

Abstract
Understanding the functional relevance of G protein-coupled receptor (GPCR) homodimerization has been limited by the insufficient tools to assess asymmetric signaling occurring within dimers comprised of the same receptor type. We present unmatched bivalent ligands (UmBLs) to study the asymmetric function of melanocortin homodimers. UmBLs contain one agonist and one antagonist pharmacophore designed to target a melanocortin homodimer such that one receptor is occupied by an agonist and the other receptor by an antagonist pharmacophore. First-in-class biased UmBLs (BUmBLs) targeting the human melanocortin-4 receptor (hMC4R) were discovered. The BUmBLs displayed biased agonism by potently stimulating cAMP signaling (EC50 ~ 2 to 6 nM), but minimally activating the β-arrestin recruitment pathway (≤55% maximum signal at 10 µM). To our knowledge, we report the first single-compound strategy to pharmacologically target melanocortin receptor allosteric signaling that occurs between homodimers that can be applied straightforwardly in vitro and in vivo to other GPCR systems.
Table of Content Graphic
Introduction
G protein-coupled receptors (GPCRs) are highly sought after drug targets in the pharmaceutical industry with approximately 30–40% of drugs targeting them (~34% of all US FDA approved drugs as of 2017).1–3 Classically, medicinal chemists targeted GPCRs as monomeric units; however increasing evidence has supported GPCRs form dimers with themselves (homodimers) and with other GPCRs (heterodimers).4–5 Targeting GPCR homodimers’ and heterodimers’ distinct and exploitable functions may yield a revolution in GPCR targeting therapeutics. Although ligands targeting heterodimers have shown much promise in both in vitro and in vivo preclinical studies,6–11 there has been limited development of ligands targeting the allosterism that can occur within homodimers.
Pharmacologically targeting homodimers possess a unique conundrum: How do you target and detect a homodimer when the two receptors comprising it are structurally similar, usually respond to the same ligands, and appear to have the same propensity to signal in standard cell culture assays? Various groups have devised clever strategies around these problems to demonstrate the functional consequences of asymmetric homodimers.12–30 Some groups focus on demonstrating subtle changes in pharmacology suggesting allosteric interaction within homodimers utilizing strategically designed in vitro experiments, and other groups exploited receptor mutation strategies in order to differentiate between the two protomers making up the dimer.12–28 For example, Han and coworkers in 2009 combined different receptor-G protein fusions and various mutant receptors to demonstrate allosteric modulation within a dopamine homodimer.12 They reported that the D2 dopamine receptor homodimers are maximally activated upon a single agonist binding a single protomer in the dimer pair. When a second agonist binds the second protomer, it blunts the signal. If an inverse agonist binds the second protomer, it enhances the signal beyond the single agonist alone.12
In a different strategy, Teitler and coworkers developed pseudo-irreversible inactivators and reactivators that can be used to block only one of the protomers within the dimer pair in order to demonstrate the crosstalk within wild type serotonin homodimers.14 This approach can and has been used to demonstrate the allosteric regulation within homodimers in native tissue samples. Application of this technique in vivo would be difficult given the multiple dosing regimen necessary and, therefore, would have very limited therapeutic applications.14 Although these reports provide critical proof of the relevancy and functional significance of asymmetric signaling homodimers, the techniques employed are limited by their use of receptor mutations or subtle pharmacological differences that make adaption of the approaches to in vivo applications difficult and therapeutic applications inexecutable. Ideally, a medicinal chemistry approach is needed to target and exploit allosteric communication between homodimers with a single chemical entity that could be used to examine the in vivo effects of asymmetric GPCR homodimers to study their potential as therapeutic targets.
One approach of pharmacologically targeting GPCR dimers is utilizing bivalent ligands. This approach was pioneered by Portoghese and coworkers targeting the opioid receptors, and was also reported early on by Conn and coworkers.31–34 Heterobivalent ligands featuring pharmacophores for two different receptor types have been utilized to exploit allosteric interactions within heterodimers to develop ligands with novel pharmacological profiles, tissue selectivity, and different functional effects.6–9, 35–36 However, to our knowledge, no one has exploited the allosteric communication that may occur between homodimers with bivalent ligands to produce novel pharmacologies. In the current report, we exploit the allosteric communication between human melancorotin-4 receptor (hMC4R) homodimers with melanocortin unmatched bivalent ligands (MUmBLs) to produce biased agonists. Unmatched bivalent ligands (UmBLs) have an agonist pharmacophore on one side of the bivalent ligand connected to an antagonist pharmacophore through an inert linker. We use the term UmBLs to separate this class of ligands from heterobivalent ligands that also have different pharmacophores on each side of the bivalent ligand, but are usually used to target different receptor types. This UmBL design has been proposed and reported previously, however, to our knowledge it has not been used to successfully exploit asymmetric signaling of GPCR homodimers.37–39 An attempt at the UmBL strategy was reported by Kühhorn and coworkers. They reported UmBLs targeting the dopamine D2 receptors that had some agonist efficacy (13% maximal) and did not induce receptor internalization suggesting biased agonism may be possible with this design strategy, although they conclude “ligand bias” was not accomplished.37
We have previously described both agonist and antagonist homobivalent ligands targeting the melanocortin receptor system.40–41 Ligands targeting the melanocortin system have been implicated as potential therapeutics or used as pharmacological probes for a wide range of disease states including cancer,42–46 skin pigmentation disorders,47 social disorders,48–49 sexual function disorders,50–52 Alzheimer’s disease,53–54 cachexia,55–59 and obesity.40, 60–62 All five melanocortin receptor subtypes (MC1-5R) signal through the Gαs protein signaling pathway. In this pathway, an agonist binding to the GPCR activates cAMP signal transduction pathways and also results in the recruitment of β-arrestin.63 The melanocortin-3 receptor (MC3R) and melanocortin-4 receptor (MC4R) in particular have been elucidated to play roles in energy homeostasis.60–62, 64–65 Ligands for the MC4R were under intense clinical development to treat obesity and related metabolic disorders; however these ligands were reported to have undesirable effects such as increasing blood pressure66 or inducing male erections.67 It is hypothesized that ligands that target melanocortin homodimers may have unique effects from the current monovalent approaches, and may, therefore circumvent some side effects.
We have previously shown that an agonist homobivalent ligand produces a distinct in vivo pharmacological profile compared to its monovalent counterpart suggesting that targeting putative melanocortin dimers may have physiological relevancy.41 Furthermore, biased ligands would be valuable pharmacological probes to elucidate which signaling pathway is responsible for the various melanocortin dependent effects (i.e. lowered food intake vs increased blood pressure). In the current study, we report the design and synthesis of MUmBLs to target asymmetrically signaling melanocortin homodimers. The ligands discovered with this underappreciated medicinal chemistry strategy had a biased agonist pharmacology not reported in the literature to date. They potently activated the cAMP signaling pathway with minimal activation of the β-arrestin recruitment pathway. The current study provides novel molecular probes for the melanocortin receptors as well as an in vitro proof-of-concept of using the biased unmatched bivalent ligand (BUmBL) design strategy to target asymmetrically signaling homodimers. This innovative design strategy could be applied to various GPCR systems for the creation of biased ligands.
Results
Design and Synthesis of MUmBLs
Homobivalent ligands targeting melanocortin receptors have previously resulted in increased binding affinity (~14 to 25-fold) consistent with a synergistic binding mode arising from receptor dimer binding.38, 40–41, 46, 68–75 In spite of increased binding affinities, we have observed much smaller fold increases in functional potencies of agonist homobivalent ligands when assessed via cAMP-based functional assays (3- to 5-fold).40 Brabez and coworkers have noted similar effects with agonist melanocortin bivalent ligands in which cAMP accumulation was not as dramatically increased with synergistic multivalent binding.46 One possibility for the incongruity between binding affinity increases and functional signaling increases with bivalent ligands may be due to allosterism between the melanocortin receptors within homodimers.40 Such asymmetric signaling within GPCR homodimers has previously been reported for a variety of systems including the vasopressin,28 dopamine,12 adenosine,26 metabotropic glutamate,19 and serotonin receptors.13
A new paradigm can be hypothesized in which one receptor within the melanocortin homodimer might be responsible for cAMP signaling and the other receptor might be responsible for signaling through a different cellular pathway (e.g. β-arrestin recruitment pathway) (Figure 1A–B). It would then follow that the increased binding would not necessarily result in an increase in functional agonist activity observed in a cAMP assay, since the effect of the second binding event is not detected by this cellular assay paradigm. Furthermore, there have been reports of asymmetry within melanocortin homodimers in both binding experiments and functional assays.27, 76–78 In order to exploit this possibility of asymmetric homodimers, we designed and synthesized MUmBLs that contained the known agonist melanocortin moiety His-DPhe-Arg-Trp on one side of the molecule,79–80 and the known MC3R and MC4R antagonist moiety His-DNal(2ʹ)-Arg-Trp81–82 on the other side of the molecule connected by three different previously validated linker systems (Table 1).38, 40, 70, 83
Figure 1.

Hypothesized interaction of ligands with asymmetrically signaling melanocortin homodimers. A) Monovalent agonist ligands (blue circle) could occupy both receptors and result in both cAMP signaling and β-arrestin recruitment. B) Agonist homobivalent ligands (blue circle connected with black linker) could result in similar functional cAMP assays as monomeric ligands in spite of increased binding affinities due to asymmetric signaling. C) The working paradigm herein in which biased unmatched bivalent ligands (BUmBLs) containing an agonist pharmacophore (blue circle) and antagonist pharmacophore (red octagon) are postulated to result in biased signaling by agonizing one signaling pathway while antagonizing the other pathway when bound to the asymmetrically signaling homodimer.
Table 1.
MUmBL functional data at the hMC4R
| Compound | Structure | cAMP Signaling | β-Arrestin Recruitment |
|
|---|---|---|---|---|
| EC50 (nM) Mean±SEM |
EC50 (nM) Mean±SEM |
|||
| Agonist Bivalent Ligands + Controls | NDP-MSH |
|
0.06±0.01 | 120±50 |
| MTII |
|
0.03±0.005 | 1.6±0.8 | |
|
| ||||
| CJL-1–14 |
|
1.8±0.2 | 290±150 | |
| CJL-5-35-4 |
|
0.67±0.03 | 55±16 | |
| CJL-1–116 |
|
2.0±0.3 | 140±70 | |
| CJL-5-35-1 |
|
5.5±0.7 | 1600±600 | |
| CJL-1–41 |
|
1.1±0.07 | 1500±500 | |
| CJL-1–31 |
|
0.95±0.08 | 1900±600 | |
| CJL-1–87 |
|
0.57±0.05 | 130±30 | |
| CJL-5–72 |
|
1.2±0.3 | 280±40 | |
|
| ||||
| Antagonist Bivalent Ligands + Controls | CJL-1–80 |
|
50% at 10 µM pA2=7.82±0.03 | 30% at 10 µM |
| CJL5-35-5 |
|
55% at 10 µM pA2=8.12±0.03 | 15% at 10 µM | |
| CJL-1–132 |
|
50% at 10 µM pA2=7.87±0.14 | 20% at 10 µM | |
| CJL-5-35-2 |
|
55% at 10 µM pA2=7.28±0.10 | 30% at 10 µM | |
| CJL-1–140 |
|
70% at 10 µM pA2=7.80±0.09 | 10% at 10 µM | |
|
| ||||
| Unmatched Bivalent Ligands | CJL-1–63 |
|
1.9±0.2 | 55% at 10 µM |
| CJL-5–58 |
|
1.9±0.5 | 45% at 10 µM | |
| CJL-1–124 |
|
4.7±1.0 | 20% at 10 µM | |
| CJL-5–74 |
|
5.9±3.2 | 25% at 10 µM | |
| CJL-1–14+CJL-1–80 |
|
1.9±0.2 | 40% at 10 µM | |
The cAMP signaling potency was determined by AlphaScreen™ assays. The β-arrestin recruitment potency was determined by PRESTO-Tango assays. The reported errors are the standard error of the mean (SEM) determined from at least three independent experiments. Changes less than 3-fold were considered to be within the inherent experimental assay error. The % symbol represents amount of maximal signal observed at 10 µM compared to control NDP-MSH maximal signal.
