for >16 hours. After incubation, the Folch partitions were performed by adding 200 μL water, 400 μL methanol and 800 μL chloroform. The mixtures were vortexed and left equilibrating for 2 hrs, then centrifuged at 100 × g for 1 min. The upper glycan-containing phases were collected and dried by CentriVap concentrator, while the lower phase containing the nonpolar ceramides could be discarded. The upper phases were desalted by active charcoal micro Spin Columns. The columns were washed 3 times with solution B (85%/15%/0.1% acetonitrile/water/trifluoroacetic acid) and centrifuged at 400 × g for 2 min. This was followed by washing the columns 3 times with solution A (5%/95%/0.1% acetonitrile/water/trifluoroacetic acid) and centrifuged at 400 g for 2 min. Subsequently, the samples were dissolved in 250 μL solution A, vortexed, loaded onto the columns and centrifuged for 2 min at 400 × g. The collected flowthroughs were reapplied to the columns and centrifuged for 2 min at 400 × g. This step was repeated one more time. Subsequently, the columns were washed with 2× 200 μL solution A, and centrifuged for 2 min at 400 g. Samples were eluted with 200 μL 50%/50%/0.1% acetonitrile/water/trifluoroacetic acid into new centrifuge tubes. The columns were centrifuged briefly for 15 s at 400 × g and were left to equilibrate for ~10 min, then centrifuged at 400 × g for 2 min. Another 200 μL 50%/50%/0.1% acetonitrile/water/trifluoroacetic acid was added and centrifuged for 1.5 min at 800 g. The 50%/50%/0.1% acetonitrile/water/trifluoroacetic acid eluates were pooled and dried in a CentriVap concentrator. The glycosphingolipid oligosaccharide recovery was followed by reduction and permethylation, as described below.
N-glycan recovery
The N-glycan denaturation and release was performed as previously described[35]. To the acetone precipitation pellets, 200 μL 10 mM sodium phosphate buffer (pH 7.5) with 0.1% SDS was added, followed by vortexing and incubation at 37°C for 10 min. The pellets were dissolved by pipetting, β-mercaptoethanol (0.1%), which was added prior to incubation at 60°C for 30-60 min. After incubation, the samples were cooled to room temperature before adding 10 μL10% Nonidet-P40 solution. The mixtures were equilibrated for 10 min at room temperature, followed by addition of 0.5 mU of PGNase F and incubation at 37°C for ≥16 hrs.
To separate the proteins from glycans, 10,000 MWCO filters (Amicon Ultra Centrifugal filters-0.5 ml, Milipore, Billerica, MA) were prepared by adding 400 μL water and centrifuging for 10 min at 9,300 × g, followed by 400 μL10 mM sodium phosphate buffer (pH 7.5) and centrifuging for 10 min at 9300 × g. Subsequently, 200 μL 10 mM sodium phosphate buffer (pH 7.5) was first added to the centrifugal filters and then the PGNase F-digested sample was added and mixed by pipetting. The centrifugal filters were centrifuged for 15 min at 14.000 × g, followed by addition of 2× 400 μL 10 mM sodium phosphate buffer (pH 7.5), and centrifuged at 14,000 × g for 15 min. The first flowthrough and sodium phosphate buffer flowthrough containing the N-glycans were pooled and dried with a CentriVap concentrator. The centrifugal filters were then turned upside down and centrifuged for 15 min at 14,000 × g to elute the proteins. The protein fractions were continued in the O-glycan preparation segment of the scheme (Figure 1).
The dried N-glycan samples were desalted by active charcoal SpinColumns. The columns were washed 3× with solution B (85%/15%/0.1% acetonitrile/water/trifluoroacetic acid), centrifuged at 400 × g for 2 min, followed by washing the columns 3× with solution A (5%/95%/0.1% acetonitrile/water/trifluoroacetic acid), and centrifuged at 400 × g for 2 min. Subsequently, the N-glycan samples were dissolved in 250 μL solution A, vortexed, loaded onto the columns and centrifuged for 2 min at 400 × g. The flowthroughs were collected, reapplied to the columns and centrifuged for 2 min at 400 × g. This step was then repeated one more time before washing the column with 2× 200 μL solution A, and centrifuged for 2 min at 400 × g. New centrifuge tubes were used and 200 μL 50%/50%/0.1% acetonitrile/water/trifluoroacetic acid was added to elute the N-glycans. First, the columns were centrifuged briefly for 15 s at 400 × g, permitting them to equilibrate for ~10 min and then centrifuged at 400 × g for 2 min. Subsequently, another 200 μl 50%/50%/0.1% acetonitrile/water/trifluoroacetic acid was added and centrifuged for 1.5 min at 800 × g. The two 50%/50%/0.1% acetonitrile/water/trifluoroacetic acid eluates were pooled and dried in a CentriVap concentrator. The N-glycan preparation was continued by reduction and permethylation as described later.
O-glycan recovery
The protein fractions obtained with the aid of spin filter columns were supplemented with 5 μg trypsin and left at room temperature for 4 h. Subsequently, 20 μg pronase was added and incubated at 55 °C for 48 hrs. The O-glycosylated peptides were purified by amino Ultra SpinColumns (Harvard apparatus) as described previously [35]. The columns were washed 3× with buffer B (10%/90%/0.1% acetonitrile, water, trifluoroacetic acid) and 3× with buffer A (90%/10%/0.1% acetonitrile, water, trifluoroacetic acid) and centrifuged for 1 min at 200 × g. 250 μL volumes of buffer A were added to the digested protein samples and vortexed before applying samples to the columns. The columns were centrifuged for 1 min at 200 × g, the flowthroughs were collected and reapplied to the column. The samples were reapplied and centrifuged two more times to ensure retention on the column. Following that, the columns were washed with 2× 300 μL buffer A and centrifuged for 1 min at 200 × g. The O-glycopeptide fractions were eluted by adding 300 μL buffer B, centrifuged at 200 × g for ~10 s, left equilibrating for 10 min, then centrifuged for 1 min at 200 × g. Another 300 μL buffer B was added and centrifuged for 1.5 min at 800 × g. The two buffer B eluate fractions were pooled and dried by a CentriVap concentrator.
The O-glycopeptides were permethylated (see Permethylation section below) the same way as glycosphingolipid oligosaccharides and N-glycans. However, the permethylation was also used here to release the O-glycans from the peptides[36] and the glycans were thus not reduced first.
Reduction
Reduction was performed as previously described [35]. The glycosphingolipid and N-glycan samples were subjected to this procedure by adding 10 μL of 10 mg/mL aqueous borane-ammonia complex, vortexing, briefly spinning down the samples and then incubating them at 60°C for 1 h. The samples were cooled to room temperature and, after 100 μL methanol was added, they were dried in a CentriVap concentrator. Another 100 μL methanol was added, dried and repeated once more to remove remaining borane-ammonia complex. The sample preparation continued with permethylation, as described below.
