

Abstract
Erythrocytes bind circulating immune complexes (IC) and facilitate IC clearance from the circulation. Chronic hepatitis C virus (HCV) infection is associated with IC-related disorders. In this study we investigated the kinetics and mechanism of HCV and HCV-IC binding to and dissociation from erythrocytes. Cell culture-produced HCV was mixed with erythrocytes from healthy blood donors and erythrocyte–associated virus particles were quantified. Purified complement proteins, complement-depleted serum, and complement receptor antibodies were used to investigate complement-mediated HCV-erythrocyte binding. Purified HCV-specific immunoglobulin G from a chronic HCV-infected patient was used to study complement-mediated HCV-IC-erythrocyte binding. Binding of HCV to erythrocytes increased 200 to 1,000 fold after adding complement active human serum in the absence of antibody. Opsonization of free HCV occurred within 10 minutes and peak binding to erythrocytes was observed at 20-30 minutes. Complement protein C1 was required for binding, while C2, C3 and C4 significantly enhanced binding. Complement receptor 1 (CR1, CD35) antibodies blocked the binding of HCV to erythrocytes isolated from chronically infected HCV patients and healthy blood donors. HCV-ICs significantly enhanced complement-mediated binding to erythrocytes compared to unbound HCV. Dissociation of complement-opsonized HCV from erythrocytes depended on the presence of Factor I. HCV released by Factor I bound preferentially to CD19+ B cells compared to other leukocytes. Conclusion: These results demonstrate that complement mediates the binding of free and IC-associated HCV to CR1 on erythrocytes, and provide a mechanistic rationale for investigating the differential phenotypic expression of HCV-IC-related disease.
Keywords: HCV, complement, erythrocytes, CD35, Immune Complex
Introduction
Hepatitis C virus (HCV) infection is a major global health burden, with up to 150 million people chronically infected.(1) Chronic HCV infection is among the most common causes of liver cirrhosis and hepatocellular carcinoma (HCC), and is the primary reason for liver transplantation in the United States (US).(2-5) Although the liver is the main site of HCV replication and injury, a broad range of HCV-related extrahepatic clinical manifestations exist and are often associated with immune complex (IC) containing viral particles. (6-8)
One of the important innate immune surveillance functions of the complement system is to promote the binding of immune complexes to complement receptor type 1 (CR1). (9) Since erythrocytes contain over 90% of the circulating CR1 pool, they may play an important role in immune complex clearance.(10) Erythrocyte CR1 (E-CR1) can bind complement proteins C3b, C4b and C1q.(11-14) E-CR1 facilitates IC removal from the circulation and prevents IC deposition by shuttling ICs and complement opsonized particles to the phagocytic systems of the liver and spleen.(15) Dysregulation of E-CR1 may play a role in the pathogenesis of IC-mediated diseases. Several reports have shown that patients with systemic lupus erythematosus (SLE), autoimmune hemolytic anemia and Sjögren’s syndrome had reduced E-CR1 levels.(16-18) Defects in complement activation and erythrocyte-mediated IC clearance are thought to be central to pathogenesis in SLE.(19)
We and another group previously demonstrated that free HCV particles from chronically infected patients could be complement-opsonized and bound to CR1 on B lymphocytes.(20-22) Chronic HCV is associated with IC-related diseases such as mixed cryoglobulinemia, membranous glomerulonephritis and non-Hodgkin lymphoma. In this study, we investigated the kinetics and mechanism of HCV and HCV-IC binding to and dissociation from human erythrocytes.
Patients and Methods
Detailed descriptions of our patients and methods used for cell culture, in vitro RNA synthesis, HCV production in cell culture, conversion of plasma to serum and PBMCs isolation were described in our previous studies.(20, 22) Ethics committees of the American Red Cross and the NIH approved the study protocol in accordance with the Declaration of Helsinki and the study has been reviewed annually by an NIH Institutional Review Board (NIH Protocol 91-CC-0017). All subjects provided written informed consent to participate in the study. Methods for in vitro HCV binding assays with complement-depleted sera were described in the Supporting Information.
Isolation of human erythrocytes
Buffy coats from healthy blood donors or whole blood from chronically infected HCV patients were obtained for isolation of erythrocytes by using Ficoll-Pague density gradient centrifugation method. After removing the plasma layer and interphase layer, the rest of the Ficoll layer and the top layer of erythrocytes were also removed. The remaining erythrocytes were washed twice with 1 × PBS, pH 7.4 by centrifugation at 470× g and 210× g for 10 minutes each at room temperature (25°C) for the first wash and second wash, respectively. After centrifugation, removed the supernatant along with the top thin layer of cells from erythrocytes in each wash. The erythrocytes were further washed twice with RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 unit/mL penicillin, and 100 μg/mL streptomycin (complete RPMI medium) and collected by centrifugation at 210× g for 10 minutes at 25°C. Finally, the erythrocytes were resuspended in complete RPMI 1640 medium, counted, and adjusted the cells concentrations to 1 × 109 E/mL.
HCV production in cell culture
The growth of human hepatoma cell line Huh7.5.1 and the preparation of full-length HCV1a (H77S) RNA were performed as previously described (22) with minor modifications. Briefly, 42 μg of HCV1a (H77S) full-length RNA was transfected into 1.2 × 107 Huh 7.5.1 cells in two 25×150mm culture dishes by using mRNA boost reagent and TranslT-mRNA reagent (Mirus, MIR2250) according to the manufacturer’s instructions. Eight hours after transfection, the transfection culture medium was removed and the cells were washed once with complete DMEM medium without antibiotics and cultured in 50 mL of the same medium per dish for 16 hours. Cells were then trypsinized and seeded into 25 × 150 mm culture dishes at 5.0 × 106 cells per dish with 50 mL complete DMEM medium. The virus producing cells were continuously sub-cultured every 2-3 days for 21 days post transfection by seeding 5 × 106 cells per 25 × 150mm culture dish with 50 mL complete DMEM medium. Before each sub-culturing, the culture supernatant was collected and filtered through 0.45 μm sterile filtration units. The filtrates were aliquoted and stored at −80°C before use. Typically, the genomic copy number of HCV in the supernatant was 1.0-3.0 × 1 07 copies per mL, and the culture supernatants collected between days 8 and days 21 were used in this study.