Because bivalent ligands presumably occupy both orthosteric sites in a receptor dimer due to synergistic binding, a MUmBL is postulated to occupy one receptor within a dimer pair with an agonist pharmacophore and the other receptor within the same dimer with an antagonist pharmacophore (Figure 1C). This assumes approximately equal binding affinities of the pharmacophores, and low enough concentrations of ligand so that intermolecular competition does not occur. The MUmBLs should favor a bivalent binding mode over a monovalent binding mode (due to increased binding affinity of bivalent ligands for dimers supported previously in competitive binding experiments.)40 This should shift the equilibrium towards occupation of one receptor with an agonist scaffold and the other receptor with an antagonist scaffold in each homodimer, but other binding states probably exist in equilibrium (Supplemental Fig. 1). It should also be noted, that we are currently assuming that only orthosteric binding is occurring, because allosteric binding has never been reported with either the His-DPhe-Arg-Trp or the His-DNal(2ʹ)-Arg-Trp scaffolds even though they both have been extensively studied as tetrapeptides and are present in standard control ligands NDP-MSH and SHU9119.80–81, 84–85 Both of these tetrapeptides are also direct competitors of 125I-NDP-MSH and 125I-AGRP(87–132) binding that further validates an orthosteric binding mode.40–41, 86
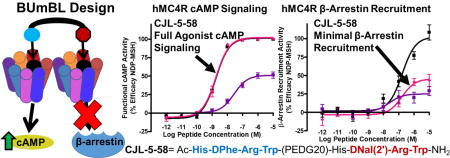
Ligands CJL-1–124, CJL-5–74, and CJL-1–63 feature the His-DPhe-Arg-Trp scaffold on the C-terminus and the His-DNal(2ʹ)-Arg-Trp scaffold on the N-terminus (Table 1). Since the molecules are not symmetric, the opposite composition of CJL-1–124 was designed and synthesized with the His-DNal(2ʹ)-Arg-Trp scaffold on the C-terminus and His-DPhe-Arg-Trp scaffold on the N-terminus in compound CJL-5–58. In particular, the construction of CJL-5–58 with the PEDG20 linker system was selected because this linker system was previously shown to be optimal in homobivalent ligands compared to the PEDG20-PEDG20 or Pro-Gly linker systems at the mMC4R.38, 40 Also, the PEDG20 linker was previously reported to be more metabolically stable than the Pro-Gly linker system and would allow an easier transition to in vivo applications.41 In vitro mouse serum stability assay results showed CJL-5–58 had similar serum stability (half-life = 6.9 h) as the previously reported CJL-1–87 confirming the PEDG20 linker selection (Supplemental Fig. 2).41
Bivalent melanocortin ligands featuring agonist and antagonist scaffolds were synthesized previously and were reported to possess increased binding affinity.38 These ligands provide evidence that MUmBLs can bind hMC4R homodimers to achieve synergistic binding, but no functional activity was evaluated.38, 87 Therefore, the functional significance of asymmetric homodimers could not be detected.
In the current study, all compounds were synthesized using standard Fmoc chemistry utilizing solid-phase synthesis methodology.88–90 A split resin approach for control ligands and bivalent ligands was performed as previously described.40 Possible degradation of the PEDG20 linker in a 3 h cleavage has previously been reported, and a maximum 1.5 h cleavage was suggested for PEDG20 containing compounds.40 Observing the words of caution from this previous report, all compounds in this study were synthesized with little difficulty. The ligands were purified to >95% by semi-preparative RP-HPLC and their mass was confirmed by ESI-MS (Supplemental Table 1). The synthesis of all compounds besides CJL-5-35-2, CJL-1–63, CJL-5–58, CJL-1–124, and CJL-5–74 was reported previously and were utilized currently as reported. The original characterization of these compounds and their pharmacology at the mouse melanocortin receptors, but not the human receptors, can be found in this previous report.40
MUmBLs are Biased Agonists at the hMC4R
Upon agonist stimulation, melanocortin receptors are known to signal through a Gαs-protein mediated signaling pathway that results in intracellular cAMP accumulation. Agonist stimulation of the melanocortin receptors also results in β-arrestin recruitment and receptor desensitization.63, 91–92 In order to evaluate the ligands’ efficacy and potency to stimulate cAMP signaling, ALPHAScreen™ cAMP Assay Technology was utilized to assess live HEK293 cells stably expressing human (h)MC4R.93–94 All ligands that contained the His-DPhe-Arg-Trp pharmacophore, including the MUmBLs, were single-digit or sub-nanomolar agonists in the cAMP assay (Table 1, Figure 2A). The most potent ligand (besides control ligand NDP-MSH) was the bivalent ligand CJL-1–87 that had an EC50 of 570 pM and was 3-fold more potent than its monovalent counterpart CJL-1–14. This result was similar to that previously observed with CJL-1–87 at the mouse (m)MC4R.40 The ligands that only contained the His-DNal(2ʹ)-Arg-Trp antagonist scaffold were not able to elicit a full agonist response when tested up to 10 µM, and all resulted in an antagonist pharmacology when analyzed via a Schild analysis (Table 1).95 Homobivalent ligand CJL-1–140 with two His-DNal(2ʹ)-Arg-Trp scaffolds resulted in 70% cAMP accumulation of that observed with NDP-MSH at 10 µM which is also consistent with previous reports at the mouse receptors.40 These results suggest that there is minimal species variation within the monovalent and homobivalent ligands currently tested.
Figure 2.

Illustrations of the in vitro functional pharmacology of MUmBLs at the MC4R. A) The cAMP signaling potency at the hMC4R was determined by AlphaScreen® assays. B) and C) The β-arrestin recruitment potency at the hMC4R was determined by PRESTO-Tango assays. The
 symbol represents the two monovalent tetrapeptides Ac-His-DPhe-Arg-Trp-NH2 and Ac-His-DNal(2’)-Arg-Trp-NH2 assayed together each at the indicated M concentration such that pharmacophore concentration is the same as the bivalent pharmacophore concentration. Functional cAMP data was normalized as discussed in experimental section to show tradition dose response curve with increasing response at increasing agonist concentrations. D) Ligand induced response on bioluminescence resonance energy transfer (BRET) signal using the mMC4R-NanoLuc and mMC4R-HaloTag homodimer. Maximal BRET signal (100%) was defined as the signal measured when assay buffer (represented as A) was added. Each ligand was dosed at 10−5, 10−7 and 10−9 M. Significance was determined using a one-way ANOVA to determine overall significance upon treatment followed by a Bonferroni post-hoc test to compare each ligand concentration to assay buffer control (A). * p<0.05, ** p<0.01, *** p<0.001. Data shown as the mean ± standard error of the mean (SEM) determined from three independent experiments.
symbol represents the two monovalent tetrapeptides Ac-His-DPhe-Arg-Trp-NH2 and Ac-His-DNal(2’)-Arg-Trp-NH2 assayed together each at the indicated M concentration such that pharmacophore concentration is the same as the bivalent pharmacophore concentration. Functional cAMP data was normalized as discussed in experimental section to show tradition dose response curve with increasing response at increasing agonist concentrations. D) Ligand induced response on bioluminescence resonance energy transfer (BRET) signal using the mMC4R-NanoLuc and mMC4R-HaloTag homodimer. Maximal BRET signal (100%) was defined as the signal measured when assay buffer (represented as A) was added. Each ligand was dosed at 10−5, 10−7 and 10−9 M. Significance was determined using a one-way ANOVA to determine overall significance upon treatment followed by a Bonferroni post-hoc test to compare each ligand concentration to assay buffer control (A). * p<0.05, ** p<0.01, *** p<0.001. Data shown as the mean ± standard error of the mean (SEM) determined from three independent experiments.
The MUmBLs (i.e. CJL-1–63, CJL-5–58, CJL-1–124, and CJL-5–74) were all single digit nanomolar potent agonists at the hMC4R. For comparison with the MUmBLs and as a control, an equal mixture of tetrapeptides CJL-1–14 + CJL-1–80 was assayed. In order to give the best comparison to the MUmBLs, 1 nM of the tetrapeptide mixture contained 1 nM CJL-1–14 and 1 nM CJL-1–80 (for a final concentration of 2 nM total peptide). This would be directly comparable to 1 nM of a MUmBL when looking at final pharmacophore concentration. This tetrapeptide mixture resulted in an agonist dose response curve with an EC50 of 1.9±0.2 nM. From this data, it appears that antagonist scaffold His-DNal(2ʹ)-Arg-Trp is not capable of affecting the cAMP agonist pharmacology of His-DPhe-Arg-Trp agonist scaffold when mixed in equal portions.
Theoretically, if both the agonist scaffold and antagonist scaffold compete equally for binding, then at 100% receptor occupancy 50% of the receptors would be occupied by agonist tetrapeptide scaffold and 50% would be occupied by the antagonist tetrapeptide scaffold (Supplemental Fig. 1 A–B, E–J). This likelihood of 50:50 binding should be amplified by the synergistic bivalent binding mode.40 Based on this assumption of 50:50 binding, the MUmBLs full cAMP agonist pharmacology would be achieved by only 50% receptor occupancy by the agonist scaffold at the receptors, since the antagonist scaffold would be occupying approximately 50% of the receptors. This is consistent with both the spare receptor theory,96–97 and the hypothesis presented above for asymmetric signaling homodimers in which ~50% of the receptors are responsible for β-arrestin recruitment and 50% are responsible for cAMP signaling (Figure 1). In practice, the MUmBLs may be binding to the melanocortin receptor monomers, dimers, and/or higher-order oligomers and may not be binding in exactly equal amounts of agonist scaffold and antagonist scaffold due to intermolecular competition (Supplemental Fig. 1). However, the synergistic binding previously achieved would only be observed if bivalent ligands are binding at a ratio of one MUmBL per dimer (two receptors) and the equilibrium should favor this bivalent binding mode (Supplemental Fig. 1G–I).
It was, therefore, hypothesized that the second binding event within the GPCR dimer may be responsible for a different functional response not detected in the cAMP functional assays. It has previously been observed that β-arrestin recruitment of one protomer within the AT1 angiotensin receptor homodimer can be allosterically regulated by selective stimulation of the other protomer.22 In order to examine if β-arrestin recruitment to the hMC4R was regulated differently by MUmBLs versus agonist or antagonist homobivalent ligands, we utilized the PRESTO-Tango assay developed by Roth and colleagues.98–99 The PRESTO-Tango technology is an open-source resource that has been utilized to identify ligands for orphan receptors based upon β-arrestin2 recruitment. This assay has previously been validated at the hMC4R reporting that agonist stimulation results in β-arrestin2 recruitment.98 In agreement with these results, classic monovalent agonist ligands result in the recruitment of β-arrestin and high signal (Table 1, Figure 2B–C). The classical melanocortin control agonists NDP-MSH, MTII and the tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 all resulted in maximal β-arrestin recruitment with MTII being the most potent ligand. The linker control and homobivalent ligands that featured only the His-DPhe-Arg-Trp pharmacophore all resulted in maximal β-arrestin recruitment relative to the NDP-MSH control. Among the linker controls, compound CJL-5-35-4 with the PEDG20 linker on the C-terminus resulted in a 5-fold increase in β-arrestin recruitment compared to the tetrapeptide CJL-1–14. This ligand also resulted in a 3-fold increase in the cAMP signaling assay. The other PEDG20 linker compound CJL-1–116 resulted in less than a 3-fold increase in β-arrestin recruitment potency compared to CJL-1–14. The His-DPhe-Arg-Trp-based ligands that utilized the Pro-Gly linker system did result in a decrease in the potency for β-arrestin recruitment in spite of them retaining their full cAMP signaling functional potency.