Permethylation
Permethylation was carried out as previously described [35]. First, the permethylation reactors were created by taking a 20 mL glass vial, filled with roughly ¼ with NaOH beads, and followed with acetonitrile. By using a cut 1-mL pipette tip, the NaOH beads were transferred to empty Micro SpinColumns (HarvardApparatus). Approximately ¾ of the column volume was filled up with the NaOH beads. The columns were centrifuged at 200 × g for 1 min to remove acetonitrile and then washed with 3× ~300 μl DMF (dimethylformamide) and centrifuged for 1 min at 200 × g.
The reduced glycan samples were supplemented with 5 μL water, vortexed and centrifuged for 30 s at 600 × g. Next, 65 μL DMF and 35 μL methyl iodide were added to the samples and mixed. The samples were applied onto the permethylation reactors, and the reactors were put in the original sample-centrifugation tubes, which were sealed by putting a cap on them. The reactors were set horizontally, slightly elevated and left to react for 20 min. Afterwards, the caps were removed and the permethylation reactors were centrifuged for 1.5 min at 200 × g. The flowthroughs were reapplied to the reactors after 35 μL methyl iodide was added and mixed. Caps were used to close the reactors and they were set horizontally, slightly elevated, and left to react for another 20 min. The caps were removed and reactors were centrifuged for 1.5 min at 2,300 × g. Then, 400 μL chloroform and 1 mL 0.5 M NaCl were added to the flowthroughs and shaken gently by hand for 2 min before centrifuging the samples at 600 × g for 30 s. The upper aqueous phases were discarded and 1 mL water was added to the lower chloroform phases. The mixtures were gently shaken by hand for 2 min and centrifuged for 30 s at 600 × g. The upper aqueous phases were discarded again, while 1 mL water was added and gently shaken by hand for 2 min. The mixtures were centrifuged for 30 s at 2,600 rpm and the upper aqueous phases were discarded. The chloroform phases were dried in a CentriVap concentrator and stored in a freezer until analysis.
Mass-Spectrometric Analyses
MALDI-MS
The permethylated glycosphingolipid-derived oligosaccharides, N-glycan and O-glycan preparations were dissolved in 5 μL 50%/50% methanol/water solution. Half-microliter samples were spotted onto a MALDI plate and left to dry for 10 min. Next, 0.5 μL of the matrix (10 mg/ml DHB in 50%/50% methanol/water) with a final concentration of 1 mM sodium acetate was added on top of the spots and dried under vacuum for 2 min. The spots were examined by an Applied Biosystems 4800 MALDI-TOF/TOF mass spectrometer (ABSciex, LLC, Framingham, MA) in the positive-ion reflector mode. The laser intensity was set to 4,100 with a light intensity of 20 (arbitrary units). Obtained spectra were processed through baseline correction and noise filter (0.7) using the software Data Explorer (ABSciex, LLC). The proportion of each glycan was normalized as total percentage of the intensity of all glycans.
LC-MS
Permethylated glycans were chromatographically separated using a Waters Acquity M class UPLC system (Waters Corporation, Milford, MA) and detected with a Finnigan LTQ linear ion trap (Thermo Electron Co., San Jose, CA) equipped with a nanoelectrospray source. Briefly, the glycolipid-originated glycans and O-glycans were dissolved in 20-100 μL 1:1 H2O: acetonitrile and 1 μL (equivalent to ~10 μg protein starting material) sample was loaded and separated on a 200 × 0.075 mm capillary column packed in-house with 3 μm porous graphitized carbon particles (Hypercarb, Thermo Scientific, Runcorn, U. K.) with an increasing concentration of acetonitrile (buffer A H20 0.1% formic acid; buffer B acetonitrile 0.1% formic acid). The gradient was performed with a flow of 0.5μL/min at 75 °C in a stepwise manner: 0-10 min at 10% B, 10-40 min 10-30% B, 40-70 min 30-60% B, 70-100 min 60-100% B, 100-120 min 100% B, 120-130min 100-10% B, 130-150 min 10-10% B. The ions were detected in the positive-ion mode with a spray voltage set to 2.0 kV. The aliquots of permethylated N-glycans were separated on the carbon columns with an increasing concentration of isopropanol (buffer A H20 0.1% formic acid; buffer B isopropanol 0.1% formic acid) prior to MS detection with more gentle ionization settings, essentially as described by Smith et al [14]. Briefly, the capillary temperature was reduced from 300°C to 150°C and the Tube voltage was reduced from 210V to 55V. The gradient was performed with a flow of 0.5μL/min at 75 °C in a stepwise manner: 0-10 min at 20% B, 10-40 min 20-50% B, 40-70 min 50-70% B, 70-100 min 70-95% B, 100-120 min 95% B, 120-130min 95-20% B, 130-180 min 20% B.
Data analysis
The MALDI mass spectra were baseline-corrected, a noise filter was applied and exported to text files. The data were normalized (percentage of ion intensities for all glycans listed in a peak list file) with PeakCalc v.2.0 (in-house developed software). After normalization, the data from three spots were averaged and used for statistical analysis using PeakCalc 2.0 and Microsoft Excel 2013 (Microsoft, WA). The LC-ESI-MS raw files were visualized with Xcalibur software 2.0.7 (Thermo Scientific). GlycoWorkbench software [37] was used for drawing of glycan cartoons and annotation of MS/MS spectra. LC-MS figures were drawn in Illustrator (Adobe Systems, San Jose, CA), while the graphs were made with Microsoft Excel and Microsoft Powerpoint.
Results and Discussion
Among the most important goals of glycoscience is to provide highly sensitive and structurally informative analytical techniques for a wide range of investigators in biological and medical fields. While the real complexity of glycomes can be overwhelming, independent profiling of N-glycans, O-glycans and glycosphingolipid-derived glycans has been mostly enabled by advances in mass spectrometry and its related techniques of separation science and microderivatization, which ultimately could be combined to achieve very high sensitivity. Such glycan profiles provide the first-hand information about differences between controls and variations of different biological experiments or the clinical samples obtained from control patients vs. disease individuals in biomarker discovery studies. For example, MALDI-MS glycan profiling may generate a set of tentatively assigned structures, annotated according to their m/z values. Depending on the goals of a particular investigation, more precise structural investigations through MS/MS may be feasible for major glycan profile components. However, for selected minor profile components, specific enrichment techniques may still be required for their positive identification.
The main objective of this study was to develop a protocol to consecutively release and profile glycans from three main classes of glycoconjugates: glycosphingolipids, N-glycans and O-glycans.
To test the feasibility of this protocol for both tissues and biological fluids, three different types of biological materials were arbitrarily examined: human blood serum; human ascites fluid; and mouse brain cortex. A complete and definitive structural assignment of all glycan components in these samples was not yet within our scope, although we were able to confirm structurally most glycans identified in our previous studies. Moreover, we demonstrate, through selected examples, the feasibility of in-depth studies of the corresponding glycan pools. It should be noted that no large amounts of glycosphingolipids were expected to be found in serum or ascites fluid, as these components are mainly found in cellular membranes. However, some minor amounts could potentially be detected in sera and ascites fluids due to cell “shedding” in the form of, for example. micelles [38]. The samples were analyzed in a parallel triplicate of each sample type. Five hundred microgram protein starting material was used for each human ascites fluid and the mouse cortex samples, while 50μl of human blood serum was employed for extraction.