HCV binding to human erythrocytes
In our standard binding assay, 3 mL of virus (1 to 3 × 107 genomic copies for HCV genotype 1a), was mixed with 100 μL of serum (about 20-25 CH50 units) to initiate complement activation (22) and then incubated at 25°C for 30 minutes. Next, 2 mL of erythrocytes (5 × 108 cells total) were added to the mixture and incubated for 15 minutes. The reaction was carried out in 15 mL sterile tubes with occasional mixing. After incubation, EDTA was added to a final concentration of 20 mM, and the reaction mixtures were immediately cooled down for 5 minutes in an ice bath. The cells were pelleted by centrifugation at 470 × g for 6 minutes at room temperature, and washed three times with 10 mL 1 × phosphate buffered saline (pH 7.4). The cell pellets were resuspended by pipetting, brought to a 200 μL volume with 1 × PBS, and then mixed with 600 μL of Blood RNA buffer (ZR Whole-Blood RNA MiniPrep kit, Zymo Research, Irvine, CA). Total RNA was isolated according to the manufacturer’s instructions, and each sample was eluted from the column with 50 μL of RNA diluent II, which contained nuclease-free water (Thermo Fisher Scientific, Waltham, MA) supplemented with 1.0 mM dithiothreitol and 200 units/mL RNasin® (Promega, Madison, WI).
HCV released from erythrocytes
In our standard assay (unless otherwise stated), HCV1a (H77s) virus (1 × 107 genomic copies total) or HCV2a (JFH1) virus (3.24 × 108 genomic copies total) in 3 mL of medium was incubated with 50 μL of Factor I-depleted serum for 30 minutes at 25°C, followed by adding 2 mL of erythrocytes (5 × 108 cells total). The reaction was carried out for 2 hours at 25°C. After incubation, EDTA was added to a final concentration of 20 mM. The cells were then pelleted by centrifugation at 470 × g for 5 minutes, washed twice with 10 mL 1× phosphate buffered saline (pH 7.4) each time, and followed by one wash with 10 mL complete RPMI medium. The washed erythrocytes were resuspended in 5 mL complete RPMI medium, and 4 μg of purified Factor I protein was added and incubated at 25°C and 37°C with variable times as indicated or at 25°C for 2 hours. The supernatants containing released virus particles from erythrocytes were collected by centrifugation at 470× g for 5 minutes at each time point or after 2 hours incubation. The viral RNA from the supernatants and cell pellets was isolated by using QIAamp Viral RNA Mini kit (Qiagen) and ZR Whole-Blood RNA MiniPrep kit (Zymo Research), respectively, according to the manufacturer’s instructions. Each sample was eluted from the column with 50 μL of RNA diluent II.
Assay for HCV binding to B cells
In vitro assay for HCV binding to B cells was conducted as described in our previous study with slight modifications. (22) Briefly, HCV2a virus or HCV1a virus (free virus in medium) released from erythrocytes by Factor I treatment in 3 mL medium was mixed with 2 mL of PBMCs (5 × 107 cells total) or pretreated PBMCs (5 × 107 cells total, at 25°C for 30 minutes with 5 μg antibodies), respectively, and incubated at 25°C for 1 hour. The reaction was carried out in 50 mL sterile tubes with occasional mixing. After incubation, the cells were pelleted by centrifugation, washed once with 10 mL of magnetic cell sorting (MACS) buffer (Miltenyi Biotec Inc., Auburn, CA) supplemented with 0.5% bovine serum albumin, centrifuged again, and resuspended in 400 μL of magnetic cell sorting (MACS) buffer supplemented with 0.5% bovine serum albumin. The cells were mixed with 100 μL of mouse antihuman CD19 or CD14 or CD3, or CD56 magnetic microbeads (Miltenyi Biotec Inc., Auburn, CA) and incubated at 4°C for 20 minutes. The cells were then diluted with 10 mL of MACS buffer, collected by centrifugation, and resuspended in 1 mL of MACS buffer. The cell suspension was applied to an MS column (Miltenyi Biotec Inc.) on a magnetic field separator. The column was washed three times with 1 mL of MACS buffer each time. The column was separated from the magnetic stand and placed on a 2 mL sterile tube. The CD19+ B cells, or CD14+ monocytes and macrophages cells, or CD3+ T cells or CD56+ NK cells were flushed from the column with 1 mL of MACS buffer, collected by centrifugation, and resuspended in 650 μL of RNeasy Plus lysis buffer (Qiagen, Chatsworth, CA) supplemented with 1% 2-mercaptoethanol. RNA isolation was carried out using the RNeasy Plus Mini kit (Qiagen) according to the manufacturer’s instructions, and each sample was eluted from the column with 50 μL of RNA diluent II.
HCV RNA quantification
HCV genomic copy numbers in purified total cellular RNA samples and/or culture medium (supernatants) were quantified using the primers and probes as described previously.(22, 23) Briefly, each sample was quantified in triplicate using a TaqMan Fast Virus 1-Step Master Mix (Thermo Fisher Scientific, Waltham, MA) with 5 μl RNA input per reaction in a 25 μl mixture. The PCR reaction conditions were: 50°C for 2 minutes, 60°C for 30 minutes, 95°C for 3 minutes, followed by 50 cycles at 95°C for 20 sec and 60°C for 1 minute using ABI 7900HT system. The copies of HCV RNA were determined by in vitro transcribed HCV1a RNA standards (20) with the Sequence Detector Software (version 2.2; Applied Biosystems) and normalized to 1 μg total RNA or per mL culture medium input.
Preparation of heat aggregated human globulin (AHG)
Human polyclonal immunoglobulin G (IgG) against HCV was isolated from a chronic HCV patient’s plasma according to a previously described method.(24) Negative control human IgG was purified from the plasma of a healthy blood donor using the same method. The production of heat aggregated human globulin was carried out as described previously.(25) The IgG solution was prepared in 0.9% NaCl at 6 mg/mL, incubated at 63°C for 30 minutes with occasional shaking, and followed by immediate cooling in an ice bath. The IgG solution was clarified by centrifugation at 1500 × g for 15 minutes at 4°C, the concentration was adjusted to 5.0 mg/mL with 0.9% NaCl solution and it was stored at −80°C until use.
Statistical analysis
Quantitative analysis of the HCV genome copy number assessed by qPCR is expressed as the mean ± standard deviation. An unpaired Student t test was used to determine statistical significance. Values of P < 0.05 were judged significant. Data were analyzed and graphs created with GraphPad Prism 7 (GraphPad Software, La Jolla, CA).
Results
Complement-mediated binding of HCV to erythrocytes
We previously developed an in vitro model system for studying HCV binding to CD19- expressing B cells from PBMCs, and found that the complement system mediates this interaction.(22)
In the current study, we first evaluated whether the most commonly used cell culture-produced HCV, viral genotypes 1a (H77s) and 2a (Japanese fulminant hepatitis 1; JFH1), could bind to erythrocytes in our model system. We performed a standard binding assay, and observed that both HCV 1a and 2a viruses became robustly bound to erythrocytes (P < 0.0001) after mixing with complement-active serum (Fig. 1A). In the absence of serum, the binding of HCV to erythrocytes was minimally detectable.
FIG. 1.