The ligands containing only the antagonist His-DNal(2ʹ)-Arg-Trp pharmacophore resulted in minimal β-arrestin recruitment consistent with a classical antagonist pharmacology. The tetrapeptide Ac-His-DNal(2ʹ)-Arg-Trp-NH2 resulted in a 30% response at 10 µM compared to the maximal efficacy of NDP-MSH. The other linker control ligands and the bivalent ligand CJL-1–140 resulted in equal or lower β-arrestin recruitment. This result was not surprising given the antagonistic nature of these compounds and that antagonist compounds have previously been reported to result in minimal β-arrestin recruitment and receptor internalization.63, 91–92
Interestingly, the MUmBLs resulted in minimal β-arrestin recruitment. The most potent MUmBL was CJL-1–63 that resulted in 55% maximal efficacy at 10 µM compared to NDP-MSH. All other MUmBLs resulted in less β-arrestin recruitment up to 10 µM concentrations. Although calculation of the EC50 values for these compounds would require making some assumption since they did not produce a sigmoidal dose response curve up the 10 µM concentration assayed, it can be estimated that the EC50 values are approximately equal to or greater than 10,000 nM. Therefore, the potency based selectivity for cAMP signaling over β-arrestin recruitment is estimated to be 3,000-fold or greater for the MUmBLs. Because these ligands still potently stimulate cAMP signaling but result in minimal β-arrestin recruitment, it supports the current hypothesis that one pharmacophore is responsible for the activation of the cAMP pathway, while the other pharmacophore is responsible for the β-arrestin recruitment. When a bivalent ligand is comprised of an agonist scaffold and an antagonist scaffold, it should favor a binding mode in which equal portions of agonist scaffold and antagonist scaffold bind to a GPCR dimer as discussed above (i.e. one MUmBL per two receptors or one dimer). The agonist pharmacophore would then signal effectively through the cAMP pathway, while the antagonist pharmacophore would block the β-arrestin recruitment pathway (Figure 1C). The current data observed with MUmBLs are not consistent with the current dogma in the field, however, these data may be explained by the asymmetric allosteric signaling within melanocortin homodimers.
Although there have been reports of biased agonists for different melanocortin pathways including cAMP, calcium mobilization, and receptor internalization,100–103 to the best of our knowledge the MUmBLs are the first melanocortin biased agonists for the cAMP pathway over the β-arrestin recruitment pathway. Biased ligands for GPCRs have been of interest for future drug development due their distinct pharmacology and functional selectivity.104–105 In particular, it is hypothesized that biased ligands stabilize a specific conformation of the receptor that favors one signaling pathway over another.104–105 We currently hypothesize similarly that the MUmBLs stabilize specific dimer conformational states (as opposed to monomeric receptor conformational states) that results in the cAMP-signaling biased agonism.
An explanation of the biased agonism is through a model for allosterically interacting receptor dimers (Figure 3).106–107 In this model, one receptor within a dimer can allosterically stabilize the other receptor within the dimer to different conformations. These different conformations are thought to be dynamic in that the receptors oscillate between the different states even with no ligand present.106 However, it is postulated that with no ligand bound, both receptors are conformationally open to cAMP signaling upon agonist stimulation (Figure 3A). After the first agonist binding event, a conformational change occurs which induces cAMP signaling pathway (Figure 3B) and this conformational change allosterically modifies the second receptor to have a propensity to signal through the β-arrestin pathway (Figure 3E). For this reason, monovalent agonist ligands, homobivalent agonist ligands, and the MUmBLs all produce full agonist cAMP induction, since the first agonist binding event is similar. After the first agonist binding event, the second receptor in the dimer is hypothesized to have structural bias for β-arrestin recruitment upon agonist binding. Therefore, the second agonist binding event results in β-arrestin recruitment (Figure 3F). This is the same for monovalent and homobivalent agonist ligands since both result in a second agonist binding event and this is observed in full β-arrestin recruitment results (Figure 2B and C). However, the MUmBLs result in an antagonist scaffold binding the second receptor instead of another agonist scaffold. The MUmBL’s antagonist tetrapeptide scaffold prevents β-arrestin recruitment that results in minimal signal in the PRESTO-Tango assay up 10 µM concentrations (Figure 3G–I).
Figure 3.

Illustrations of a previously reported model for allosteric interactions in GPCR dimers. (Durroux, 2005; Casado et al., 2007). In this model, GPCRs oscillate through different conformational states. Different conformations have different propensity to signal through cAMP or through β-arrestin. Signaling is represented by green arrows (B, C, E, F, H, I, L). Conformational changes are represented based on receptor highlighting (B, C, D, E, F, H, I, L). The binding of an agonist pharmacophore to one receptor that signals through cAMP stabilizes the second receptor’s conformation to increases its propensity to signal through the β-arrestin recruitment pathway (State E). Therefore, the second agonist binding event results in β-arrestin recruitment (State F). The BUmBL design strategy can be used to block the β-arrestin recruitment by increasing the likelihood of an antagonist pharmacophore binding the second receptor in the homodimer (States G–I). Even if the opposite binding order occurs, the antagonist blocks β-arrestin recruitment since it is already bound to the receptor after the agonist induces a conformational change (States J–L). This model assumes that the receptors are dimeric in nature, but they are likely in an equilibrium as monomers and higher-order oligomers. This models also assumes that the bivalent synergistic binding mode is favored with MUmBLs due to the decreased entropic cost of binding of the second pharmacophore. It is possible that MUmBLs compete in monovalent fashion (Supplemental Fig. 1), but then the increased binding affinity observed with bivalent ligands would not be expected.
There is an assumption above that the agonist tetrapeptide scaffold of the MUmBLs binds first before the antagonist tetrapeptide scaffold, but in practice the order of binding is not determined (Figure 3J–L). However, antagonist scaffolds restrict the GPCR from accessing conformational states that result in GPCR signaling (i.e. G-protein or β-arrestin). Therefore, even if the antagonist does bind first to the receptor dimer pair (Figure 3K), it would not induce G-protein signaling. Instead it would still require the first agonist binding event to the second receptor in the dimer pair to occur that would result in cAMP signaling (Figure 2A), and allosteric modulation to the β-arrestin ready state. However, the antagonist scaffold would already be bound to the dimer, and would block β-arrestin recruitment resulting in minimal PRESTO-Tango signal (Figure 2B, Figure 3L).
This interpretation of the data is based on the model for allosteric interaction within GPCR homodimers that has previously been reported.106–107 However, to add further complexity to the current model, it has been supported that GPCRs oscillate between monomeric, dimeric, and higher-order oligomeric states and models that include these states have also been proposed (Supplemental Fig. 1).27, 78, 106–108 Also the MUmBLs may bind in various different ways such that the antagonist scaffold displaces the agonist scaffold or vice versa resulting in more agonist scaffold binding than antagonist scaffold binding. As stated above, the bivalent binding mode should be favored due to synergistic binding, but these different confrontational states may help explain the minimal β-arrestin recruitment observed at higher concentrations of the MUmBLs. It is predicted that similar states are present in various GPCR systems, and that a biased unmatched bivalent ligand (BUmBL) design strategy could be applied to any asymmetric GPCR homodimer system for the creation of biased ligands.
This model may help explain a variety of observations in the field related to melanocortin dimerization and oligomerization. For example, Piechowski and coworkers reported in 2013 that disruption of hMC4R homodimerization leads to increased cAMP signaling capacity (or efficacy) although receptor expression was unchanged.78 This can be explained by the current model of asymmetric homodimers (Figure 1), since disruption of the dimer to monomers would increase the amount of receptors capable of accessing the cAMP signaling states (Figure 3B). In other words, with more monomers there would be less receptors being allosterically modulated to signal through β-arrestin recruitment since the asymmetric signaling could not occur. Therefore, more receptors would signal through the cAMP signaling pathway upon agonist binding increasing signaling capacity consistent with Piechowski and coworkers’ results.78
In our laboratory, initial in vivo evaluations of melanocortin ligands are performed in mice, therefore, an investigation of the ligands’ in vitro pharmacology in HEK293 cells expressing the mouse (m)MC1R, mMC3R, mMC4R, and mMC5R has been performed. Results from functional cAMP signaling assays and competitive binding experiments on the mouse melanocortin receptors along with a detailed discussion can be found in the Supplemental Information. Briefly, CJL-1–124 and CJL-5–74 showed some species variation between the mMC4R and the hMC4R in cAMP agonist activity highlighting the importance of studying lead ligands at the mouse isoform before conducting mouse studies. Compounds CJL-1–63 and CJL-5–58 showed minimal species variation in the assays performed suggesting further study in mouse models will be possible. Specifically, CJL-5–58 would be an ideal compound for further study in mouse models due to its biased agonism at the hMC4R, consistent pharmacology in cAMP signaling assays between the mouse and human receptor isoforms, and the increased serum stability of a PEDG20 linker compared to a Pro-Gly linker.41 During the in vitro investigation on the mMC4R, the ligands’ effects on receptor proximity using a bioluminescence resonance energy transfer (BRET) assay was studied and is discussed below.
MUmBLs Produce an Intermediate Effect on BRET Signal
Bioluminescence resonance energy transfer (BRET) has been routinely used to assess GPCR dimerization.109 Specifically, the MC3R and MC4R have been reported to result in high basal BRET signal supporting the formation of homodimers.41, 76, 110–111 Furthermore, BRET has been utilized to support the existence of hMC1R-hMC3R and mMC3R-mMC4R heterodimers.41, 110 It has also been suggested that ligand treatment can increase or decrease GPCR dimerization which should be detectable with changes in BRET signal.108, 112–117 However, these reports vary depending on the receptor system and ligands used.112 For example, agonist treatment at the somatostatin receptor 2 has been reported to cause the homodimers to dissociate into monomers.113 Whereas at the vasopressin V1a receptor, agonist ligand treatment had no observable effect on the dimerization ratio.114 There are also several examples of bivalent ligand treatment resulting in increased BRET signal suggesting they are inducing or increasing dimerization.108, 115–117 In previous reports focused on melanocortin receptors, no significant effect of agonist or antagonist ligand was reported for the hMC1R, hMC3R, or hMC4R homodimerization.110–111 However, in the BRET study involving the hMC4R, there does appear to be a trend towards decreasing BRET signal after agonist dosing, albeit not significant. After dosing α-MSH at 1 µM the mean BRET signal decreased by approximately 20% compared to basal BRET signal of the hMC4R.111 Because of the potential of the compounds to be modulating the dimer or oligomer state or changing the dimer conformational state, we investigated the response of BRET signal from mMC4R in response to ligand treatment (Figure 2D).
Ligands α-MSH, CJL-1–14, and CJL-1–87, that have full agonist activity in both the cAMP signaling assay (at the mMC4R and hMC4R) and the β-arrestin recruitment assay (at the hMC4R), resulted in a dose dependent decrease in BRET signal at the mMC4R (Figure 2D). Dosing these ligands at 10 µM resulted in a significant 15% reduction in BRET signal compared to basal signal. In contrast, ligands CJL-1–80 and CJL-1–140 contain only the antagonist tetrapeptide scaffold and have minimal functional agonist activity in both the cAMP signaling assay (at the mMC4R and hMC4R) and the β-arrestin recruitment assay (at the hMC4R). These antagonist-based ligands resulted in no significant changes in BRET signal from basal levels at the concentrations assayed at the mMC4R. In addition, the equal tetrapeptide mixture of agonist CJL-1–14 and antagonist CJL-1–80 resulted in no significant changes from basal signal. The MUmBLs, CJL-1–124 and CJL-5–58, resulted in a significant effect in which dosing 10 µM of ligand resulted in approximately an 8% reduction in BRET signal compared to basal signal at the mMC4R (Figure 2D).