It should be noted that the primary purpose of this methodologically oriented study was to demonstrate the feasibility of recording sequentially three different glycoconjugate groups originated from different types of biological specimens. The following figures and graphs are thus not intended to compare them with each other, and any structural or quantitative similarities thus may be entirely coincidental. The acquired glycan profiles, however, all demonstrate the merits of this overall analytical scheme and its compatibility with the current MS detection approaches, although the absolute structural identification of some tentatively assigned components remains to be a task of future studies.
Figure 2 demonstrates the profiling results of N-glycans from the three different sample types. A total of 97 sodiated ions were detected and tentatively annotated with their m/z values based on at least one or combinations of the following information: our in-house structural database of serum glycans, MS/MS spectra from LC-MS and knowledge of available glycosyltransferases, and previous publications. The MALDI-MS N-glycan profile of the human blood serum and ascites fluid have comparable distribution and relative abundance of the major glycans, which is consistent with the previous reports[39, 40]. As expected, the mouse cortex MALDI-MS data for the N-glycan profile was considerably different from the serous fluid profiles, with a high percentage of high-mannose and bisecting structures (Figure 2) represented. The larger N-glycan structures also appear to be more fucosylated when compared with those from the ascites fluid and serum samples. This is in agreement with the earlier studies on mouse cortex [41]. The serum N-glycan numbers roughly agree with those seen in our previous investigations [8,23,40] and those reported elsewhere [42]. A supplementary table (Table S1) further lists these N-glycans, some of which were also verified through LC-ESI-MS/MS. Additionally, we display the N-glycan data from all three biological materials as their structural types in a “pie chart” in supplementary Figure S1. The minor glycans detected below m/z about 1187.6 are unlikely procedural fragments of larger glycans, but rather the paucimannosidic structures that had previously been linked to important biological activities [43–46]. They are created through distinct glycan enzymatic activities [43]. Interestingly, the mouse cortex sample exhibits a relatively high percentage of the paucimannosidic glycans (see supplementary Figure S1). Adjusting the electrospray settings to minimize potential in-source fragmentation, we could confirm several small puacimannosidic structures that did not correspond to fragments from larger N-glycans (data not shown).
Figure 2.
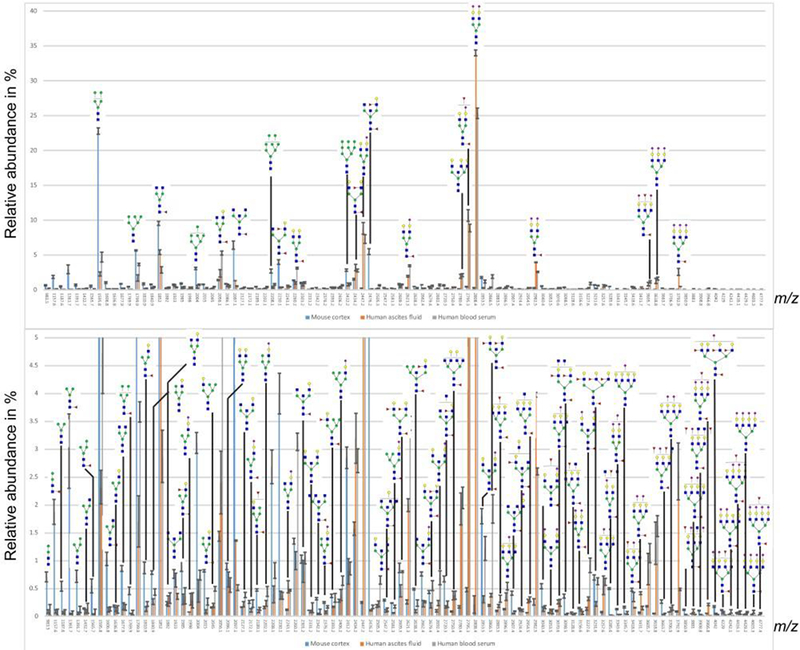
Profiles of permethylated N-glycans detected as sodiated ions in the positive-ion mode with MALDI-MS. Y-axis displays percentage of total glycans. Top graph shows the total distribution of glycans, while the bottom profile is zoomed from 5 % relative intensity.
Whereas N-glycan profiles are now relatively easy to measure in small samples of human serum, plasma or tissues due to the facile enzymatic cleavage by PNGase F, O-glycan profiling is still far from being trivial. Here, we show significant progress in this direction, as outlined in Figure 1, and clearly demonstrated in Figure 3. A total of fifty-one sodiated ions were detected and tentatively annotated with tentative structures represented by m/z values. The ascites fluid and human blood serum O-glycan profiles were qualitatively similar to each other. Seven of the 10 most abundant O-glycans were shared between the two samples.
Figure 3.
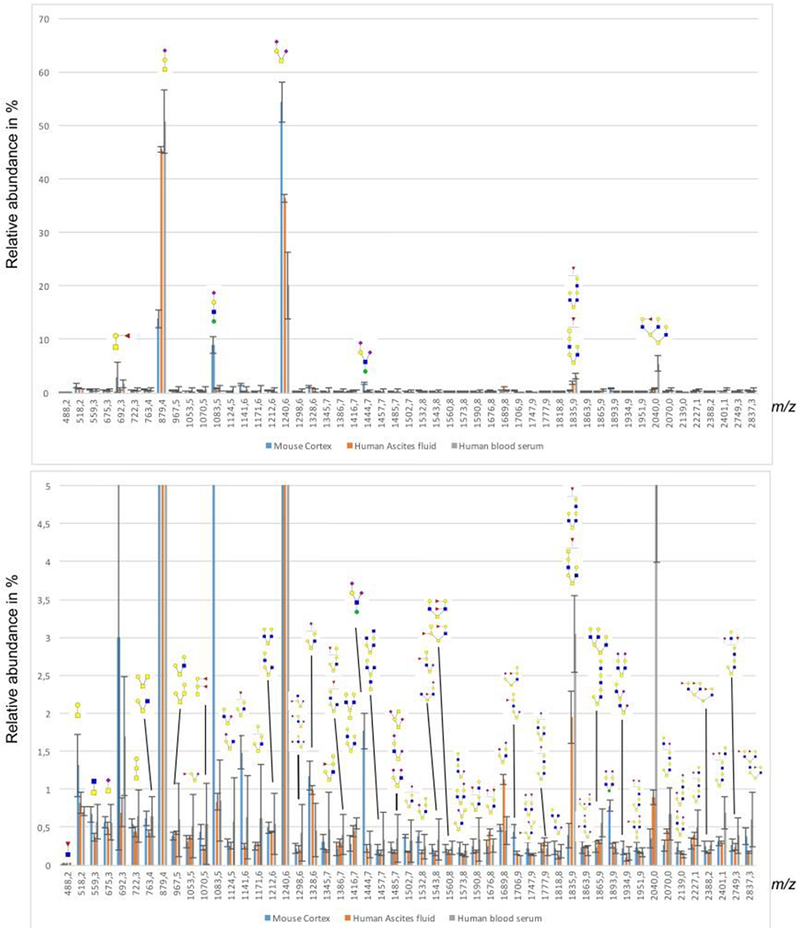
Profiles of detected permethylated O-glycans detected by MALDI-MS as sodiated ions in the positive-ion mode. Y-axis displays percentage of total glycans. Top graph shows the total distribution of glycans, while the bottom graph is zoomed from 5 % relative intensity.