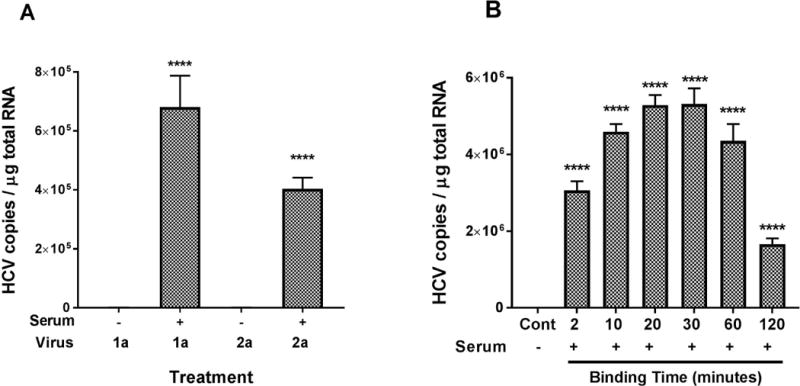
(A) Factors in serum are required for HCV genotype 1a and 2a binding to erythrocytes. Three milliliters medium containing HCV virus as indicated [HCV1a (H77s), 3 × 107 copies total; HCV2a (JFH1), 2 × 108 copies total] were incubated in the absence (100 μL medium only) or in the presence of serum (100 μL) at 25°C for 30 minutes. Then 2 mL of erythrocytes (2.5 × 108 cells/mL) in complete RPMI medium were added to the reaction mixture and incubated further at 25°C for 15 minutes. Finally, a final concentration of 20 mM EDTA was added to stop the reaction, and the reaction mixture was immediately cooled for 5 minutes in an ice bath. Erythrocytes were collected by centrifugation and washed three times with 1× PBS, pH7.4. Total RNA isolation from erythrocytes and quantification of HCV RNA were performed as described in the Methods section. HCV associated with erythrocytes was significantly higher for both genotypes in the presence of serum. (B) Kinetic profiles for HCV binding to erythrocytes. For the optimal binding time, 3 mL medium containing HCV1a virus (1.5 × 107 copies total) were treated in the presence of serum (100 μL) at 25°C for 30 minutes. The erythrocytes (5 × 108 cells total) in 2 mL RPMI medium were then added and the incubation continued at 25°C with variable times as stated. A control (Cont) experiment without serum (100 μL medium only) was performed for each assay, and incubated at 25°C for the longest time point as indicated. After adding EDTA to a final concentration of 20 mM to stop the reaction, and the reaction mixture was immediately cooled for 5 minutes in an ice bath. Erythrocytes were collected by centrifugation and washed three times with 1× PBS, pH7.4. Total RNA isolation from erythrocytes and quantification of HCV RNA were performed as described in the Methods section. HCV associated with erythrocytes was significantly higher at each time point in the presence of serum. Each value represents the mean ± standard deviation of six determinations. The data are representative of two independent experiments using erythrocytes from at least two different healthy donors. **** P < 0.0001, compared to absence of serum; control (serum absent).
Kinetics of HCV binding to erythrocytes
To determine the optimal binding time, time-course experiments were performed. Opsonized HCV1a virus bound to erythrocytes within 2 minutes (Fig. 1B). Maximum binding of opsonized HCV to erythrocytes was observed after 20 minutes of incubation and then binding decreased significantly after 30 minutes. Similar results were obtained with HCV2a virus (Supporting Fig. S1).
Complement pathways involved in the binding of HCV to erythrocytes
To investigate which complement pathways were involved in the binding of HCV to erythrocytes, we used commercially available complement-depleted serum and purified complement proteins. C1 is a multimeric protein that initiates the classical complement pathway and is composed of a recognition subunit (C1q) and two protease subunits (C1r and C1s). When HCV binding experiments were performed with C1q-depleted serum samples, HCV binding activity was diminished to bare detectability (Fig. 2A). When C1q-depleted serum was supplemented with purified C1q protein, the binding of HCV to erythrocytes was fully restored, suggesting that C1q protein is essential for promoting the binding of HCV to erythrocytes.
FIG. 2.
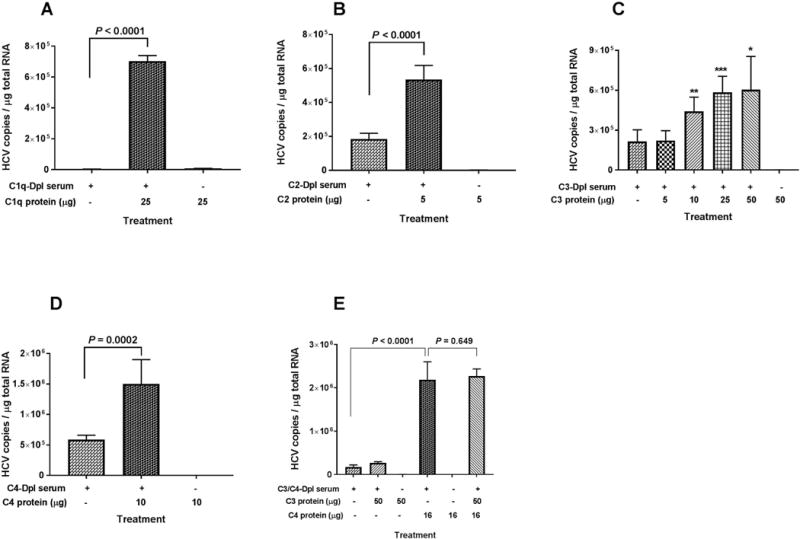
Purified complement proteins C1q, C2, C3, and C4 added to the indicated complement-depleted serum can restore HCV binding to erythrocytes. (A-E) Three milliliters of medium containing HCV1a virus (1 to 3 × 107 genomic copies total) were treated with 100 μL of the indicated complement-depleted serum sample, complement-depleted serum sample plus the indicated purified complement protein, or purified complement protein only. After 30 minutes incubation at 25°C, 2 mL of erythrocytes (5 × 108 cells total) in complete RPMI medium were added to the reaction mixture and incubated further at 25°C for 15 minutes. After adding EDTA to a final concentration of 20 mM, the erythrocytes were collected by centrifugation and washed three times with 1× PBS, pH7.4. Total RNA isolation from erythrocytes and quantification of HCV RNA were performed as described in Methods section. Each value represents the mean ± standard deviation of six determinations. The data are representative of two independent experiments using erythrocytes from at least two different healthy donors. Dpl, depleted.
In experiments with C2 or C4-depleted serum, the binding activities were reduced by 60-65% compared to the respective serum samples supplemented with purified proteins (Fig. 2B and 2D). None of the purified proteins were able to promote the binding of HCV to erythrocytes in the absence serum (Fig. 2A, 2B, and 2D).
In C3 reconstitution experiments, the binding activity of HCV to erythrocytes occurred in a dose-dependent manner when increasing concentrations of purified C3 protein were added to the C3-depleted serum (Fig. 2C). There was no binding in the absence of serum, even when higher amounts of purified C3 protein were added.