The reduction of BRET signal observed with agonist containing ligands could be the result of three different mechanisms: 1) The dimerization or oligomerization is being disrupted and moving towards a lower oligomer state (e.g. dimers to monomers). 2) A conformational change is occurring within the intact dimer or higher-order oligomer in which the NanoLuc®-donor and the HaloTag®-acceptor are being orientated such that the BRET signal is being reduced (e.g. moving further away or dipole orientation is incorrect.)118 3) A receptor trafficking event, such as receptor internalization, is resulting in lowered BRET signal. It is currently difficult to determine which of these possibilities are the driving force for the BRET signal reduction observed in our studies. Regardless, it is apparent that some sort of conformational or receptor change is occurring that effects the BRET signal that relates with ligands’ agonist activity both for cAMP and for β-arrestin recruitment.
These changes match the proposed asymmetric signaling model for MC4R homodimers (Figure 1, Figure 3). It follows from the proposed model that at basal levels in which only assay buffer is added (Figure 3A), no conformational changes have occurred. With the addition of agonist ligand and the first binding event, cAMP signaling pathway is activated and a conformational change occurs that affects BRET signal (c.a. 7–8% change) (Figure 3B or E). This is observed with all ligands that contain an agonist scaffold including α-MSH, CJL-1–14, CJL-1–87, CJL-1–124, and CJL-5–58 (Figure 2D). The second agonist binding event is hypothesized to result in an additional conformational change at the second receptor in the homodimer, and this is postulated to be responsible for the maximal observed decrease in BRET signal (c.a. 15%) (Figure 3F). This is observed with ligands α-MSH, CJL-1–14, and CJL-1–87 because they result in the second conformational change within the homodimer due to a second agonist binding event on the second receptor. However, the second receptor in the homodimer pair is postulated to be bound by an antagonist scaffold with ligands CJL-5–58 and CJL-1–124 (Figure 1C) and, therefore, the full conformational change to the homodimer unit does not occur (Figure 3I) resulting in the lack of β-arrestin recruitment (Figure 2B–C) and there is only 50% maximal change in BRET signal (i.e. 7–8% change instead of 15%) (Figure 2D).
Although further experimental work will be necessary to determine the exact dimer conformational changes that are occurring, the current studies support the hypothesis that the biased agonism observed currently with CJL-5–58 is the result of a conformational change of the dimeric state corresponding to the changes observed in the BRET signal. These conformational changes could be changes in the oligomeric number (e.g. dimers to monomers), orientation of the receptors within a dimer pair (e.g. which transmembrane helixes are interacting), or changes in the cellular location of the receptors (e.g. receptor internalization).9, 27, 78 Further elucidation of these mechanisms, as well as work looking at possible difference between the mouse and human MC4R isoforms, will be necessary.
Discussion
There is a growing amount of evidence that GPCR homodimers are functionally relevant and are pharmaceutical targets. A broadly applicable drug design strategy that targets homodimers, as opposed to monomeric receptors, would theoretically double the amount GPCR drug targets. Although various labs have presented different techniques and proofs-of-concept approaches for methods to target asymmetrically signaling GPCR homodimers,12–26, 30 these techniques would be difficult to apply broadly to multiple GPCR systems or challenging to adapt to therapeutic design and in vivo applications. The current report presents a possible design strategy that targets asymmetric homodimers that should be easily amenable to various GPCR systems. Because the current design strategy produces a single compound to target both receptors in a homodimer, it should be easily amendable to in vivo applications.
In the current report, we designed bivalent ligands that contained an agonist tetrapeptide scaffold and antagonist tetrapeptide scaffold for the MC4R. The MUmBL design strategy aims at occupying each of the two receptors within the homodimer with a different pharmacophore such that an agonist pharmacophore and an antagonist pharmacophore each occupy one of the two receptors within each homodimer. This design strategy produced biased ligands at the hMC4R in which the cAMP signaling pathway was robustly activated at nanomolar concentrations (EC50 ~ 2 to 6 nM), but the β-arrestin2 pathway was only partially activated at concentrations up to 10 µM. To our knowledge, these are the first melanocortin biased ligands favoring cAMP signaling over β-arrestin recruitment and will be valuable chemical probes to study melanocortin signaling in the disease states and disorders in which the melanocortin receptors are implicated including: cancer,42–46 skin pigmentation disorders,47 social disorders,48–49 sexual function disorders,50–52 Alzheimer’s disease,53–54 cachexia,55–59 and obesity.40, 60–62
We then functionally characterized the MUmBLs at the mouse melanocortin receptors. Two of the compounds showed species difference in which a partial agonist dose response curve was observed at the mMC4R (see Supplemental Discussion). These in vitro studies identified CJL-5–58 as the lead ligand for future evaluations due to its biased agonism at the hMC4R, consistent pharmacology in cAMP signaling assays between the mouse and human receptors, and the increased serum stability of a PEDG20 linker compared to a Pro-Gly linker.41 Further elucidation of the pharmacology of biased melanocortin ligands will be necessary, but the current report provides the melanocortin field with the first-in-class biased ligand that favors the cAMP pathway over the β-arrestin pathway.
The UmBL methodology presented currently should be applicable to various other GPCRs and can easily accommodate the plethora of well-studied and developed selective agonists and antagonists for various GPCR systems. This bivalent ligand targeting method should allow for biased ligands or unique pharmacologies at various receptors by combining known agonists and antagonists with an effective linker. Considering the wide array of GPCRs that are already reported to exist as allosterically modulated or asymmetric homodimers (including the vasopressin,28 dopamine,12 adenosine,26 metabotropic glutamate,19 and serotonin receptors13) this strategy should be broadly applicable. In order to effectively synthesize UmBLs for other receptor systems, it will be necessary to perform some standard medicinal chemistry to optimize the connection points of the linker to the pharmacophores, optimize the linker properties, and optimize the orientation of the pharmacophores. Based on studies at the mouse melanocortin receptors, it was optimal for the agonist scaffold and the antagonist scaffold to have approximately equal binding affinities (see Supplemental Information).
The exact pharmacology that may be achieved through the UmBL design strategy will be as diverse as the allosteric mechanisms between different GPCR homodimers. For example, based on the results of Han and coworkers it can be hypothesized that UmBLs targeting the dopamine D2 receptor would result in increased receptor activation beyond just monovalent agonist alone. This is because allosteric cross-talk of a second agonist protomer was shown to blunt activation, so the occupation of the second protomer with an antagonist scaffold instead of an agonist scaffold should increase signal activation.12 In contrast if the UmBL approach was applied to the metabotropic glutamate receptor, it would be hypothesized to result in lower than full receptor activation of agonist alone as Kniazeff and coworkers observed that one agonist can partially activate a dimeric unit but two agonists are required for full activation.19 Finally, it has been reported that the vasopressin V1b receptor signals through both the Gq/11-inositol phosphate (IP) and the cAMP pathways.28 It was hypothesized by Orcel and coworkers, that “the IP pathway could be activated by the binding of either one or two AVP molecules to a single receptor dimer… By contrast, cAMP production could only be turned on upon the binding of two ligands to a dimer.” Their observations and hypothesis is consistent with asymmetric homodimers such that the IP pathway is activated by the first agonist binding event and the cAMP pathway is activated second (similar to Figure 1).28 Therefore, if the UmBL design strategy was applied to ligands targeting the vasopressin V1b receptor, it would be predicted to result in biased ligands in which the agonist pharmacophore would activate the IP pathway, and the antagonist pharmacophore would block the cAMP pathway activation within the homodimer. The UmBL design approach could also be applied to GPCR systems in which asymmetry between homodimers has not been identified, or even systems in which homodimerization has not yet been observed. In these situations, designed UmBLs could be evaluated for their ability to induce signaling in multiple signaling pathways (e.g. cAMP, Ca+, kinase signaling, β-arrestin signaling, etc.) to identify asymmetrically signaling GPCR homodimers.
Conclusions
In conclusion, the UmBL design approach reported herein has been used to progress the melanocortin field by identifying the first known biased ligands for the cAMP pathway over the β-arrestin2 recruitment pathway. This report also provides functional pharmacological evidence of melanocortin asymmetric homodimerization that complements previous BRET studies supporting homodimerization,41, 110 radioligand binding studies suggesting asymmetry of melanocortin homodimers propensity to bind ligands,76–77 and functional reports demonstrating the significance of melanocortin oligomerization.27, 78 In a much broader sense, the UmBL approach provides a medicinal chemistry design strategy for the future advancement of GPCR pharmacology that can be applied to various receptor systems.
Experimental Section
Chemical Synthesis
Peptides were synthesized utilizing standard solid phase peptide synthesis and fluorenyl-9-methoxycarbonyl (Fmoc) methodologies to protect the elongating peptide chain.89, 119 A CEM Discover SPS microwave peptide synthesizer was used to expedite couplings and deprotections. A split resin technique was used to synthesize common sequences as previously described.40 The O-(N-Fmoc-3-aminopropyl)-O'-(N-diglycolyl-3-aminopropyl)-diethyleneglycol [Fmoc-NH-(PEG)2-COOH (20atoms) or Fmoc-NH2-PEDG20-COOH] was purchased from Novobiochem® EMD Millipore Corp (Billerica, MA, USA). The N,N-diisopropylethylamine (DIEA), triisopropylsilane (TIS), 1,2-ethanedithiol (EDT), piperidine, pyridine, and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich (St. Louis, MO). The 4-(2ʹ,4ʹ-dimethoxyphenyl-Fmoc-aminomethyl)phenoxyacetyl-MBHA resin [Rink-amide-MBHA (200–400 mesh), 0.35–0.37 meq/g substitution], 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and Fmoc-protected amino acids [Fmoc-Pro, Fmoc-Gly, Fmoc-His(Trt), Fmoc-DPhe, Fmoc-Arg(Pbf), Fmoc-Trp(Boc), and Fmoc-DNal(2ʹ)] were purchased from Peptides International (Louisville, KY, USA). Acetonitrile (MeCN), N,N-dimethylformamide (DMF), acetic anhydride, dichloromethane (DCM), and methanol (MeOH) were purchased from FischerScientific. All reagents were ACS grade or better and were used without further purification.
Peptides were assembled in a fritted polypropylene reaction vessel (25 mL CEM reaction vessel) on the Rink-amide-MBHA resin. A repeated two-step cycle of deprotection with 20% piperidine in DMF, then amide coupling with the Fmoc-amino acid, HBTU, and DIEA was employed until the final peptide was synthesized on resin. Excess reagents were removed between all deprotection or coupling by 3–5 washes of DMF between steps. A Kaiser/ninhydrin test was used after each deprotection or coupling step (except with Pro residues) to indicated the presence or lack of a free primary amine.120 For Pro residues, the presence or lack of a free secondary amine was indicated by a chloranil test.89, 121 Removal of the Fmoc group was achieved in a two-step process. First an initial two minute deprotection was performed outside of the microwave. Then a second aliquot of 20% piperidine was added and further deprotection was assisted by microwave heating (75°C, 30 W, 4 min).
Amide coupling was achieved by addition of 3.1-fold excess Fmoc-protected amino acids (5.1-fold for Arg) and 3-fold excess of HBTU (5-fold for Arg) in DMF added to the free amine on the elongating peptide on the resin. After which the 5-fold excess of DIEA (7-fold for Arg) was added, and the reaction was heated in the microwave synthesizer (75°C, 30 W or 50°C, 30 W for His) for five minutes (10 min for Arg). The Fmoc-NH-(PEDG20)-COOH was incorporated into the peptide using the same protocol except it was allowed to cool for at least one hour after microwave heating to ensure the reaction went to completion.