Only a few O-glycans, out of more than fifty detected, reached a relative abundance level above 1%, with the majority thus being detected at very low levels. Two glycans were detected above 1% in all three samples; namely, tentatively identified sialyl-T (NeuAcα2-3Galβ1-3GalNAc, m/z 879.4)) and di-sialyl-T (NeuAcα2-3Galβ1-3(NeuAcα2-6-)GalNAc, m/z 1240.6) (Figure 3).
The different samples show an inverse relationship between sialyl-T and di-sialyl-T, probably due to differences in the activity of ST6GalNAc III and IV sialyltransferases, which use the sialyl-T as a substrate to add a sialic acid to the GalNAc in a α2-6 linkage[47], thus depleting the substrate sialyl-T. A number of core-mannosylated O-glycans could be detected in relatively high abundance in the mouse cortex, including tentative core M1-based Sia-Hex-HexNAc-Hex (m/z 1083.5) and Sia-Hex-(Sia)HexNAc-Hex (m/z 1444.7) as well as the core M2-based Sia-Hex-HexNAc(Sia-Hex-HexNAc)-Hex (m/z 1893.9). The sequences of the mannosylated structures were further confirmed through LC-MS/MS, with one example shown for the pentasaccharide Sia-Hex-(Sia)HexNAc-Hex, as will be shown below in the LC-MS analysis section. The cortex sample also contained a few more glycans above the 1% mark, including tentative core 1 (m/z 518.2) and fucosylated core 1 (m/z 692.3) and fucosylated (m/z 1141.6) and sialylated/extended core 2 (m/z 1328.6) structures. Since the O-glycans are released and derivatized after a microfiltration step that removes released N-glycans, there is a potential risk that some proteins could be lost if the cut-off is not appropriately chosen. In this communication, we used 10kDa MWCO filters which should retain glycoproteins in serum (most glycoproteins are larger than this, [48]) while retaining acceptable filtration speed compared to other procedures.
The MALDI-MS glycan profile of glycosphingolipid-derived permethylated glycans are shown in Figure 4. In total, twelve sodiated ions from glycosphingolipid-derived glycans were detected and their tentative structures are shown in Figure 4. The ascites fluid and blood serum samples had qualitatively and quantitatively similar glycan profiles, with the main components tentatively assigned as GM3 (m/z 854.4), globotri- (m/z 697.3), globotetra- (m/z 924.4), GD3 (m/z 1215.5), GM1 (m/z 1303.6) and H5 structures. The glycosphingolipids originated from mouse cortex, on the other hand, show a profile with more complex gangliosides, such as the major peaks tentatively annotated as GM1 (m/z 1303.6) and GD1 (m/z 1664.7). Additionally, a few minor, highly sialylated glycans such as GD2 (m/z 1460.6), GT3 (m/z 1576.7), GT1 (m/z 2026.0) and HexNAc-elongated GD1 (m/z 1909.9) are also evident (Figure 4). The mouse cortex samples exhibit larger standard deviations, which is expected, as these samples originated from three different animals when compared to the ascites fluid and blood serum samples that were just sample replicates. The globoside peaks for the spectra are most likely underrepresented, since the recombinant endoglycoceramidase II used to cleave the glycan from the ceramide has been shown to have low efficiency for globoside structures[49, 50]. A possible solution to this problem would be to use the ceramide glycanase from Macrobdella decora, which has a more universal activity[51]. However, this enzyme is no longer commercially available. Another possible release strategy would be ozonolysis [52], however, that requires the availability of an ozone generator.
Figure 4.
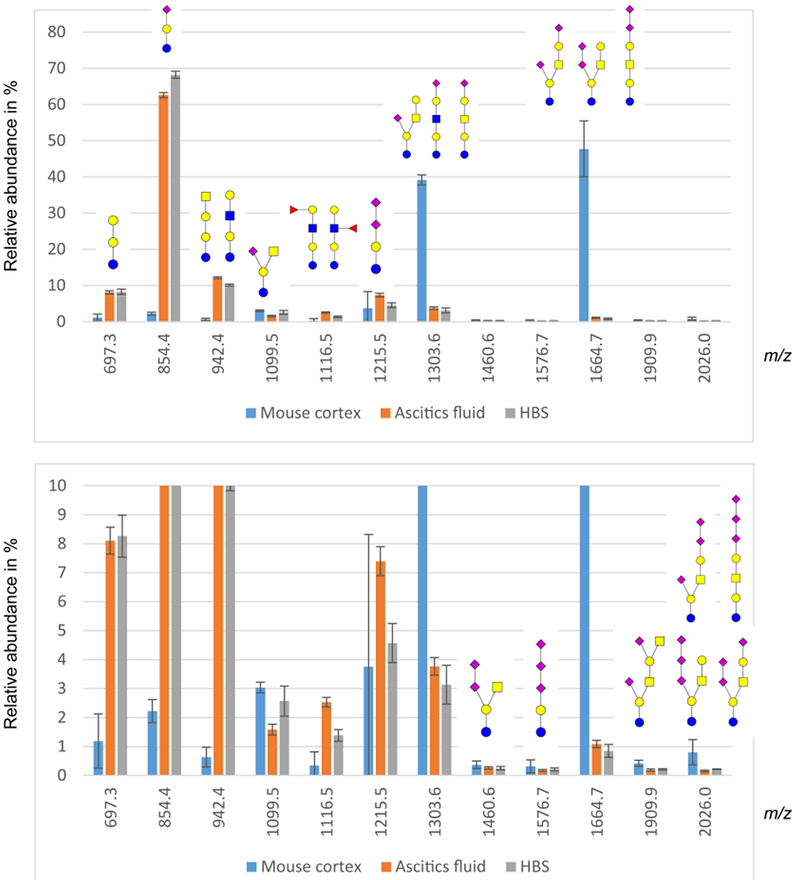
Profiles of glycosphingolipid oligosaccharides detected by MALDI-MS as sodiated ions in the positive-ion mode. Y-axis displays percentage of total glycans. Top graph shows the total distribution of glycans, while the bottom chart is zoomed from 10 % relative intensity.