In C5 reconstitution experiments, the binding activity of HCV to erythrocytes declined when increasing concentrations of C5 protein were added to the C5-depleted serum, suggested that C5 is not required for this binding (Supporting Fig. S2).
Complement receptor type 1 (CR1, CD35) on erythrocytes can bind C3b and C4b. It was thus of interest to explore which complement activation fragments of these molecules play a critical role in HCV binding to erythrocytes. To address this question, complement C3/C4 reconstitution experiments were performed. We observed that addition of C3 to C3/C4-depleted serum did not affect binding of HCV to erythrocytes, while addition of C4 significantly increased binding (Fig. 2E). There was no significant change when we compared binding after addition of C3 and C4 to the addition of C4 alone (Fig. 2E). These results suggest that complement fragment C4b is important to the association of HCV with erythrocytes, most likely via CR1 (see below), whereas complement fragment C3b does not appear to play a significant role.
Receptors involved in the binding of HCV to erythrocytes
To confirm that the binding of opsonized HCV to erythrocytes occurred via CR1, the opsonized HCV particles were mixed with erythrocytes pre-incubated with antibodies (6 μg) against different complement receptors and cell surface receptors (Fig. 3), including CD11b (part of complement receptor 3), CD21 (complement receptor 2), CD32 (receptor for the Fc fragment of immunoglobulin G), CD47 (integrin-associated protein, IAP), and CD35 (complement receptor 1). Only anti-CD35 significantly blocked HCV binding to erythrocytes. Anti-CD35 blocking activity was demonstrated at various concentrations of antibody ranging from 1 to 10 μg (Supporting Fig. S3).
FIG. 3.
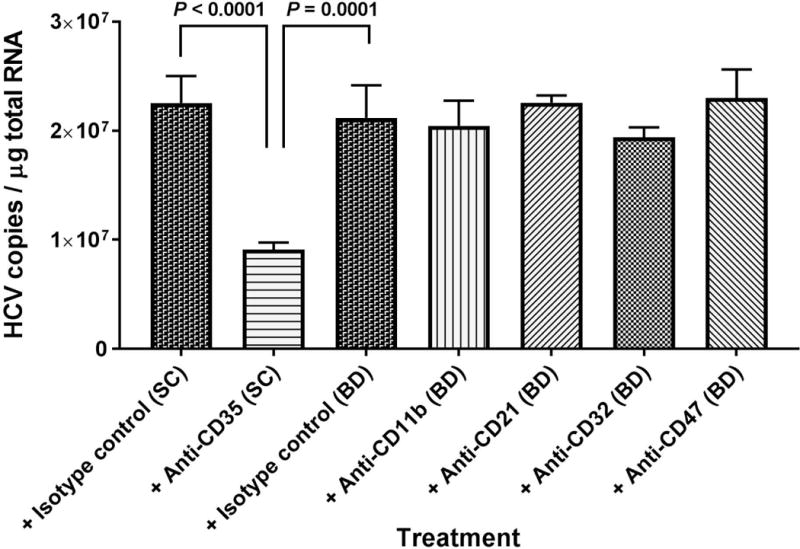
Antibodies against CR1 (anti-CD35) block binding of HCV to erythrocytes. The erythrocytes (5 × 108 cells total) in 2 mL of RPMI medium were mixed with 6 μg antibodies as stated and incubated at 25°C for 30 minutes. A mixture of complement opsonized HCV1a virus particles (pre-incubation at 25°C for 30 minutes, 1.5 × 107 copies total) were added and incubated further at 25°C for 15 minutes. After adding EDTA to a final concentration of 20 mM, the erythrocytes were collected by centrifugation and washed three times with 1× PBS, pH7.4. Total RNA isolation from erythrocytes and quantification of HCV RNA were performed as described in the Methods section. Each value represents the mean ± standard deviation of six determinations. The data are representative of two independent experiments using erythrocytes from at least two different healthy donors. SC, Santa Cruz; BD, BD Biosciences.
Effect of anti-CD35 (CR1) antibody on the binding of HCV to erythrocytes from patients chronically infected with HCV
To investigate whether complement-mediated binding of HCV to erythrocytes occurred on red cells derived from HCV-infected patients, we performed binding experiments with erythrocytes isolated from four patients with chronic HCV in the presence or absence of antibodies against CR1 (anti-CD35). Complement-opsonized HCV particles prepared by our in vitro methods efficiently bound to erythrocytes from HCV-infected patients and pretreatment with anti-CR1 antibodies significantly blocked binding activity (Fig. 4A-D).
FIG. 4.
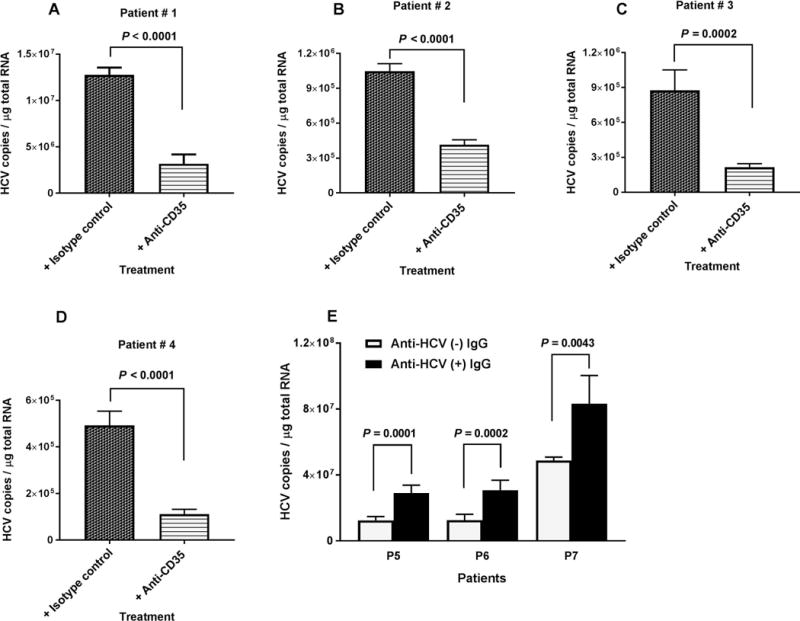
Effects of anti-CD35 antibody and HCV specific polyclonal antibodies on HCV binding to erythrocytes. (A-D) Erythrocytes from four chronically infected HCV patients in 2 mL of RPMI medium (5 × 108 cells total) were incubated with 4 to 6 μg antibodies as indicated at 25°C for 30 minutes, followed by adding 3 mL of complement opsonized HCV1a virus (pre-incubation for 30 minutes at 25°C, 1 to 1.5 × 107 genomic copies total) and incubating further at 25°C for 15 minutes. (E) Three milliliters of HCV1a (1 × 107 genomic copies total) were incubated with heat aggregated IgG (100 μg/mL) as indicated at 25°C for 30 minutes, followed by mixing with 100 μL serum and incubating at 25°C for 30 minutes. Then, 2 mL erythrocytes (2.5 × 108 cells per mL), isolated from three chronically infected HCV patients were added. The reaction was carried out at room temperature (25°C) for 15 minutes. After adding EDTA to a final concentration of 20 mM, the erythrocytes were collected by centrifugation and washed three times with 1× PBS, pH7.4. Total RNA isolation from erythrocytes and quantification of HCV RNA were performed as described in the Methods section. Each value represents the mean ± standard deviation of six determinations.