Acetylation was achieved on resin after the final Fmoc deprotection by addition of 3:1 mixture of acetic anhydride to pyridine and were mixed at room temperature with bubbling nitrogen for 30 minutes. Before cleavage, all peptides were washed with DCM at least 3 times and dried in a desiccator. Side chain deprotection and resin cleavage was simultaneously accomplished via addition of 8 mL of a cleavage cocktail (91% TFA, 3% EDT, 3% TIS, 3% water) for 1.5–3 hours. Peptides were precipitated from cleavage solution using cold (4°C) anhydrous diethyl ether. The cloudy mixture of peptides was vortexed and centrifuged at 4°C and 4000 RPM for 4 minutes (Sorval Super T21 high-speed centrifuge swinging bucket rotor). The supernatant was discarded. The crude peptide pellet was then washed with cold (4 °C) diethyl ether and centrifuged. This process was repeated until no thiol aroma was present (usually 3 times) and the peptides were dried overnight in a desiccator.
A Shimadzu chromatography system with a photodiode array detector and a semipreparative RP-HPLC C18 bonded silica column (Vydac 218TP1010, 1 cm × 25 cm) were used to purify 5–20 mg sample of crude peptide by RP-HPLC. The solvent system for purification was either MeCN or MeOH in 0.1% aqueous TFA. Purified fractions were collected and peptides were concentrated in vacuo and lyophilized. A purity of 95% or greater was confirmed by analytical RP-HPLC at 214 nm in two diverse solvent systems (10% MeCN in 0.1 % TFA/water and a gradient to 90% MeCN over 35 min; and 10% MeOH in 0.1 % TFA/water and a gradient to 90% MeOH over 35 minutes at a flow rate of 1.5 mL/min) using an analytical Vydac C18 column (Vydac 218TP104). ESI-MS was used to confirm the correct molecular mass (University of Minnesota Department of Chemistry Mass Spectrometry Laboratory) (Supplemental Table 1).
Cell Culture
HEK293 cells for the ALPHAScreen assay, competitive binding assay, PRESTO-Tango assay, and BRET assay were maintained in humidified atmosphere of 95% air and 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% newborn calf serum (NCS), and 1% penicillin/streptomycin. Stable cell lines were generated with wild type mMC1R, mMC4R, mMC5R, hMC4R-Flag, and mMC3R-Flag DNA in pCDNA3 expression vector (20 µg) using the calcium phosphate transfection method.122 Stable populations were selected for using G418 selection (0.7–1.0 mg/mL) and used in bioassays unless indicated otherwise. In vitro experimental ligands were dissolved to a 10−2 M stock in DMSO and stored at −20 °C. Subsequent dilutions were performed in each assay’s specific buffer to achieve the final concentration in the well. The ligands were assayed as TFA salts.
AlphaScreen® cAMP Functional Bioassay
The AlphaScreen® cAMP technology (PerkinElmer Life Sciences, Cat #6760625M) was utilized to measure cAMP signaling after ligand stimulation in HEK293 cells stably expressing the mMC1R, mMC3R, mMC4R, hMC4R and mMC5R. The AlphaScreen® assay was performed as described by manufacturer. This method has been previously utilized by our lab,40, 123 and it is described briefly below.
On the day of the assay, cells were 70–95% confluent in 10 cm plates. Cells were removed from plates using Gibco® Versene solution and pelleted by centrifugation (Sorvall Super T21 high speed centrifuge, swinging bucket rotor) at 800 RPM for five minutes. Media was gently aspirated and cells were resuspended in Dulbecco’s phosphate buffered saline solution (DPBS 1× [−] without calcium and magnesium chloride, Gibco ® Cat # 14190-144). A 10 µL aliquot of cell suspension was counted manually using a hemocytometer after addition of Trypan blue dye (BioRad). Cells were again pelleted by centrifugation, and DPBS was gently aspirated. The pelleted cells were then resuspended in a solution of freshly made stimulation buffer (Hank’s Balanced Salt Solution [HBSS 10× [−] sodium bicarbonate] and [−] phenol red, Gibco®], 0.5 mM isobutylmethylxanthine [IBMX], 5 mM HEPES buffer solution [1M, Gibco®], 0.1% bovine serum albumin [BSA] in Milli-Q water, pH=7.4) and anti-cAMP acceptor beads (1.0 unit per well, AlphaScreen®). A cell/acceptor bead solution was added manually to each well of a 384 well microplate (OptiPlate-384; PerkinElmer) for final concentrations of 10,000 cells/well and 1.0 Unit anti-cAMP acceptor beads/well. The cells were then stimulated with ligand diluted in stimulation buffer to achieve their final concentrations in the well ranging from 10−13 to 10−4 M. The stimulated plates were incubated in a dark laboratory drawer at room temperature for two hours.
Meanwhile, a biotinylated cAMP/streptavidin donor bead working solution was made by adding biotinylated cAMP (1 Unit/well, AlphaScreen®) and streptavidin donor beads (1 Unit/well, AlphaScreen®) to a lysis buffer (10% Tween-20, 5 mM HEPES buffer solution [1M, Gibco®], 0.1% bovine serum albumin [BSA] in Milli-Q water, pH=7.4). After the two hour stimulation, the biotinylated cAMP/ Streptavidin donor bead working solution was added to each well under green light and mixed well by pipetting up and down. The cells were incubated for another two hours at room temperature in a dark drawer at room temperature. The plate was then read on an EnSpireTM Alpha plate reader using a pre-normalized assay protocol set by the manufacturer. Assays were performed with duplicate data points on each plate and repeated in at least three independent experiments. Each plate contained a control ligand dose response (NDP-MSH, α-MSH, or ɣ2-MSH), a 10−4 M forskolin positive control, and a no ligand assay buffer negative control.
Dose response curves were analyzed using the PRISM program (v4.0; GraphPad Inc.). Potency EC50 values (concentration that caused 50% maximal signal) were calculated by a nonlinear regression method. Schild regression analysis was used assess antagonist properties and to calculate pA2 values [pA2 = −log(Ki)]. In the Schild analysis, antagonist ligands were tested in a dose-dependent manner to inhibit NDP-MSH agonist receptor stimulation.95 To be consistent with functional data being represented as an increasing response with increasing concentration and because the AlphaScreen® assay is a competition assay, a transformation was carried out for illustration purposes to normalize data to control compounds and flip dose response curves as previously described.40, 123
PRESO-Tango (β-arrestin2 Recruitment) Assay
The PRESTO-Tango assay was developed by Kroeze and coworkers for identifying biologically active compounds by the rapid screening for most of the entire druggable GPCRome.98–99 The plasmids and assay technology was kindly provided by the Bryan Roth laboratory (University of North Carolina at Chapel Hill) and are now available through ADDGENE (Kit # 1000000068). Briefly, HTLA cells (HEK293 cells that stably express a tTA-dependent luciferase reporter and a β-arrestin 2 –TEV fusion gene and were kindly provided by Richard Axel99) were maintained in DMEM supplemented with 10% FBS, 100 U/mL penicillin, 100 µg/mL streptomycin, 2 µg/mL puromycin, and 100 µg/mL hygromycin B in humidified atmosphere of 95% air and 5% CO2 at 37 °C.
The first day of the assay, HTLA cells were plated at approximately 1×106 cells per 10 cm plate and grown to 20–40% confluency. On the second day, cells were transiently transfected using the calcium phosphate method with 4 µg/plate of hMC4R PRESTO-Tango plasmid construct and incubated 15–24 hours in humidified atmosphere of 97% air and 3% CO2 at 35 °C.98, 122 The third day, cells were removed from 10 cm plates using Gibco® Versene solution and pelleted by centrifugation (Sorvall Super T21 high speed centrifuge, swinging bucket rotor) at 800 rpm for five minutes at room temperature. Cells were manually counted using a hemocytometer and resuspended in 1% dialyzed FBS and 1% penicillin/streptomycin in DMEM to a final concentration of 400,000 cells/mL. Cells were plated into 384-well white wall and clear bottom microplate (ViewPlate-384 TC, PerkinElmer Cat # 6007480) for a final concentration of 20,000 cells/well and incubated in 5% CO2 at 37 °C. On the fourth day, cells were stimulated by ligands diluted to the appropriate in well concentrations (i.e. 10−12 to 10−5 M) in filter-sterilized assay buffer (20 mM HEPES, 1× HBSS, water, titrated to pH 7.4 with 1 N NaOH). Stimulated cells were incubated for 18 hours in 5% CO2 at 37 °C. On the fifth day, the assay buffer and cell medium was removed by aspiration. Then 20 µL of Bright-Glo (Promega, Cat # N1661) diluted 20-fold in assay buffer was added to each well and incubated to 15–20 minutes. After incubation, luminescence was then read on an EnSpire™ Alpha plate reader using a pre-normalized assay protocol for luminescence set by the manufacturer. Assays were performed with duplicate data points on each plate and repeated in at least three independent experiments. Dose response curves were analyzed using the PRISM program (v4.0; GraphPad Inc.). Potency EC50 values (concentration that caused 50% maximal signal) were calculated by a nonlinear regression method.
Bioluminscence Resonance Energy Transfer (BRET) Studies
The NanoBRET™ Protein:Protein Interaction System was utilized according to manufacturer’s instructions to examine the association and proximity of the melanocortin receptors as previously reported.41 Briefly, the plasmids were constructed to incorporate the NanoLuc® fusion protein and the HaloTag® fusion protein onto the C-terminus of the mMC4R of the plasmids described above. Proper cell membrane expression and ligand binding have previously been supported by competitive binding experiments.41 The specificity of signal has also previously been shown.41 On the first day, cells were plated into 6 well plates in the morning. In the afternoon of the same day, cells were transiently transfected with mMC4R-NanoLuc® fusion protein and the MC4R-HaloTag® fusion protein by adding FuGene6 Transfection (8 µL/well, Promega), DNA (2 µg/well) in OptiMem medium (Invitrogen) at a total volume of 100 µL/well. The ratio of donor NanoLuc® to acceptor HaloTag® DNA has previously been optimized and a ratio of 1 Receptor-NanoLuc® plasmid: 4 Receptor-HaloTag® plasmid was utilized for all experiments.41 Cells were incubated with transfection reagent overnight at 5% CO2 at 37° C. One day after the transfection, cells were re-plated into 96-well black clear bottom plates (Cat # 3603, Corning Life Sciences) at 30,000 cells in 90 µL of assay buffer (4% FBS in OptiMem).
To each well, 1 µL of 0.1 mM HaloTag® NanoBRET™ 618 ligand was added and incubated 18–24 h at 5% CO2 at 37° C. As a negative control, each assay also included; “no acceptor controls” in which 1 µL of DMSO was added instead of 618 ligand rendering the BRET relay system incomplete. This provides the background signal and was subtracted from the final experimental signal. Plates were then developed 48 to 60 hours after transfection. Two hours before the plates were developed, 10 µL of a 10× aliquot of the ligand diluted in assay buffer was added to each well to yield the final in well concentration (10−5, 10−7, or 10−9 M) of each compound. For the assay buffer control, 10 µL of assay buffer was added instead of compound. To develop plates, 25 µL of 5× solution of NanoBRET™ Nano-Glo® Substrate in Opti-MEM® was added to each well. Plates were then read within 10 min on a FlexStation® 3 plate reader (Molecular Devices) at the donor emission wavelength (460 nm) and acceptor emission wavelength (618 nm). The milli BRET Units (mBUs) were calculated by dividing the acceptor emission of 618 nm by the donor emission at 460 nm and multiplying it by 1000. The standard error of the mean (SEM) was derived from at least three independent experiments.
Data Analysis
The data was analyzed utilizing PRISM program (v 4.0; GraphPad Inc.). Statistical significance was considered p < 0.05. The results are not corrected for net peptide content, although all the peptides examined in this study were determined to have approximately equal peptide content as determined by using Beers Law. Data analysis is discussed in more detail at the end of each assays’ experimental section.