While MALDI-MS combines here conveniently with permethylation in profiling different glycan groups at high sensitivity, this approach falls short of identifying isomeric structures, unless additional derivatization is performed as was, for example, previously shown for the case of sialylated N-glycans [24, 53, 54]. Unfortunately, this step does not fit our overall analytical scheme and additional approaches must be developed. When using LC-ESI-MS techniques, the isomerism problem can sometimes be addressed through the use of appropriately chosen (selective) LC-columns that may resolve isomeric species. However, once the glycans of interest are permethylated, the choice of selectivity-based separations becomes limited, with the possible exception of graphitized carbon columns [30].
As a minor extension of this work, we tested the use of graphitized carbon LC columns on the aliquots of extracts resulting from the reported analytical scheme, with the main emphasis on demonstrating isomeric distinction. Permethylated N-glycan isomeric separations at high temperatures were recently reported [55] using graphitized carbon columns. Here, our LC-MS analyses of the permethylated glycosphingolipid-originated glycans and O-glycans (Figures 5 and 7) were performed in order to show the major glycans in more detail (e. g., possible presence of isomers). It was noticed that during both LC-MS and MALDI-MS the acetone precipitation improved the quality of the O-glycan spectra, containing less interfering impurities. The separated major O-glycan components in mouse cortex were detected as protonated ions m/z 496, 712, 857, 1061 and 1218. Similar structures could be found for the ascites fluid and blood serum, however, the m/z 1061 and 712 ions representing O-mannose-based structures were not detected (Figure 5). The peaks were tentatively assigned by MS2 to the following structures: T-antigen (m/z 496), sialyl-T (m/z 857), disialyl-T (m/z 1218), “classical tetrasaccharide” Siaα2,3Galβ1,4GlcNAcβ1,2Manα (m/z 1061) and a pentasaccharide representing the “classical tetrasaccharide” with an additional sialic acid attached to the GlcNAc (m/z 712). The latter structure is unusual and we thus provide the annotated MS/MS spectrum in Figure 6. While the MS/MS fragments indicate that the sialic acid is linked to GlcNAc, we could not determine its linkage type. A pentasaccharide with the same sequence was previously reported on the murine brain-specific protein known as phosphacan[56].
Figure 5.
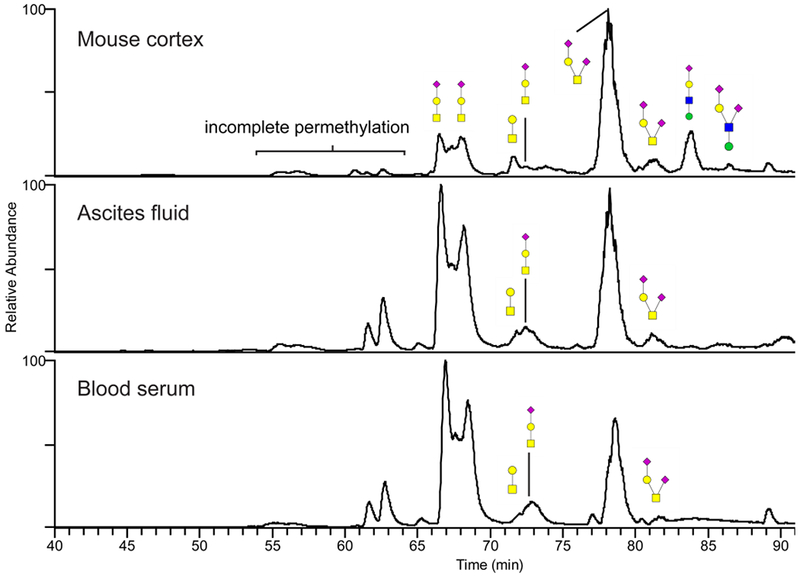
Annotated base-peak chromatograms of permethylated O-glycans released from mouse brain, human ascites fluid and serum. The glycans were separated on a porous graphitized carbon column maintained at 75°C prior to their MS detection as protonated ions in the positive-ion mode. The annotation uses the color cartoon nomenclature according to Varki et al. [57]. A group of incompletely permethylated glycans migrated faster through the column and were observed as a set of minor peaks preceding the main components. These minor peaks are marked in the chromatograms.
Figure 7.
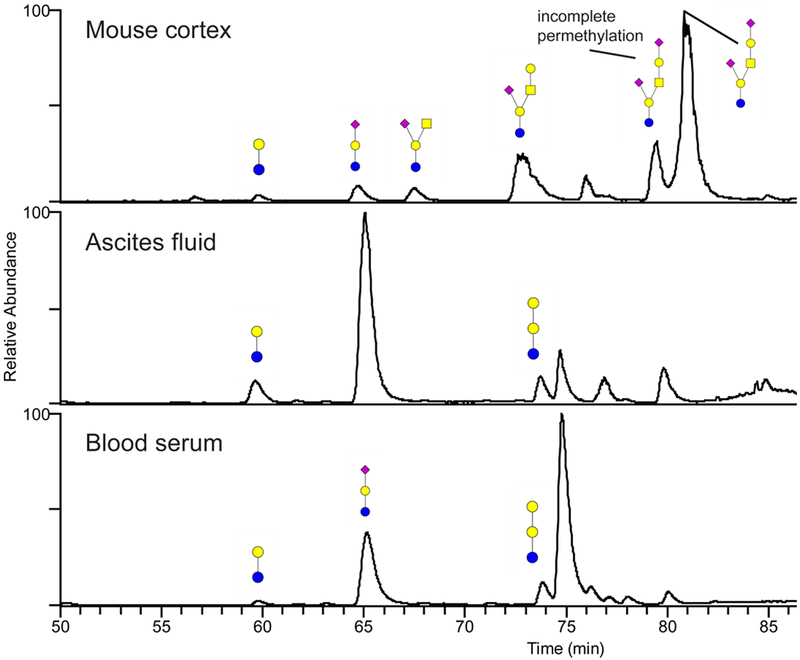
Annotated base-peak chromatograms from the permethylated glycosphingolipid oligosaccharides released from mouse brain, human ascites fluid and serum. The glycans were separated on a porous graphitized carbon column maintained at 75°C prior to MS detection as protonated ions in the positive-ion mode. The annotation uses color cartoon nomenclature according to Varki et al. [57]. Some incompletely permethylated glycans were observed preceding the major peaks in the chromatogram, most markedly for the two peaks around 80 minutes in mouse cortex. The incompletely permethylated glycans were verified with MS/MS.
Figure 6.