Effects of immune complexes consisting of HCV-specific antibodies on the binding of HCV to erythrocytes
The above experiments were mainly focused on antibody-independent activation of the complement system by HCV. However, most published studies (26, 27) have described antibody-dependent binding between erythrocyte CR1 and circulating ICs. To investigate the role of HCV-specific antibodies in the binding of HCV to erythrocytes, we first incubated HCV with IgG from a healthy blood donor or with HCV-specific IgG from a patient chronically infected with HCV who had high titer neutralizing antibody activity (50% reduction of JFH1 infectivity at 1 μg/mL IgG) to HCV as measured by an in vitro infectivity assay. After incubation with antibody, complement-active serum was added to opsonize HCV particles, and binding activity to erythrocytes was then measured and compared. Complement-mediated binding of HCV to erythrocytes was significantly enhanced in the presence of antigen-specific antibodies (Fig. 4E). From binding experiments that restored C1q protein to C1q-depleted serum we found that in the presence of HCV antibodies, HCV binding to erythrocytes was significantly enhanced compared to binding in the absence of HCV antibodies (Supporting Fig. S4).
Factor I-mediated detachment of opsonized HCV from erythrocytes
Factor I release of C3b and C4b from CR1 on erythrocytes resulted in an observed peak concentration of erythrocyte-bound HCV within 30 minutes and then a significant decline thereafter (Fig. 1B). In the absence of Factor I, the concentration of erythrocyte-bound HCV increased continuously throughout a 2 hour incubation period (Fig. 5A). Further, erythrocyte-bound HCV levels were high in Factor I-depleted serum, but were significantly lower after Factor I was added (Fig. 5B). Correspondingly, free HCV levels were low in Factor I-depleted serum, but were significantly higher after Factor I was repleted (Fig. 5C).
FIG. 5.
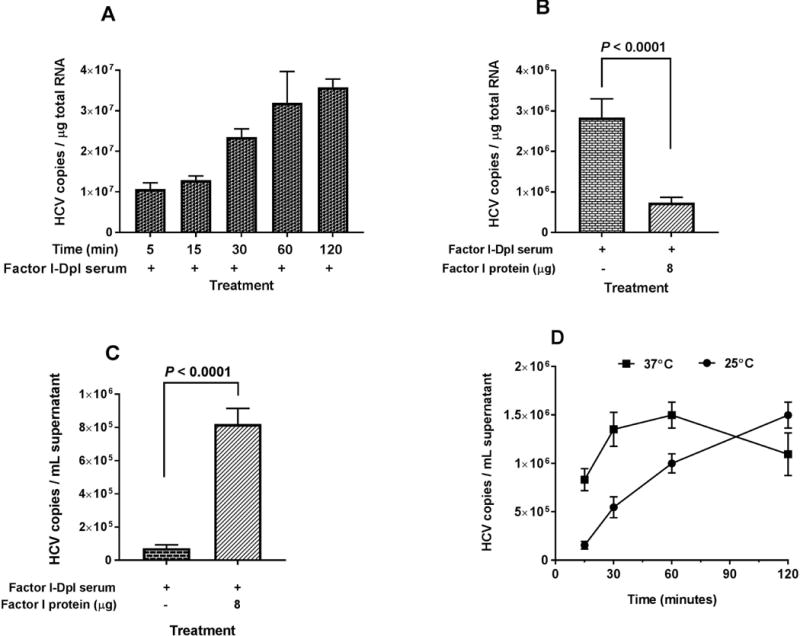
Effects of Factor I on HCV association with erythrocytes. (A) Three milliliters of HCV1a virus (1 × 107 genomic copies total) were incubated with 50 μL Factor I-depleted serum (titrated previously) for 30 minutes at 25°C, followed by mixing with 2 mL of erythrocytes (5 × 108 cells total) and incubating with variable times at 25°C as indicated. After adding EDTA to a final concentration of 20 mM, erythrocytes were collected by centrifugation and washed three times with 1× PBS, pH7.4. Total RNA isolation from erythrocytes and quantification of HCV RNA were performed as described in the Methods section. (B-C) Six milliliters of HCV2a virus (6 × 108 genomic copies total) were incubated with 100 μL Factor I-depleted serum for 30 minutes at 25°C, followed by adding 4 mL of erythrocytes (1 × 109 cells total) and incubating at 25°C for 2 hours. After several washing steps, the reaction was carried out in the presence of Factor I (8 μg) for 2 hours at 25°C. After adding EDTA to a final concentration of 20 mM, the cells and supernatants were collected by centrifugation. Erythrocyte-bound HCV and free HCV released into the medium were measured as described in the Methods section. (D) Three milliliters of HCV1a virus (1 × 107 genomic copies total) were incubated with 50 μL Factor I-depleted serum (titrated previously) for 30 minutes at 25°C, followed by adding 2 mL of erythrocytes (5 × 108 cells total) and incubating at 25°C for 2 hours. After several washing steps, the reaction was carried with Factor I (4 μg) at 25°C and 37°C at various times. The supernatants were collected by centrifugation. Free HCV released into the medium, was measured as described in the Methods section. The data were expressed for erythrocyte-bound RNA as HCV copies/μg total RNA and for free, released HCV RNAs as HCV copies/mL supernatant. Each value represents the mean ± standard deviation of six determinations (A and D) and twelve determinations (B and C). The data are representative of two independent experiments using erythrocytes from at least two different healthy donors.
The effects of temperature on the release of HCV from erythrocytes was also investigated by incubating at 25°C and 37°C with variable times. Factor I-mediated release of HCV from erythrocytes occurred more efficiently at 37°C than at 25°C (Fig. 5D).
HCV retained infectivity to cultured hepatoma cell line (Huh 7.5.1 cells) after spontaneous or Factor I-mediated release from erythrocytes (Supporting Fig. S5).
HCV released from erythrocytes by Factor I binds to CD19+ B cells through CR2
The release of opsonized HCV from erythrocytes was Factor I-dependent, indicating that C3b/C4b-fragments were further processed into C3d/C4d on the opsonized HCV particles. Next, we investigated which subset of cell populations in PBMCs would be the target of HCV particles attached with C3d/C4d fragments. HCV particles released from erythrocytes by Factor I treatment as described in the previous section were incubated with PBMCs for 1 hour. Subsequently the cells were fractionated into CD19+ B cells, CD14+ monocytes-macrophage cells, CD3+ T cells, and CD56+ NK cells. Among these subset populations, only CD19+ expressing B cells significantly captured the opsonized HCV (Fig. 6A).