Supplementary Material
Acknowledgments
We would like to thank Katlyn Fleming and Dr. Mark Ericson for their insights while preparing this manuscript. We would also like to thank Dr. Radleigh Santos, Dr. Skye R. Doering, Dr. Rodney Johnson, and Dr. Philip Portoghese for their detailed discussion and help preparing this manuscript. This work has been supported by NIH Grant R01DK091906 (C.H.-L.). C.J.L. and A.T.Z. was provided support from the University of Minnesota Doctoral Dissertation Fellowship. C.J.L. was provided additional the University of Minnesota College of Pharmacy Olsteins Graduate Fellowship. We would also like to acknowledge the receipt of a 2017 Wallin Neuroscience Discovery Fund Award.
Footnotes
Author Contributions: Competitive binding assays and AlphaScreen assays were performed by C.J.L., K.T.F., and S.M.S. PRESTO-Tango assays and BRET assays were performed by K.T.F. Radiolabeled compounds were prepared by R.C.S. All experimental compounds were synthesized and prepared by C.J.L. Serum stability studies were performed by C.J.L. and A.T.Z. The manuscript was written by C.J.L. with contributions from all authors.
Conflict of Interest: The authors declare no competing financial interests.
Supporting Information: Detailed Description of Characterization of cAMP Signaling at the Mouse Melanocortin Receptors (S2–S3); Detailed Description of 125I-NDP-MSH Competitive Binding Assays at the Mouse Melanocortin Receptors (S4–S5); Methods for 125I-NDP-MSH Competitive Binding Affinity Studies (S6); Methods for Serum Stability Assays (S7); The analytical data for peptides synthesized currently (S8); Summary of cAMP functional experiments at the mMC1R, mMC3R, mMC4R, and mMC5R (S9) Summary of competitive binding experiments at the mMC1R, mMC3R, and mMC4R (S10); Different possible binding states of the MUmBLs (S11); In vitro mouse serum stability assay to aid in the design of the MUmBLs (S12); Illustrations of the in vitro functional pharmacology of the MUmBLs at the mMC1R, mMC3R, mMC4R, and mMC5R (S13); Illustrations of the competitive binding experiments against 125I-NDP-MSH at the mMC3R and mMC4R (S14); Illustrations of the competitive binding experiments against 125I-NDP-MSH at the mMC1R (S15); HPLC chromatograms of all compounds reported in the study (S16–S52).
References
- 1.Rask-Andersen M, Almen MS, Schioth HB. Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discovery. 2011;10:579–590. doi: 10.1038/nrd3478. [DOI] [PubMed] [Google Scholar]
- 2.Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, Overington JP. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discovery. 2017;16:19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discovery. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferre S, Casado V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X. G protein-coupled receptor oligomerization revisited: Functional and pharmacological perspectives. Pharmacol. Rev. 2014;66:413–434. doi: 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferre S. The GPCR heterotetramer: challenging classical pharmacology. Trends Pharmacol. Sci. 2015;36:145–152. doi: 10.1016/j.tips.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl. Acad. Sci. U. S. A. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smeester BA, Lunzer MM, Akgun E, Beitz AJ, Portoghese PS. Targeting putative mu opioid/metabotropic glutamate receptor-5 heteromers produces potent antinociception in a chronic murine bone cancer model. Eur. J. Pharmacol. 2014;743:48–52. doi: 10.1016/j.ejphar.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Le Naour M, Akgun E, Yekkirala A, Lunzer MM, Powers MD, Kalyuzhny AE, Portoghese PS. Bivalent ligands that target mu opioid (MOP) and cannabinoid1 (CB1) receptors are potent analgesics devoid of tolerance. J. Med. Chem. 2013;56:5505–5513. doi: 10.1021/jm4005219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akgün E, Javed MI, Lunzer MM, Powers MD, Sham YY, Watanabe Y, Portoghese PS. Inhibition of inflammatory and neuropathic pain by targeting a mu opioid receptor/chemokine receptor5 heteromer (MOR-CCR5) J. Med. Chem. 2015;58:8647–8657. doi: 10.1021/acs.jmedchem.5b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Portoghese PS, Akgun E, Lunzer MM. Heteromer induction: An approach to unique pharmacology? ACS Chem. Neurosci. 2017;8:426–428. doi: 10.1021/acschemneuro.7b00002. [DOI] [PubMed] [Google Scholar]
- 11.Peterson CD, Kitto KF, Akgun E, Lunzer MM, Riedl MS, Vulchanova L, Wilcox GL, Portoghese PS, Fairbanks CA. Bivalent ligand that activates mu opioid receptor and antagonizes mGluR5 receptor reduces neuropathic pain in mice. Pain. 2017;158:2431–2441. doi: 10.1097/j.pain.0000000000001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat. Chem. Biol. 2009;5:688–695. doi: 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pellissier LP, Barthet G, Gaven F, Cassier E, Trinquet E, Pin JP, Marin P, Dumuis A, Bockaert J, Baneres JL, Claeysen S. G protein activation by serotonin type 4 receptor dimers: Evidence that turning on two protomers is more efficient. J. Biol. Chem. 2011;286:9985–9997. doi: 10.1074/jbc.M110.201939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teitler M, Klein MT. A new approach for studying GPCR dimers: Drug-induced inactivation and reactivation to reveal GPCR dimer function in vitro, in primary culture, and in vivo. Pharmacol. Ther. 2012;133:205–217. doi: 10.1016/j.pharmthera.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comps-Agrar L, Kniazeff J, Norskov-Lauritsen L, Maurel D, Gassmann M, Gregor N, Prezeau L, Bettler B, Durroux T, Trinquet E, Pin JP. The oligomeric state sets GABA(B) receptor signalling efficacy. EMBO J. 2011;30:2336–2349. doi: 10.1038/emboj.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pin JP, Kniazeff J, Liu J, Binet V, Goudet C, Rondard P, Prezeau L. Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J. 2005;272:2947–2955. doi: 10.1111/j.1742-4658.2005.04728.x. [DOI] [PubMed] [Google Scholar]
- 17.Hlavackova V, Goudet C, Kniazeff J, Zikova A, Maurel D, Vol C, Trojanova J, Prezeau L, Pin JP, Blahos J. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J. 2005;24:499–509. doi: 10.1038/sj.emboj.7600557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prezeau L, Goudet C, Kniazeff J, Hlavackova V, Malhaire F, Maurel D, Acher F, Blahos J, Pin JP. Binding of a single positive allosteric modulator per dimer is sufficient for full enhancement of metabotropic glutamate receptor activity. Neuropharmacology. 2005;49:267–267. [Google Scholar]
- 19.Kniazeff J, Bessis AS, Maurel D, Ansanay H, Prezeau L, Pin JP. Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat. Struct. Mol. Biol. 2004;11:706–713. doi: 10.1038/nsmb794. [DOI] [PubMed] [Google Scholar]
- 20.Kniazeff J, Saintot PP, Goudet C, Liu J, Charnet A, Guillon G, Pin JP. Locking the dimeric GABA(B) G-protein-coupled receptor in its active state. J. Neurosci. 2004;24:370–377. doi: 10.1523/JNEUROSCI.3141-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zylbergold P, Hebert TE. A division of labor: asymmetric roles for GPCR subunits in receptor dimers. Nat. Chem. Biol. 2009;5:608–609. doi: 10.1038/nchembio0909-608. [DOI] [PubMed] [Google Scholar]
- 22.Szalai B, Barkai L, Turu G, Szidonya L, Varnai P, Hunyady L. Allosteric interactions within the AT(1) angiotensin receptor homodimer: Role of the conserved DRY motif. Biochem. Pharmacol. 2012;84:477–485. doi: 10.1016/j.bcp.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Damian M, Martin A, Mesnier D, Pin JP, Baneres JL. Asymmetric conformational changes in a GPCR dimer controlled by G-proteins. EMBO J. 2006;25:5693–5702. doi: 10.1038/sj.emboj.7601449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brock C, Oueslati N, Soler S, Boudier L, Rondard P, Pin JP. Activation of a dimeric metabotropic glutamate receptor by intersubunit rearrangement. J. Biol. Chem. 2007;282:33000–33008. doi: 10.1074/jbc.M702542200. [DOI] [PubMed] [Google Scholar]
- 25.Sartania N, Appelbe S, Pediani JD, Milligan G. Agonist occupancy of a single monomeric element is sufficient to cause internalization of the dimeric beta2-adrenoceptor. Cell. Signal. 2007;19:1928–1938. doi: 10.1016/j.cellsig.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Gracia E, Moreno E, Cortes A, Lluis C, Mallol J, McCormick PJ, Canela EI, Casado V. Homodimerization of adenosine A(1) receptors in brain cortex explains the biphasic effects of caffeine. Neuropharmacology. 2013;71:56–69. doi: 10.1016/j.neuropharm.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 27.Chapman KL, Findlay JB. The melanocortin 4 receptor: Oligomer formation, interaction sites and functional significance. Biochim. Biophys. Acta. 2013;1828:535–542. doi: 10.1016/j.bbamem.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 28.Orcel H, Albizu L, Perkovska S, Durroux T, Mendre C, Ansanay H, Mouillac B, Rabie A. Differential coupling of the vasopressin V1b receptor through compartmentalization within the plasma membrane. Mol. Pharmacol. 2009;75:637–647. doi: 10.1124/mol.108.049031. [DOI] [PubMed] [Google Scholar]
- 29.Iglesias A, Cimadevila M, Cadavid MI, Loza MI, Brea J. Serotonin-2A homodimers are needed for signalling via both phospholipase A2 and phospholipase C in transfected CHO cells. Eur. J. Pharmacol. 2017;800:63–69. doi: 10.1016/j.ejphar.2017.02.028. [DOI] [PubMed] [Google Scholar]
- 30.Lane JR, Donthamsetti P, Shonberg J, Draper-Joyce CJ, Dentry S, Michino M, Shi L, Lopez L, Scammells PJ, Capuano B, Sexton PM, Javitch JA, Christopoulos A. A new mechanism of allostery in a G protein-coupled receptor dimer. Nat. Chem. Biol. 2014;10:745–752. doi: 10.1038/nchembio.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conn PM, Rogers DC, Stewart JM, Niedel J, Sheffield T. Conversion of a gonadotropin-releasing hormone antagonist to an agonist. Nature. 1982;296:653–655. doi: 10.1038/296653a0. [DOI] [PubMed] [Google Scholar]
- 32.Blum JJ, Conn PM. Gonadotropin-releasing hormone stimulation of luteinizing hormone release: A ligand-receptor-effector model. Proc. Natl. Acad. Sci. U. S. A. 1982;79:7307–7311. doi: 10.1073/pnas.79.23.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Portoghese PS, Ronsisvalle G, Larson DL, Yim CB, Sayre LM, Takemori AE. Opioid agonist and antagonist bivalent ligands as receptor probes. Life Sci. 1982;31:1283–1286. doi: 10.1016/0024-3205(82)90362-9. [DOI] [PubMed] [Google Scholar]
- 34.Erez M, Takemori AE, Portoghese PS. Narcotic antagonistic potency of bivalent ligands which contain beta-naltrexamine. Evidence for bridging between proximal recognition sites. J. Med. Chem. 1982;25:847–849. doi: 10.1021/jm00349a016. [DOI] [PubMed] [Google Scholar]
- 35.Le Naour M, Lunzer MM, Powers MD, Kalyuzhny AE, Benneyworth MA, Thomas MJ, Portoghese PS. Putative kappa opioid heteromers as targets for developing analgesics free of adverse effects. J. Med. Chem. 2014;57:6383–6392. doi: 10.1021/jm500159d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hiller C, Kuhhorn J, Gmeiner P. Class A G-protein-coupled receptor (GPCR) dimers and bivalent ligands. J. Med. Chem. 2013;56:6542–6559. doi: 10.1021/jm4004335. [DOI] [PubMed] [Google Scholar]
- 37.Kuhhorn J, Gotz A, Hubner H, Thompson D, Whistler J, Gmeiner P. Development of a bivalent dopamine D-2 receptor agonist. J. Med. Chem. 2011;54:7911–7919. doi: 10.1021/jm2009919. [DOI] [PubMed] [Google Scholar]