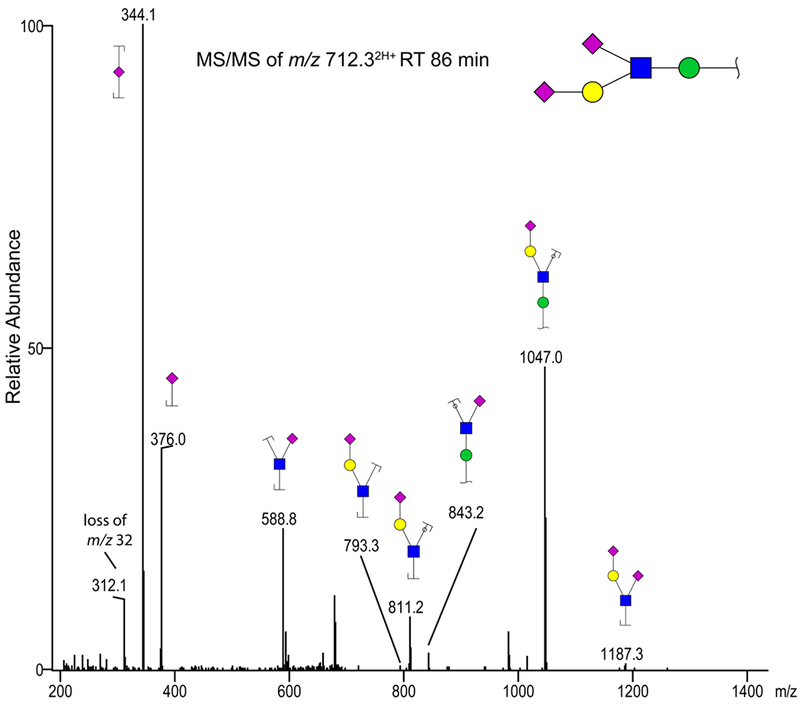
MS/MS spectrum of the mouse brain-derived O-glycan ion m/z 712.32H+ due to a peak eluting at 86 min. We suggest that this ion represents a permethylated O-glycan with the sequence of Sia-Hex(Sia-HexNAc)-Hex. The annotation uses color cartoon nomenclature according to Varki et al. [57]; a loss of m/z 32 corresponding to an O-methyl group.
In this work, we also observe that the permethylated sialyl-T structure was chromatographically separated into several peaks which were indistinguishable from each other when based on their MS/MS fragments (see Figure 5 for chromatograms, MS/MS data not shown). The different peaks may represent reducing-end α and β anomer glycans, which can be separated in a native form on such columns
The permethylated glycosphingolipid oligosaccharides were also analyzed by LC-MS, with their structures tentatively annotated through MS2. In the mouse cortex, the structures of lactosylceramide (m/z 472), GM3 (m/z 831), GM2 (m/z 1077), GM1 (m/z 1281) and GD1a (m/z 822) were detected. As for the ascites fluid and blood serum, only lactosylceramide (m/z 472), GM3 (m/z 831) and globotriaosylceramide (m/z 674) were identified (Figure 7).
Overall, the LC-MS profiles appear less abundant in comparison to the MALDI-MS data. We do not readily have explanation for this, except that some polymeric contaminants (inadvertently introduced into the samples through the extraction steps [58] might increase the baseline noise.
While the analysis of permethylated glycans yields a good signal in MS, a downside with permethylation is that the sulfated structures become suppressed [59]. This could potentially be avoided by using other types of derivatization or simply analyzing the glycans in their native or reduced form. The O-glycans could then be released by the more commonly used β-elimination[60, 61], but this is likely to necessitate much larger initial samples.
Conclusions
There is still a considerable need to arrive at precise glycan structures for the benefits of their functional understanding. Whereas the analyses of N-glycans from the glycoproteins contained in physiological fluids and tissues have been enhanced significantly during the last decades due to MS advances, the same does not hold for the methodologically more demanding O-glycans and glycolipids. In the analyses of complex biological mixtures, MALDI-MS of permethylated glycans permits a fairly comprehensive profiling of structures at high sensitivity and respectable dynamic concentration range. As shown in this communication, a sequential isolation of N- and O-glycans, together with the glycans originated from glycosphingolipids, can now be performed from a single biological sample. The described protocol basically preserves performance and analytical advantages of MALDI/MS-based glycan profiling when performed separately for three glycan groups. The results of high-sensitivity profiling typically yield suggested cartoon structures matching their expected m/z ratios. However, additional avenues toward a more complete in-depth structural elucidation must still be developed. These will undoubtedly need to perform additional preconcentration steps prior to MS investigations. We have shown here selected examples of isomeric resolution of permethylated glycans in the LC-MS runs using graphitized carbon columns. Overall, our analytical scheme appears suitable for profiling studies seeking a fuller coverage of different glycomes.
Supplementary Material
Acknowledgments
This work was supported by the grants from the National Institute of General Medical Sciences, U.S. Department of Health (NIH R21GM118340 and NIH R01GM106084). We thank Dr. Sachiko Koyama for providing the mouse tissue samples and Dr. Daniela Matei for providing the ascites fluids, and Dr. Helana Soini for help in preparing the manuscript. Reporting on experimental details has been prepared in accordance with MIRAGE (The Minimum Information Required for a Glycomics Experiment) sample preparation (dx.doi.org/10.3762/mirage.1) and MS guidelines (dx.doi.org/10.3762/mirage.2).
References
- 1.Apweiler R, Hermjakob H, Sharon N: On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 (1999) [DOI] [PubMed] [Google Scholar]
- 2.Schachter H, Freeze HH: Glycosylation diseases: Quo vadis? Biochim. Biophys. Acta 1792, 925–930 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hart GW, Copeland RJ: Glycomics hits the big time. Cell. 143, 672–676 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gray GM, Yardley HJ: Lipid compositions of cells isolated from pig, human, and rat epidermis. J. Lipid. Res 16, 434–440 (1975) [PubMed] [Google Scholar]
- 5.Barone A, Säljö K, Benktander J, Blomqvist M, Månsson J-E, Johansson BR, Mölne J, Aspegren A, Björquist P, Breimer ME, Teneberg S: Sialyl-lactotetra, a novel cell surface marker of undifferentiated human pluripotent stem cells. J. Biol. Chem 289, 18846–18859 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wright AJ, Andrews PW: Surface marker antigens in the characterization of human embryonic stem cells. Stem Cell Res 3, 3–11 (2009) [DOI] [PubMed] [Google Scholar]
- 7.Natunen S, Satomaa T, Pitkanen V, Salo H, Mikkola M, Natunen J, Otonkoski T, Valmu L: The binding specificity of the marker antibodies Tra-1-60 and Tra-1-81 reveals a novel pluripotency-associated type 1 lactosamine epitope. Glycobiology. 21, 1125–1130 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snyder CM, Alley WR Jr., Campos MI, Svoboda M, Goetz JA, Vasseur JA, Jacobson SC, Novotny MV: Complementary glycomic analyses of sera derived from colorectal cancer patients by MALDI-TOF-MS and microchip electrophoresis. Anal. Chem 88, 9597–9605 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goto Y, Uematsu S, Kiyono H: Epithelial glycosylation in gut homeostasis and inflammation. Nat. Immunol 17, 1244–1251 (2016) [DOI] [PubMed] [Google Scholar]
- 10.Pinho SS, Reis CA: Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer. 15, 540–555 (2015) [DOI] [PubMed] [Google Scholar]