FIG. 6.
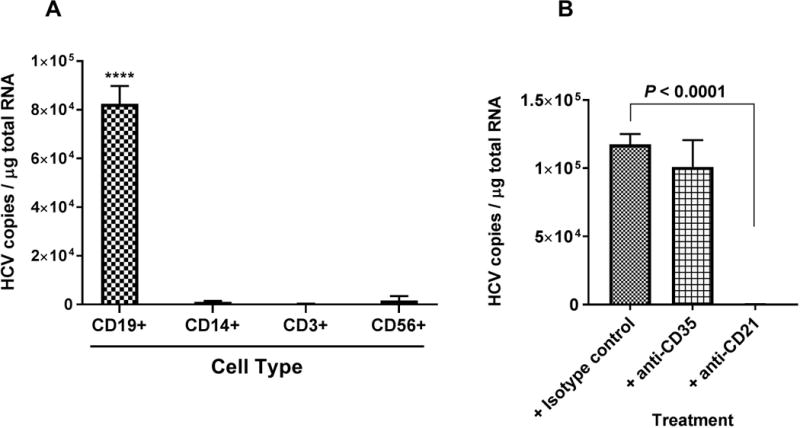
Factor I-mediated release of HCV virus from erythrocytes can bind to B cells. (A) HCV2a virus particles released from erythrocytes by Factor I treatment, preferentially bind to CD19+ B cells. Cell type: CD19+ B cells; CD14+ monocytes and macrophages; CD3+ T cells; CD56+ NK cells. ****P < 0.0001, when HCV binding to CD19+ B cells was compared to other cell types. (B) HCV1a virus particles released from erythrocytes by Factor I treatment bind to CD19+ B cells via CR2. The experimental details were described in the Methods sections. Each value represents the mean ± standard deviation of six determinations. The data are representative of two independent experiments using PBMCs from two different healthy donors.
Both complement receptor 1 (CR1/CD35) and complement receptor 2 (CR2/CD21) are expressed on the surface of B cells. To determine which receptor plays a major role in mediating the binding of HCV particles released from erythrocytes by Factor I, PBMCs were treated with antibodies against cell surface receptors such as CR1 (Ab: anti-CD35) or CR2 (Ab: anti-CD21), followed by incubation with Factor I released HCV particles and isolation CD19+ B cells using magnetic microbeads. Only anti-CD21 showed strong inhibition of the binding of HCV to B cells (P < 0.0001) compared to isotype control (Fig. 6B).
Discussion
In this study, we demonstrated that free HCV is opsonized by complement and binds to CR1 on erythrocytes (E-CR1); the presence of HCV-specific antibody significantly increased this binding. Furthermore, complement protein C1 was required while C2, C3 and C4 enhanced binding. Finally, dissociation of complement-opsonized HCV from E-CR1 was mediated by Factor I, and the released HCV was preferentially bound to CD19+ B cells via CR2 compared to other leukocytes.
Erythrocytes are known to shuttle complement-opsonized particles and ICs via E-CR1 to secondary lymphoid organs and to facilitate IC clearance from the circulation.(10) We previously found that free HCV was bound to CR1 on B cells through a complement-mediated mechanism.(22) Since 90% of the CR1 pool exists on erythrocytes and HCV is associated with IC-related diseases, we were interested in investigating whether complement could also mediate free and antibody-bound HCV attachment to CR1 on erythrocytes.
Using our assay system with cell culture-produced HCV, both HCV genotypes 1a and 2a (H77s and JFH1, respectively) became robustly bound to erythrocytes with up to 1,000 fold increased affinity in the presence of complement. In the absence of serum, virtually no erythrocyte-bound HCV was detected. Subsequent investigation of the opsonization time and binding kinetics revealed that the association of opsonized HCV particles with erythrocytes was consistent with reports on the binding of HIV-specific-ICs and other ICs to erythrocytes.(27)
The near absence of HCV binding to erythrocytes in C1q-depleted serum demonstrated that the classical pathway of complement activation was crucial in our model system. However, C1q protein alone was not sufficient to mediate HCV binding to erythrocytes, since there was negligible binding activity observed when either free HCV or antibody-bound HCV was mixed with C1q in the absence of other complement proteins (Supporting Fig. S4). This result was different from other non-HCV in vitro models (13, 14, 26) and may be related to HCV-binding dynamics or to differences in assay systems. In complement factor depletion and reconstitution experiments, we observed that complement proteins C2, C3 and C4 enhanced HCV-erythrocyte binding, but C2, C3 or C4 alone were not sufficient for binding (Fig. 2B-D). Further, the results of C3/C4 depletion and reconstitution experiments suggest a hierarchical role in the association of HCV with erythrocytes. In complement C3 depleted serum (C4 present) HCV binding was significantly enhanced by repletion of C3 in a dose-dependent manner (Fig. 2C). However, in C3/C4 depleted serum, the repletion of high concentration C3 (C4 absent) did not enhance binding. Both C3 and C4 activation fragments, C3b and C4b, bind to CR1, suggesting that C3b plays a less prominent role than C4b for HCV-E-CR1 binding. This is further supported by the observation that after restoring both C3 and C4 proteins to C3/C4-depleted serum, HCV-erythrocyte binding wasn’t significantly different from when C4 was restored alone (Fig. 2E).
Lower C3 and C4 levels have been reported in patients with chronic HCV when compared to healthy controls and when compared to patients with non-HCV-related liver disease.(28-30) Chronic HCV-associated immune complexes may consume complement resulting in acquired hypocomplementemia. For instance, profoundly decreased C4 levels in chronic HCV patients with mixed cryoglobulinemia has been reported.(31) Conversely, HCV may directly reduce complement levels through repression of C3 and C4 mRNA expression levels in hepatocytes.(28, 30) It is currently not clear if decreased C3 and C4 levels in HCV infection are a result of HCV-related immune complex consumption, of HCV repression of C3 and C4, or both and whether these decreases result in clinically relevant deficiency. In our complement reconstitution experiments we showed that HCV binding to erythrocytes was nearly absent when C3 and C4 were depleted (Fig. 2E). If erythrocytes play a significant role in clearance of chronic HCV-related ICs, then our results would suggest that low C3 and C4 in HCV-infected patients may precede and predispose patients to immune complex-related disease.
To confirm the adherence of opsonized HCV to erythrocytes occurred via CR1 (CD35), we used anti-CD35 monoclonal antibody which is known to block the C3b/C4b binding sites. Whether using erythrocytes from healthy blood donors or HCV-infected patients, HCV binding to erythrocytes was blocked significantly more by anti-CD35 than by antibodies to other receptors (Fig. 3). Antibodies to CR1 similarly blocked complement-mediated binding of HIV to erythrocytes in a prior study.(26)
We observed a large decrease in the binding time of opsonized HCV to erythrocytes after 1-2 hours of incubation (Fig. 1B). Similar findings were observed in the detachment of complement-coated bacteria and complement-opsonized HIV from erythrocytes and were attributed to complement Factor I activity.(27, 32, 33) Indeed, in the absence of Factor I, the binding activity of opsonized HCV to erythrocytes continuously increased beyond 2 hours of incubation (Fig. 5A).