- 38.Fernandes SM, Lee YS, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent ligands for binding to the human melanocortin-4 receptor. Bioorg. Med. Chem. 2014;22:6360–6365. doi: 10.1016/j.bmc.2014.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol. Rev. 2010;62:701–725. doi: 10.1124/pr.110.002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lensing CJ, Freeman KT, Schnell SM, Adank DN, Speth RC, Haskell-Luevano C. An in vitro and in vivo investigation of bivalent ligands that display preferential binding and functional activity for different melanocortin receptor homodimers. J. Med. Chem. 2016;59:3112–3128. doi: 10.1021/acs.jmedchem.5b01894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lensing CJ, Adank DN, Wilber SL, Freeman KT, Schnell SM, Speth RC, Zarth AT, Haskell-Luevano C. A direct in vivo comparison of the melanocortin monovalent agonist Ac-His-DPhe-Arg-Trp-NH2 versus the bivalent agonist Ac-His-DPhe-Arg-Trp-PEDG20-His-DPhe-Arg-Trp-NH2: A bivalent advantage. ACS Chem. Neurosci. 2017;8:1262–1278. doi: 10.1021/acschemneuro.6b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu LP, Josan JS, Vagner J, Caplan MR, Hruby VJ, Mash EA, Lynch RM, Morse DL, Gillies RJ. Heterobivalent ligands target cell-surface receptor combinations in vivo. Proc. Natl. Acad. Sci. U. S. A. 2012;109:21295–21300. doi: 10.1073/pnas.1211762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Josan JS, Handl HL, Sankaranarayanan R, Xu L, Lynch RM, Vagner J, Mash EA, Hruby VJ, Gillies RJ. Cell-specific targeting by heterobivalent ligands. Bioconjugate Chem. 2011;22:1270–1278. doi: 10.1021/bc1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barkey NM, Tafreshi NK, Josan JS, De Silva CR, Sill KN, Hruby VJ, Gillies RJ, Morse DL, Vagner J. Development of melanoma-targeted polymer micelles by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. J. Med. Chem. 2011;54:8078–8084. doi: 10.1021/jm201226w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brabez N, Saunders K, Nguyen KL, Jayasundera T, Weber C, Lynch RM, Chassaing G, Lavielle S, Hruby VJ. Multivalent interactions: Synthesis and evaluation of melanotropin multimers-tools for melanoma targeting. ACS Med. Chem. Lett. 2013;4:98–102. doi: 10.1021/ml300312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brabez N, Lynch RM, Xu LP, Gillies RJ, Chassaing G, Lavielle S, Hruby VJ. Design, synthesis, and biological studies of efficient multivalent melanotropin ligands: Tools toward melanoma diagnosis and treatment. J. Med. Chem. 2011;54:7375–7384. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach JC, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FPJ, Sijbrands EJG, de Rooij FWM, Lebwohl M, Naik H, Goding CR, Wilson JHP, Desnick RJ. Afamelanotide for erythropoietic protoporphyria. N. Engl. J. Med. 2015;373:48–59. doi: 10.1056/NEJMoa1411481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Penagarikano O, Lazaro MT, Lu XH, Gordon A, Dong HM, Lam HA, Peles E, Maidment NT, Murphy NP, Yang XW, Golshani P, Geschwind DH. Exogenous and evoked oxytocin restores social behavior in the Cntnap2 mouse model of autism. Sci. Transl. Med. 2015;7:271ra8. doi: 10.1126/scitranslmed.3010257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barrett CE, Modi ME, Zhang BC, Walum H, Inoue K, Young LJ. Neonatal melanocortin receptor agonist treatment reduces play fighting and promotes adult attachment in prairie voles in a sex-dependent manner. Neuropharmacology. 2014;85:357–366. doi: 10.1016/j.neuropharm.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uckert S, Bannowsky A, Albrecht K, Kuczyk MA. Melanocortin receptor agonists in the treatment of male and female sexual dysfunctions: results from basic research and clinical studies. Expert Opin. Invest. Drugs. 2014;23:1477–1483. doi: 10.1517/13543784.2014.934805. [DOI] [PubMed] [Google Scholar]
- 51.Clayton AH, Althof SE, Kingsberg S, DeRogatis LR, Kroll R, Goldstein I, Kaminetsky J, Spana C, Lucas J, Jordan R, Portman DJ. Bremelanotide for female sexual dysfunctions in premenopausal women: A randomized, placebo-controlled dose-finding trial. Womens Health (Lond Engl) 2016;12:325–337. doi: 10.2217/whe-2016-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kingsberg S, Jordan R, Clayton A, Krychman M. Bremelanotide for hypoactive sexual desire disorder: Analyses from a phase 2b dose-ranging study. J. Sex. Med. 2015;12:389–389. [Google Scholar]
- 53.Giuliani D, Neri L, Canalini F, Calevro A, Ottani A, Vandini E, Sena P, Zaffe D, Guarini S. NDP-alpha-MSH induces intense neurogenesis and cognitive recovery in Alzheimer transgenic mice through activation of melanocortin MC4 receptors. Mol. Cell. Neurosci. 2015;67:13–21. doi: 10.1016/j.mcn.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 54.Giuliani D, Bitto A, Galantucci M, Zaffe D, Ottani A, Irrera N, Neri L, Cavallini GM, Altavilla D, Botticelli AR, Squadrito F, Guarini S. Melanocortins protect against progression of Alzheimer's disease in triple-transgenic mice by targeting multiple pathophysiological pathways. Neurobiol. Aging. 2014;35:537–547. doi: 10.1016/j.neurobiolaging.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 55.Joppa MA, Ling N, Chen C, Gogas KR, Foster AC, Markison S. Central administration of peptide and small molecule MC4 receptor antagonists induce hyperphagia in mice and attenuate cytokine-induced anorexia. Peptides. 2005;26:2294–2301. doi: 10.1016/j.peptides.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 56.Deboer MD, Marks DL. Cachexia: Lessons from melanocortin antagonism. Trends Endocrinol. Metab. 2006;17:199–204. doi: 10.1016/j.tem.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 57.Doering SR, Todorovic A, Haskell-Luevano C. Melanocortin antagonist tetrapeptides with minimal agonist activity at the mouse melanocortin-3 receptor. ACS Med. Chem. Lett. 2015;6:123–127. doi: 10.1021/ml500340z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ericson MD, Wilczynski A, Sorensen NB, Xiang Z, Haskell-Luevano C. Discovery of a beta-hairpin octapeptide, c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro], mimetic of agouti-related protein(87–132) [AGRP(87–132)] with equipotent mouse melanocortin-4 receptor (mMC4R) antagonist pharmacology. J. Med. Chem. 2015;58:4638–4647. doi: 10.1021/acs.jmedchem.5b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fleming KA, Ericson MD, Freeman KT, Adank DN, Lunzer MM, Wilber SL, Haskell-Luevano C. Structure-activity relationship studies of a macrocyclic AGRP-mimetic scaffold c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] yield potent and selective melanocortin-4 receptor antagonists and melanocortin-5 receptor inverse agonists that increase food intake in mice. ACS Chem. Neurosci. doi: 10.1021/acschemneuro.7b00495. [Online early access]. Published Online: January 24, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Irani BG, Xiang Z, Yarandi HN, Holder JR, Moore MC, Bauzo RM, Proneth B, Shaw AM, Millard WJ, Chambers JB, Benoit SC, Clegg DJ, Haskell-Luevano C. Implication of the melanocortin-3 receptor in the regulation of food intake. Eur. J. Pharmacol. 2011;660:80–87. doi: 10.1016/j.ejphar.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat. Genet. 1999;21:119–122. doi: 10.1038/5070. [DOI] [PubMed] [Google Scholar]
- 62.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 63.Shinyama H, Masuzaki H, Fang H, Flier JS. Regulation of melanocortin-4 receptor signaling: Agonist-mediated desensitization and internalization. Endocrinology. 2003;144:1301–1314. doi: 10.1210/en.2002-220931. [DOI] [PubMed] [Google Scholar]
- 64.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 65.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nat. Genet. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 66.Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O'Rahilly S, Farooqi IS. Modulation of blood pressure by central melanocortinergic pathways. N. Engl. J. Med. 2009;360:44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 67.Van der Ploeg LH, Martin WJ, Howard AD, Nargund RP, Austin CP, Guan X, Drisko J, Cashen D, Sebhat I, Patchett AA, Figueroa DJ, DiLella AG, Connolly BM, Weinberg DH, Tan CP, Palyha OC, Pong SS, MacNeil T, Rosenblum C, Vongs A, Tang R, Yu H, Sailer AW, Fong TM, Huang C, Tota MR, Chang RS, Stearns R, Tamvakopoulos C, Christ G, Drazen DL, Spar BD, Nelson RJ, MacIntyre DE. A role for the melanocortin 4 receptor in sexual function. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carrithers MD, Lerner MR. Synthesis and characterization of bivalent peptide ligands targeted to G-protein-coupled receptors. Chem. Biol. 1996;3:537–542. doi: 10.1016/s1074-5521(96)90144-1. [DOI] [PubMed] [Google Scholar]
- 69.Elshan NGRD, Jayasundera T, Anglin BL, Weber CS, Lynch RM, Mash EA. Trigonal scaffolds for multivalent targeting of melanocortin receptors. Org. Biomol. Chem. 2015;13:1778–1791. doi: 10.1039/c4ob02094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash EA, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent NDP-alpha-MSH(7) peptide ligands for binding to the human melanocortin receptor 4 (hMC4R) Bioconjugate Chem. 2007;18:1101–1109. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: Homooligomers of alpha-MSH. Bioorg. Med. Chem. Lett. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 72.Bowen ME, Monguchi Y, Sankaranarayanan R, Vagner J, Begay LJ, Xu L, Jagadish B, Hruby VJ, Gillies RJ, Mash EA. Design, synthesis, and validation of a branched flexible linker for bioactive peptides. J. Org. Chem. 2007;72:1675–1680. doi: 10.1021/jo062276g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jagadish B, Sankaranarayanan R, Xu L, Richards R, Vagner J, Hruby VJ, Gillies RJ, Mash EA. Squalene-derived flexible linkers for bioactive peptides. Bioorg. Med. Chem. Lett. 2007;17:3310–3313. doi: 10.1016/j.bmcl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dehigaspitiya DC, Navath S, Weber CS, Lynch RM, Mash EA. Synthesis and bioactivity of MSH4 oligomers prepared by an A + B strategy. Tetrahedron Lett. 2015;56:3060–3065. doi: 10.1016/j.tetlet.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dehigaspitiya DC, Anglin BL, Smith KR, Weber CS, Lynch RM, Mash EA. Linear scaffolds for multivalent targeting of melanocortin receptors. Org. Biomol. Chem. 2015;13:11507–11517. doi: 10.1039/c5ob01779c. [DOI] [PubMed] [Google Scholar]
- 76.Kopanchuk S, Veiksina S, Mutulis F, Mutule I, Yahorava S, Mandrika I, Petrovska R, Rinken A, Wikberg JE. Kinetic evidence for tandemly arranged ligand binding sites in melanocortin 4 receptor complexes. Neurochem. Int. 2006;49:533–542. doi: 10.1016/j.neuint.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 77.Kopanchuk S, Veiksina S, Petrovska R, Mutule I, Szardenings M, Rinken A, Wikberg JE. Co-operative regulation of ligand binding to melanocortin receptor subtypes: Evidence for interacting binding sites. Eur. J. Pharmacol. 2005;512:85–95. doi: 10.1016/j.ejphar.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 78.Piechowski CL, Rediger A, Lagemann C, Muhlhaus J, Muller A, Pratzka J, Tarnow P, Gruters A, Krude H, Kleinau G, Biebermann H. Inhibition of melanocortin-4 receptor dimerization by substitutions in intracellular loop 2. J. Mol. Endocrinol. 2013;51:109–118. doi: 10.1530/JME-13-0061. [DOI] [PubMed] [Google Scholar]