- 11.Alley WR Jr., Madera M, Mechref Y, Novotny MV: Chip-based reversed-phase liquid chromatography-mass spectrometry of permethylated N-linked glycans: a potential methodology for cancer-biomarker discovery. Anal. Chem 82, 5095–5106 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mann BF, Goetz JA, House MG, Schmidt CM, Novotny MV: Glycomic and proteomic profiling of pancreatic cyst fluids identifies hyperfucosylated lactosamines on the N-linked glycans of overexpressed glycoproteins. Mol. Cell. Proteomics. 11, M111.015792 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaunitz S, Nagy G, Pohl NL, Novotny MV: Recent advances in the analysis of complex glycoproteins. Anal. Chem 89, 389–413 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox JT, Kronewitter SR, Shukla AK, Moore RJ, Smith RD, Tang K: High sensitivity combined with extended structural coverage of labile compounds via nanoelectrospray ionization at subambient pressures. Anal. Chem 86, 9504–9511 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu X, Jiang Y, Chen Y, Huang Y, Costello CE, Lin C: Detailed glycan structural characterization by electronic excitation dissociation. Anal. Chem 85, 10017–10021 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma C, Zhang Q, Qu J, Zhao X, Li X, Liu Y, Wang PG: A precise approach in large scale core-fucosylated glycoprotein identification with low- and high-normalized collision energy. J. Proteomics. 114, 61–70 (2015) [DOI] [PubMed] [Google Scholar]
- 17.Alley WR Jr., Mann BF, Novotny MV: High-sensitivity analytical approaches for the structural characterization of glycoproteins. Chem. Rev 113, 2668–2732 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Novotny MV, Alley WR Jr.: Recent trends in analytical and structural glycobiology. Curr. Opin. Chem. Biol 17, 832–840 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang S, Zhang H: Glycomic analysis of glycans released from glycoproteins using chemical immobilization and mass spectrometry. Curr. Protoc. Chem. Biol 6, 191–208 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bladergroen MR, Reiding KR, Hipgrave Ederveen AL, Vreeker GC, Clerc F, Holst S, Bondt A, Wuhrer M, van der Burgt YE: Automation of high-throughput mass spectrometry-based plasma N-glycome analysis with linkage-specific sialic acid esterification. J. Proteome Res 14, 4080–4086 (2015) [DOI] [PubMed] [Google Scholar]
- 21.Zhou H, Morley S, Kostel S, Freeman MR, Joshi V, Brewster D, Lee RS: Universal solid-phase reversible sample-prep for concurrent proteome and N-glycome characterization. J. Proteome Res 15, 891–899 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun S, Shah P, Eshghi ST, Yang W, Trikannad N, Yang S, Chen L, Aiyetan P, Hoti N, Zhang Z, Chan DW, Zhang H: Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat. Biotechnol 34, 84–88 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Snyder CM, Zhou X, Karty JA, Fonslow BR, Novotny MV, Jacobson SC: Capillary electrophoresis-mass spectrometry for direct structural identification of serum N-glycans. J. Chromatogr. A. 1523, 127–139 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alley WR Jr., Novotny MV: Glycomic analysis of sialic acid linkages in glycans derived from blood serum glycoproteins. J. Proteome Res 9, 3062–3072 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujitani N, Furukawa J.-i., Araki K, Fujioka T, Takegawa Y, Piao J, Nishioka T, Tamura T, Nikaido T, Ito M, Nakamura Y, Shinohara Y: Total cellular glycomics allows characterizing cells and streamlining the discovery process for cellular biomarkers. Proc. Natl. Acad. Sci. U.S.A 110, 2105–2110 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babu P, North SJ, Jang-Lee J, Chalabi S, Mackerness K, Stowell SR, Cummings RD, Rankin S, Dell A, Haslam SM: Structural characterisation of neutrophil glycans by ultra sensitive mass spectrometric glycomics methodology. Glycoconjugate J 26, 975 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qin H, Cheng K, Zhu J, Mao J, Wang F, Dong M, Chen R, Guo Z, Liang X, Ye M, Zou H: Proteomics analysis of O-GalNAc glycosylation in human serum by an integrated strategy. Anal. Chem 89, 1469–1476 (2017) [DOI] [PubMed] [Google Scholar]
- 28.Karlsson H, Larsson JMH, Thomsson KA, Härd I, Bäckström M, Hansson GC: High-throughput and high-sensitivity nano-LC/MS and MS/MS for O-glycan profiling. In: Packer NH, Karlsson NG (eds.). Glycomics: Methods and Protocols, Humana Press, Totowa, NJ, (2009) [DOI] [PubMed] [Google Scholar]
- 29.Kang P, Mechref Y, Kyselova Z, Goetz JA, Novotny MV: Comparative glycomic mapping through quantitative permethylation and stable-isotope labeling. Anal. Chem 79, 6064–6073 (2007) [DOI] [PubMed] [Google Scholar]
- 30.Costello CE, Contado-Miller JM, Cipollo JF: A glycomics platform for the analysis of permethylated oligosaccharide alditols. J. Am. Soc. Mass Spectrom 18, 1799–1812 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gizaw ST, Ohashi T, Tanaka M, Hinou H, Nishimura S: Glycoblotting method allows for rapid and efficient glycome profiling of human Alzheimer’s disease brain, serum and cerebrospinal fluid towards potential biomarker discovery. Biochim. Biophys. Acta. 1860, 1716–1727 (2016) [DOI] [PubMed] [Google Scholar]
- 32.Koles K, van Berkel PH, Pieper FR, Nuijens JH, Mannesse ML, Vliegenthart JF, Kamerling JP: N- and O-glycans of recombinant human C1 inhibitor expressed in the milk of transgenic rabbits. Glycobiology. 14, 51–64 (2004) [DOI] [PubMed] [Google Scholar]
- 33.Garcia AD, Chavez JL, Mechref Y: Rapid and sensitive LC-ESI-MS of gangliosides. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 947-948, 1–7 (2014) [DOI] [PubMed] [Google Scholar]
- 34.Karlsson H, Halim A, Teneberg S: Differentiation of glycosphingolipid-derived glycan structural isomers by liquid chromatography/mass spectrometry. Glycobiology. 20, 1103–1116 (2010) [DOI] [PubMed] [Google Scholar]
- 35.Zou G, Benktander JD, Gizaw ST, Gaunitz S, Novotny MV: Comprehensive analytical approach toward glycomic characterization and profiling in urinary exosomes. Anal. Chem 89, 5364–5372 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goetz JA, Novotny MV, Mechref Y: Enzymatic/chemical release of O-glycans allowing MS analysis at high sensitivity. Anal. Chem 81, 9546–9552 (2009) [DOI] [PubMed] [Google Scholar]