Factor I is a plasma serine protease that proteolytically cleaves C3b and C4b in the presence of co-factors such as CR1, releasing these complement proteins and any associated pathogens and/or immune complexes from the surface of erythrocytes. In this study, erythrocyte-bound opsonized HCV particles released by Factor I preferentially bound to CD19+ B cells in peripheral blood mononuclear cells (PBMCs) through CR2 (Fig. 6). Factor I deficiency results in the depletion of serum C3 levels and is associated with increased infections.(34) However, the correlation of the functional activity of differential Factor I serum levels with erythrocyte IC clearance capacity and/or IC-related disease or the role of CD19+ B cell binding of dissociated HCV have not been investigated. Therefore, Factor I release of HCV from erythrocytes and binding to CD19+ B cells are of uncertain significance.
In summary, this study provides the first direct evidence that complement opsonized free and IC-bound HCV can bind to erythrocytes via E-CR1. Whether binding of free HCV to erythrocytes is related to HCV clearance or pathogenesis has not been investigated. Mixed cryoglobulinemia (Type II) is among the most common IC diseases associated with chronic HCV infection. While nearly half of patients with HCV have detectable cryoglobulins.(35, 36) less than 10% develop clinically apparent disease.(36, 37) We’ve demonstrated that HCV-IC bind to erythrocytes, and it is therefore plausible that factors affecting the HCV-IC-erythrocyte interaction could influence the expression of clinically evident IC-related disease.
Supplementary Material
Acknowledgments
We thank Dr. Francis Chisari for providing Huh-7.5.1 cells, Dr. Stanley Lemon for providing H77s plasmids, and Dr. Jake Liang for providing the JFH1 virus.
Financial support: This research was supported by the National Institutes of Health (NIH) Clinical Center, part of the NIH Intramural Research Program.
List of Abbreviations
- HCV
hepatitis C virus
- JFH1
Japanese fulminant hepatitis 1
- IC
Immune complex
- PBMCs
peripheral blood mononuclear cells
- CD
cluster of differentiation
- MACS
magnetic cell sorting
- PCR
polymerase chain reaction
- PBS
phosphate-buffered saline
- BSA
bovine serum albumin
- Dpl
depleted
- CR1/CD35
complement receptor 1
- CR2/CD21
complement receptor 2
- E-CR1
erythrocyte complement receptor 1
- qPCR
real-time quantitative RT-PCR
Footnotes
Manuscript ID number: HEP-18-0151
Potential conflict of interest: Nothing to report.
References
- 1.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 2.Burra P, De Martin E, Zanetto A, Senzolo M, Russo FP, Zanus G, Fagiuoli S. Hepatitis C virus and liver transplantation: where do we stand? Transpl Int. 2016;29:135–152. doi: 10.1111/tri.12642. [DOI] [PubMed] [Google Scholar]
- 3.Campos-Varela I, Lai JC, Verna EC, O’Leary JG, Todd Stravitz R, Forman LM, Trotter JF, et al. Hepatitis C genotype influences post-liver transplant outcomes. Transplantation. 2015;99:835–840. doi: 10.1097/TP.0000000000000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim WR, Terrault NA, Pedersen RA, Therneau TM, Edwards E, Hindman AA, Brosgart CL. Trends in waiting list registration for liver transplantation for viral hepatitis in the United States. Gastroenterology. 2009;137:1680–1686. doi: 10.1053/j.gastro.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biggins SW, Bambha KM, Terrault NA, Inadomi J, Shiboski S, Dodge JL, Gralla J, et al. Projected future increase in aging hepatitis C virus-infected liver transplant candidates: a potential effect of hepatocellular carcinoma. Liver Transpl. 2012;18:1471–1478. doi: 10.1002/lt.23551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Araki K, Nagashima H, Tsuji T. Detection and characterization of circulating immune complexes during acute exacerbation of chronic viral hepatitis. Clin Exp Immunol. 1982;47:520–526. [PMC free article] [PubMed] [Google Scholar]
- 7.Hijikata M, Shimizu YK, Kato H, Iwamoto A, Shih JW, Alter HJ, Purcell RH, et al. Equilibrium centrifugation studies of hepatitis C virus: evidence for circulating immune complexes. J Virol. 1993;67:1953–1958. doi: 10.1128/jvi.67.4.1953-1958.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sansonno D, Iacobelli AR, Cornacchiulo V, Lauletta G, Distasi MA, Gatti P, Dammacco F. Immunochemical and biomolecular studies of circulating immune complexes isolated from patients with acute and chronic hepatitis C virus infection. Eur J Clin Invest. 1996;26:465–475. doi: 10.1046/j.1365-2362.1996.162317.x. [DOI] [PubMed] [Google Scholar]
- 9.Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. Philadelphia: Elsevier; 2018. Effector mechanisms of humoral immunity; pp. 281–297. [Google Scholar]
- 10.Walport MJ, Lachmann PJ. Erythrocyte complement receptor type 1, immune complexes, and the rheumatic diseases. Arthritis Rheum. 1988;31:153–158. doi: 10.1002/art.1780310201. [DOI] [PubMed] [Google Scholar]
- 11.Schreiber RD, Pangburn MK, Muller-Eberhard HJ. C3 modified at the thiolester site: acquisition of reactivity with cellular C3b receptors. Biosci Rep. 1981;1:873–880. doi: 10.1007/BF01114821. [DOI] [PubMed] [Google Scholar]
- 12.Ross GD, Newman SL, Lambris JD, Devery-Pocius JE, Cain JA, Lachmann PJ. Generation of three different fragments of bound C3 with purified factor I or serum. II. Location of binding sites in the C3 fragments for factors B and H, complement receptors, and bovine conglutinin. J Exp Med. 1983;158:334–352. doi: 10.1084/jem.158.2.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity. 1997;7:345–355. doi: 10.1016/s1074-7613(00)80356-8. [DOI] [PubMed] [Google Scholar]
- 14.Tas SW, Klickstein LB, Barbashov SF, Nicholson-Weller A. C1q and C4b bind simultaneously to CR1 and additively support erythrocyte adhesion. J Immunol. 1999;163:5056–5063. [PubMed] [Google Scholar]
- 15.Birmingham DJ. Erythrocyte complement receptors. Crit Rev Immunol. 1995;15:133–154. doi: 10.1615/critrevimmunol.v15.i2.20. [DOI] [PubMed] [Google Scholar]