- 79.Haskell-Luevano C, Hendrata S, North C, Sawyer TK, Hadley ME, Hruby VJ, Dickinson C, Gantz I. Discovery of prototype peptidomimetic agonists at the human melanocortin receptors MC1R and MC4R. J. Med. Chem. 1997;40:2133–2139. doi: 10.1021/jm960840h. [DOI] [PubMed] [Google Scholar]
- 80.Haskell-Luevano C, Holder JR, Monck EK, Bauzo RM. Characterization of melanocortin NDP-MSH agonist peptide fragments at the mouse central and peripheral melanocortin receptors. J. Med. Chem. 2001;44:2247–2252. doi: 10.1021/jm010061n. [DOI] [PubMed] [Google Scholar]
- 81.Holder JR, Bauzo RM, Xiang Z, Haskell-Luevano C. Structure-activity relationships of the melanocortin tetrapeptide Ac-His-DPhe-Arg-Trp-NH(2) at the mouse melanocortin receptors: Part 2 modifications at the Phe position. J. Med. Chem. 2002;45:3073–3081. doi: 10.1021/jm010524p. [DOI] [PubMed] [Google Scholar]
- 82.Chen M, Georgeson KE, Harmon CM, Haskell-Luevano C, Yang YK. Functional characterization of the modified melanocortin peptides responsible for ligand selectivity at the human melanocortin receptors. Peptides. 2006;27:2836–2845. doi: 10.1016/j.peptides.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 83.Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gillies RJ, Hruby VJ. Solid-phase synthesis of heterobivalent ligands targeted to melanocortin and cholecystokinin receptors. Int. J. Pept. Res. Ther. 2008;14:293–300. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-phenylalanine-alpha-melanocyte-stimulating hormone - A highly potent alpha-melanotropin with ultralong biological-activity. Proc. Natl. Acad. Sci. U.S.A. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hruby VJ, Lu DS, Sharma SD, Castrucci AD, Kesterson RA, Alobeidi FA, Hadley ME, Cone RD. Cyclic lactam alpha-melanotropin analogs of Ac-Nle(4)-Cyclo[Asp(5),D-Phe(7)Lys(10)] alpha-melanocyte-stimulating hormone-(4–10)-NH2 with bulky aromatic-amino-acids at position-7 show high antagonist potency and selectivity at specific melanocortin receptors. J. Med. Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 86.Ericson MD, Lensing CJ, Fleming KA, Schlasner KN, Doering SR, Haskell-Luevano C. Bench-top to clinical therapies: A review of melanocortin ligands from 1954 to 2016. Biochim. Biophys. Acta. 2017;1863:2414–2435. doi: 10.1016/j.bbadis.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fernandes SM, Sankaranarayanan R, Handl HL, Josan J, Vagner J, Xu L, Mash E, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent ligands with NDP-alpha-MSH and SHU9119 for melanocortin 4 receptor. Biopolymers. 2007;88:619–619. [Google Scholar]
- 88.Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. J. Am. Chem. Soc. 1970;92:5748–5749. [Google Scholar]
- 89.Stewart JM, Young JD. Solid Phase Peptide Synthesis. 2. Pierce Chemical Co.; Rockford, Il: 1984. [Google Scholar]
- 90.Merrifield RB. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc. 1963;85:2149–2154. [Google Scholar]
- 91.Cai MY, Varga EV, Stankova M, Mayorov A, Perry JW, Yamamura HI, Trivedi D, Hruby VJ. Cell signaling and trafficking of human melanocortin receptors in real time using two-photon fluorescence and confocal laser microscopy: Differentiation of agonists and antagonists. Chem. Biol. Drug Des. 2006;68:183–193. doi: 10.1111/j.1747-0285.2006.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gao ZH, Lei DC, Welch J, Le K, Lin J, Leng S, Duhl D. Agonist-dependent internalization of the human melanocortin-4 receptors in human embryonic kidney 293 cells. J. Pharmacol. Exp. Ther. 2003;307:870–877. doi: 10.1124/jpet.103.055525. [DOI] [PubMed] [Google Scholar]
- 93.Xiang Z, Proneth B, Dirain ML, Litherland SA, Haskell-Luevano C. Pharmacological characterization of 30 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists, synthetic agonists, and the endogenous agouti-related protein antagonist. Biochemistry. 2010;49:4583–4600. doi: 10.1021/bi100068u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xiang Z, Litherland SA, Sorensen NB, Proneth B, Wood MS, Shaw AM, Millard WJ, Haskell-Luevano C. Pharmacological characterization of 40 human melanocortin-4 receptor polymorphisms with the endogenous proopiomelanocortin-derived agonists and the agouti-related protein (AGRP) antagonist. Biochemistry. 2006;45:7277–7288. doi: 10.1021/bi0600300. [DOI] [PubMed] [Google Scholar]
- 95.Schild HO. pA, a new scale for the measurement of drug antagonism. Br. J. Pharmacol. Chemother. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stephenson RP. A modification of receptor theory. Br. J. Pharmacol. Chemother. 1956;11:379–393. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Takeyasu K, Uchida S, Wada A, Maruno M, Lai RT, Hata F, Yoshida H. Experimental evidence and dynamic aspects of spare receptor. Life Sci. 1979;25:1761–1771. doi: 10.1016/0024-3205(79)90480-6. [DOI] [PubMed] [Google Scholar]
- 98.Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguere PM, Sciaky N, Roth BL. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015;22:362–U328. doi: 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, Lee KJ. The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U. S. A. 2008;105:64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nickolls SA, Fleck B, Hoare SRJ, Maki RA. Functional selectivity of melanocortin 4 receptor peptide and nonpeptide agonists: Evidence for ligand-specific conformational states. J. Pharmacol. Exp. Ther. 2005;313:1281–1288. doi: 10.1124/jpet.105.083337. [DOI] [PubMed] [Google Scholar]
- 101.Breit A, Buch TRH, Boekhoff I, Solinski HJ, Damm E, Gudermann T. Alternative G protein coupling and biased agonism: New insights into melanocortin-4 receptor signalling. Mol. Cell. Endocrinol. 2011;331:232–240. doi: 10.1016/j.mce.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 102.Yang Z, Tao YX. Biased signaling initiated by agouti-related peptide through human melanocortin-3 and -4 receptors. Biochim. Biophys. Acta. 2016;1862:1485–1494. doi: 10.1016/j.bbadis.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 103.Yang LK, Tao YX. Biased signaling at neural melanocortin receptors in regulation of energy homeostasis. Biochim. Biophys. Acta. 2017;1863:2486–2495. doi: 10.1016/j.bbadis.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Violin JD, Crombie AL, Soergel DG, Lark MW. Biased ligands at G-protein-coupled receptors: Promise and progress. Trends Pharmacol. Sci. 2014;35:308–316. doi: 10.1016/j.tips.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 105.Marti-Solano M, Schmidt D, Kolb P, Selent J. Drugging specific conformational states of GPCRs: Challenges and opportunities for computational chemistry. Drug Discovery Today. 2016;21:625–631. doi: 10.1016/j.drudis.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 106.Durroux T. Principles: A model for the allosteric interactions between ligand binding sites within a dimeric GPCR. Trends Pharmacol. Sci. 2005;26:376–384. doi: 10.1016/j.tips.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 107.Casado V, Cortes A, Ciruela F, Mallol J, Ferre S, Lluis C, Canela EI, Franco R. Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: The receptor-dimer cooperativity index. Pharmacol. Ther. 2007;116:343–354. doi: 10.1016/j.pharmthera.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 108.Tabor A, Weisenburger S, Banerjee A, Purkayastha N, Kaindl JM, Hubner H, Wei LX, Gromer TW, Kornhuber J, Tschammer N, Birdsall NJM, Mashanov GI, Sandoghdar V, Gmeiner P. Visualization and ligand-induced modulation of dopamine receptor dimerization at the single molecule level. Sci. Rep. 2016;6:33233. doi: 10.1038/srep33233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pfleger KD, Seeber RM, Eidne KA. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat. Protoc. 2006;1:337–345. doi: 10.1038/nprot.2006.52. [DOI] [PubMed] [Google Scholar]
- 110.Mandrika I, Petrovska R, Wikberg J. Melanocortin receptors form constitutive homo- and heterodimers. Biochem. Biophys. Res. Commun. 2005;326:349–354. doi: 10.1016/j.bbrc.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 111.Nickolls SA, Maki RA. Dimerization of the melanocortin 4 receptor: A study using bioluminescence resonance energy transfer. Peptides. 2006;27:380–387. doi: 10.1016/j.peptides.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 112.Cottet M, Faklaris O, Maurel D, Scholler P, Doumazane E, Trinquet E, Pin JP, Durroux T. BRET and time-resolved FRET strategy to study GPCR oligomerization: From cell lines toward native tissues. Front. Endocrinol. (Lausanne) 2012;3:92. doi: 10.3389/fendo.2012.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grant M, Collier B, Kumar U. Agonist-dependent dissociation of human somatostatin receptor 2 dimers: A role in receptor trafficking. J. Biol. Chem. 2004;279:36179–36183. doi: 10.1074/jbc.M407310200. [DOI] [PubMed] [Google Scholar]
- 114.Albizu L, Cottet M, Kralikova M, Stoev S, Seyer R, Brabet I, Roux T, Bazin H, Bourrier E, Lamarque L, Breton C, Rives ML, Newman A, Javitch J, Trinquet E, Manning M, Pin JP, Mouillac B, Durroux T. Time-resolved FRET between GPCR ligands reveals oligomers in native tissues. Nat. Chem. Biol. 2010;6:587–594. doi: 10.1038/nchembio.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zheng Y, Akgun E, Harikumar KG, Hopson J, Powers MD, Lunzer MM, Miller LJ, Portoghese PS. Induced association of mu opioid (MOP) and type 2 cholecystokinin (CCK2) receptors by novel bivalent ligands. J. Med. Chem. 2009;52:247–258. doi: 10.1021/jm800174p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Journe AS, Habib SAM, Dodda BR, Morcos MNF, Sadek MS, Tadros SAA, Witt-Enderby PA, Jockers R, Zlotos DP. N1-linked melatonin dimers as bivalent ligands targeting dimeric melatonin receptors. Medchemcomm. 2014;5:792–796. [Google Scholar]
- 117.Russo O, Berthouze M, Giner M, Soulier JL, Rivail L, Sicsic S, Lezoualc'h F, Jockers R, Berque-Bestel I. Synthesis of specific bivalent probes that functionally interact with 5-HT(4) receptor dimers. J. Med. Chem. 2007;50:4482–4492. doi: 10.1021/jm070552t. [DOI] [PubMed] [Google Scholar]
- 118.Broussard JA, Rappaz B, Webb DJ, Brown CM. Fluorescence resonance energy transfer microscopy as demonstrated by measuring the activation of the serine/threonine kinase Akt. Nat. Protoc. 2013;8:265–281. doi: 10.1038/nprot.2012.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl amino-protecting group. J. Org. Chem. 1972;37:3404–3409. [Google Scholar]
- 120.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 121.Christensen T. Qualitative test for monitoring coupling completeness in solid-phase peptide-synthesis using chloranil. Acta Chem. Scand. 1979;33:763–766. [Google Scholar]
- 122.Chen CA, Okayama H. Calcium phosphate-mediated gene transfer: a highly efficient transfection system for stably transforming cells with plasmid DNA. Biotechniques. 1988;6:632–638. [PubMed] [Google Scholar]
- 123.Singh A, Tala SR, Flores V, Freeman K, Haskell-Luevano C. Synthesis and pharmacology of alpha/beta(3)-peptides based on the melanocortin agonist Ac-His-DPhe-Arg-Trp-NH2 Sequence. ACS Med. Chem. Lett. 2015;6:568–572. doi: 10.1021/acsmedchemlett.5b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.