- 37.Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM: GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans. J. Proteome Res 7, 1650–1659 (2008) [DOI] [PubMed] [Google Scholar]
- 38.Lauc G, Heffer-Lauc M: Shedding and uptake of gangliosides and glycosylphosphatidylinositol-anchored proteins. Biochim. Biophys. Acta. 1760, 584–602 (2006) [DOI] [PubMed] [Google Scholar]
- 39.Biskup K, Braicu EI, Sehouli J, Tauber R, Blanchard V: The ascites N-glycome of epithelial ovarian cancer patients. J. Proteomics. 157, 33–39 (2017) [DOI] [PubMed] [Google Scholar]
- 40.Alley WR, Vasseur JA, Goetz JA, Svoboda M, Mann BF, Matei DE, Menning N, Hussein A, Mechref Y, Novotny MV: N-linked glycan structures and their expressions change in the blood sera of ovarian cancer patients. J. Proteome Res 11, 2282–2300 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishii A, Ikeda T, Hitoshi S, Fujimoto I, Torii T, Sakuma K, Nakakita S, Hase S, Ikenaka K: Developmental changes in the expression of glycogenes and the content of N-glycans in the mouse cerebral cortex. Glycobiology. 17, 261–276 (2007) [DOI] [PubMed] [Google Scholar]
- 42.Aldredge D, An HJ, Tang N, Waddell K, Lebrilla CB: Annotation of a serum N-glycan library for rapid identification of structures. J. Proteome Res 11, 1958–1968 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thaysen-Andersen M, Venkatakrishnan V, Loke I, Laurini C, Diestel S, Parker BL, Packer NH: Human neutrophils secrete bioactive paucimannosidic proteins from azurophilic granules into pathogen-infected sputum. J. Biol. Chem 290, 8789–8802 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sethi MK, Kim H, Park CK, Baker MS, Paik YK, Packer NH, Hancock WS, Fanayan S, Thaysen-Andersen M: In-depth N-glycome profiling of paired colorectal cancer and non-tumorigenic tissues reveals cancer-, stage- and EGFR-specific protein N-glycosylation. Glycobiology. 25, 1064–1078 (2015) [DOI] [PubMed] [Google Scholar]
- 45.Balog CI, Stavenhagen K, Fung WL, Koeleman CA, McDonnell LA, Verhoeven A, Mesker WE, Tollenaar RA, Deelder AM, Wuhrer M: N-glycosylation of colorectal cancer tissues: a liquid chromatography and mass spectrometry-based investigation. Mol. Cell. Proteomics. 11, 571–585 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaprio T, Satomaa T, Heiskanen A, Hokke CH, Deelder AM, Mustonen H, Hagstrom J, Carpen O, Saarinen J, Haglund C: N-glycomic profiling as a tool to separate rectal adenomas from carcinomas. Mol. Cell. Proteomics. 14, 277–288 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takashima S, Kurosawa N, Tachida Y, Inoue M, Tsuji S: Comparative analysis of the genomic structures and promoter activities of mouse Siaalpha2, 3 Galbeta1, 3GalNAc GalNAcalpha 2, 6-Sialyltransferase genes (ST6GalNAc III and IV): characterization of their Spl binding sites. J. Biochem 127, 399–409 (2000) [DOI] [PubMed] [Google Scholar]
- 48.Clerc F, Reiding KR, Jansen BC, Kammeijer GS, Bondt A, Wuhrer M: Human plasma protein N-glycosylation. Glycoconjugate J 33, 309–343 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ito M, Yamagata T: Purification and characterization of glycosphingolipid-specific endoglycosidases (endoglycoceramidases) from a mutant strain of Rhodococcus sp. Evidence for three molecular species of endoglycoceramidase with different specificities. J. Biol. Chem 264, 9510–9519 (1989) [PubMed] [Google Scholar]
- 50.Li YT, Chou CW, Li SC, Kobayashi U, Ishibashi YH, Ito M: Preparation of homogenous oligosaccharide chains from glycosphingolipids. Glycoconjugate J 26, 929–933 (2009) [DOI] [PubMed] [Google Scholar]
- 51.Zhou B, Li SC, Laine RA, Huang RT, Li YT: Isolation and characterization of ceramide glycanase from the leech, Macrobdella decora. J. Biol. Chem 264, 12272–12277 (1989) [PubMed] [Google Scholar]
- 52.Nagahori N, Yamashita T, Amano M, Nishimura S: Effect of ganglioside GM3 synthase gene knockout on the glycoprotein N-glycan profile of mouse embryonic fibroblast. Chembiochem 14, 73–82 (2013) [DOI] [PubMed] [Google Scholar]
- 53.Holst S, Heijs B, de Haan N, van Zeijl RJ, Briaire-de Bruijn IH, van Pelt GW, Mehta AS, Angel PM, Mesker WE, Tollenaar RA, Drake RR, Bovee JV, McDonnell LA, Wuhrer M: Linkage-specific in situ sialic acid derivatization for N-glycan mass spectrometry imaging of formalin-fixed paraffin-embedded tissues. Anal. Chem 88, 5904–5913 (2016) [DOI] [PubMed] [Google Scholar]
- 54.Nishikaze T, Tsumoto H, Sekiya S, Iwamoto S, Miura Y, Tanaka K: Differentiation of sialyl linkage isomers by one-pot sialic acid derivatization for mass spectrometry-based glycan profiling. Anal. Chem 89, 2353–2360 (2017) [DOI] [PubMed] [Google Scholar]
- 55.Zhou S, Huang Y, Dong X, Peng W, Veillon L, Kitagawa DAS, Aquino AJA, Mechref Y: Isomeric separation of permethylated glycans by porousg graphitic carbon (PGC)-LC-MS/MS at high temperatures. Anal. Chem 89, 6590–6597 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dwyer CA, Katoh T, Tiemeyer M, Matthews RT: Neurons and glia modify receptor protein-tyrosine phosphatase zeta (RPTPzeta)/phosphacan with cell-specific O-mannosyl glycans in the developing brain. J. Biol. Chem 290, 10256–10273 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Marth JD, Bertozzi CR, Hart GW, Etzler ME: Symbol nomenclature for glycan representation. Proteomics. 9, 5398–5399 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rehulka P, Zahradnikova M, Rehulkova H, Dvorakova P, Nenutil R, Valik D, Vojtesek B, Hernychova L, Novotny MV: Microgradient separation technique for purification and fractionation of permethylated N-glycans before mass spectrometric analyses. J. Sep. Sci (2018) [DOI] [PubMed] [Google Scholar]
- 59.Lei M, Mechref Y, Novotny MV: Structural analysis of sulfated glycans by sequential double-permethylation using methyl iodide and deuteromethyl iodide. J. Am. Soc. Mass Spectrom 20, 1660–1671 [DOI] [PubMed] [Google Scholar]
- 60.Cooper CA, Packer NH, Redmond JW: The elimination of O-linked glycans from glycoproteins under non-reducing conditions. Glycoconjugate J 11, 163–167 (1994) [DOI] [PubMed] [Google Scholar]
- 61.Iyer RN, Carlson DM: Alkaline borohydride degradation of blood group H substance. Arch. Biochem. Biophys. 142, 101–105 (1971) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.