- 16.Miyakawa Y, Yamada A, Kosaka K, Tsuda F, Kosugi E, Mayumi M. Defective immune-adherence (C3b) receptor on erythrocytes from patients with systemic lupus erythematosus. Lancet. 1981;2:493–497. doi: 10.1016/s0140-6736(81)90882-5. [DOI] [PubMed] [Google Scholar]
- 17.Ross GD, Yount WJ, Walport MJ, Winfield JB, Parker CJ, Fuller CR, Taylor RP, et al. Disease-associated loss of erythrocyte complement receptors (CR1, C3b receptors) in patients with systemic lupus erythematosus and other diseases involving autoantibodies and/or complement activation. J Immunol. 1985;135:2005–2014. [PubMed] [Google Scholar]
- 18.Thomsen BS, Oxholm P, Nielsen H, Manthorpe R. Erythrocyte complement C3b receptor (CR1) levels and immune complex-induced manifestations in patients with primary Sjogren’s syndrome. Scand J Rheumatol Suppl. 1986;61:127–130. [PubMed] [Google Scholar]
- 19.Manzi S, Navratil JS, Ruffing MJ, Liu CC, Danchenko N, Nilson SE, Krishnaswami S, et al. Measurement of erythrocyte C4d and complement receptor 1 in systemic lupus erythematosus. Arthritis Rheum. 2004;50:3596–3604. doi: 10.1002/art.20561. [DOI] [PubMed] [Google Scholar]
- 20.Fujiwara K, Allison RD, Wang RY, Bare P, Matsuura K, Schechterly C, Murthy K, et al. Investigation of residual hepatitis C virus in presumed recovered subjects. Hepatology. 2013;57:483–491. doi: 10.1002/hep.25921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stamataki Z, Shannon-Lowe C, Shaw J, Mutimer D, Rickinson AB, Gordon J, Adams DH, et al. Hepatitis C virus association with peripheral blood B lymphocytes potentiates viral infection of liver-derived hepatoma cells. Blood. 2009;113:585–593. doi: 10.1182/blood-2008-05-158824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang RY, Bare P, De Giorgi V, Matsuura K, Salam KA, Grandinetti T, Schechterly C, et al. Preferential association of hepatitis C virus with CD19+ B cells is mediated by complement system. Hepatology. 2016;64:1900–1910. doi: 10.1002/hep.28842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang L, Alter HJ, Wang H, Jia S, Wang E, Marincola FM, Shih JW, et al. The modulation of hepatitis C virus 1a replication by PKR is dependent on NF-kB mediated interferon beta response in Huh7.51 cells. Virology. 2013;438:28–36. doi: 10.1016/j.virol.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meuleman P, Bukh J, Verhoye L, Farhoudi A, Vanwolleghem T, Wang RY, Desombere I, et al. In vivo evaluation of the cross-genotype neutralizing activity of polyclonal antibodies against hepatitis C virus. Hepatology. 2011;53:755–762. doi: 10.1002/hep.24171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nash JT, Davies KA. Complement and immune complexes. Methods Mol Biol. 2000;150:203–214. doi: 10.1385/1-59259-056-X:203. [DOI] [PubMed] [Google Scholar]
- 26.Horakova E, Gasser O, Sadallah S, Inal JM, Bourgeois G, Ziekau I, Klimkait T, et al. Complement mediates the binding of HIV to erythrocytes. J Immunol. 2004;173:4236–4241. doi: 10.4049/jimmunol.173.6.4236. [DOI] [PubMed] [Google Scholar]
- 27.Banki Z, Wilflingseder D, Ammann CG, Pruenster M, Mullauer B, Hollander K, Meyer M, et al. Factor I-mediated processing of complement fragments on HIV immune complexes targets HIV to CR2-expressing B cells and facilitates B cell-mediated transmission of opsonized HIV to T cells. J Immunol. 2006;177:3469–3476. doi: 10.4049/jimmunol.177.5.3469. [DOI] [PubMed] [Google Scholar]
- 28.Banerjee A, Mazumdar B, Meyer K, Di Bisceglie AM, Ray RB, Ray R. Transcriptional repression of C4 complement by hepatitis C virus proteins. J Virol. 2011;85:4157–4166. doi: 10.1128/JVI.02449-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chowdhury SJ, Karra VK, Bharali R, Kar P. Role of complement component C4 in treatment response and disease progression in chronic hepatitis C patients. J Viral Hepat. 2015;22:671–674. doi: 10.1111/jvh.12383. [DOI] [PubMed] [Google Scholar]
- 30.Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, Ray R. Hepatitis C virus proteins inhibit C3 complement production. J Virol. 2012;86:2221–2228. doi: 10.1128/JVI.06577-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonacci M, Lens S, Londono MC, Marino Z, Cid MC, Ramos-Casals M, Sanchez-Tapias JM, et al. Virologic, Clinical, and Immune Response Outcomes of Patients With Hepatitis C Virus-Associated Cryoglobulinemia Treated With Direct-Acting Antivirals. Clin Gastroenterol Hepatol. 2017;15:575–583 e571. doi: 10.1016/j.cgh.2016.09.158. [DOI] [PubMed] [Google Scholar]
- 32.Hament JM, van Dijk H, Fleer A, Aerts PC, Schoenmakers M, de Snoo MW, Dekker BH, et al. Pneumococcal immune adherence to human erythrocytes. Eur J Clin Invest. 2003;33:169–175. doi: 10.1046/j.1365-2362.2002.01109.x. [DOI] [PubMed] [Google Scholar]
- 33.Cunnion KM, Hair PS, Buescher ES. Cleavage of complement C3b to iC3b on the surface of Staphylococcus aureus is mediated by serum complement factor I. Infect Immun. 2004;72:2858–2863. doi: 10.1128/IAI.72.5.2858-2863.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nilsson SC, Sim RB, Lea SM, Fremeaux-Bacchi V, Blom AM. Complement factor I in health and disease. Mol Immunol. 2011;48:1611–1620. doi: 10.1016/j.molimm.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Ramos-Casals M, Munoz S, Medina F, Jara LJ, Rosas J, Calvo-Alen J, Brito-Zeron P, et al. Systemic autoimmune diseases in patients with hepatitis C virus infection: characterization of 1020 cases (The HISPAMEC Registry) J Rheumatol. 2009;36:1442–1448. doi: 10.3899/jrheum.080874. [DOI] [PubMed] [Google Scholar]
- 36.Lunel F, Musset L, Cacoub P, Frangeul L, Cresta P, Perrin M, Grippon P, et al. Cryoglobulinemia in chronic liver diseases: role of hepatitis C virus and liver damage. Gastroenterology. 1994;106:1291–1300. doi: 10.1016/0016-5085(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 37.Pawlotsky JM, Ben Yahia M, Andre C, Voisin MC, Intrator L, Roudot-Thoraval F, Deforges L, et al. Immunological disorders in C virus chronic active hepatitis: a prospective case-control study. Hepatology. 1994;19:841–848. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.