

Abstract
Vici syndrome is a rare, under-recognised, relentlessly progressive congenital multisystem disorder characterised by five principal features of callosal agenesis, cataracts, cardiomyopathy, combined immunodeficiency and oculocutaneous hypopigmentation. In addition, three equally consistent features (profound developmental delay, progressive failure to thrive and acquired microcephaly) are highly supportive of the diagnosis. Since its recognition as a distinct entity in 1988, an extended phenotype with sensorineural hearing loss, skeletal myopathy and variable involvement of virtually any organ system, including the lungs, thyroid, liver and kidneys, have been described.
Autosomal recessive mutations in EPG5 encoding ectopic P-granules autophagy protein 5 (EPG5), a key autophagy regulator implicated in the formation of autolysosomes, were identified as the genetic cause of Vici syndrome. The eight key features outlined above are highly predictive of EPG5 involvement, with pathogenic EPG5 mutations identified in >90% of cases where six or more of these features are present. The manifestation of all eight features has a specificity of 97% and sensitivity of 89% for EPG5-related Vici syndrome. Nevertheless, substantial clinical overlap exists with other multisystem disorders, in particular congenital disorders of glycosylation and mitochondrial disorders. Clinical and pathological findings suggest Vici syndrome as a paradigm of congenital disorders of autophagy, a novel group of inherited neurometabolic conditions linking neurodevelopment and neurodegeneration due to primary autophagy defects.
Here we describe the diagnostic odyssey in a 4-year-old boy whose clinical presentation with multisystem manifestations including skeletal myopathy mimicked a mitochondrial disorder. A genetic diagnosis of Vici syndrome was made through whole genome sequencing which identified compound heterozygous variants in EPG5. We also review the myopathic presentation and morphological characterisation of previously reported cases.
Keywords: Autophagy, Corpus callosal agenesis, EPG5-related Vici syndrome, Myopathy, Secondary mitochondrial dysfunction
Introduction
Vici syndrome [OMIM242840] is a severe, progressive, neurodevelopmental disorder caused by recessively inherited mutations in the key autophagy EPG5 (ectopic P-granules autophagy protein 5) gene (Cullup et al. 2013). This multisystem disorder is classically characterised by the five cardinal features of callosal agenesis, cataracts, cardiomyopathy, combined immunodeficiency and oculocutaneous hypopigmentation (McClelland et al. 2010). Since its original description in 1988 (Vici et al. 1988), the phenotypic spectrum has continued to evolve with an increasing number of patients described and approximately 50 genetically confirmed cases reported to date (Vici et al. 1988; Al-Owain et al. 2010; Chiyonobu et al. 2002; Cullup et al. 2013; del Campo et al. 1999; Finocchi et al. 2012; McClelland et al. 2010; Miyata et al. 2007; Ozkale et al. 2012; Rogers et al. 2011; Said et al. 2012; Ehmke et al. 2014; Byrne et al. 2016a). These five manifestations in addition to three features recently described to occur almost universally in affected patients, namely, profound developmental delay, progressive failure to thrive and acquired microcephaly, are highly supportive of the diagnosis (Byrne et al. 2016a). The manifestation of all eight of these features has a specificity of 97% and a sensitivity of 89% for the presence of EPG5 mutations (Byrne et al. 2016b).
Additional variable multisystem involvement, not infrequently observed in individual patients, include sensorineural hearing loss (McClelland et al. 2010), unilateral lung hypoplasia (Al-Owain et al. 2010), skeletal myopathy (Said et al. 2012), mild dysmorphism with coarse facial features, full lips and macroglossia resembling those observed in lysosomal storage disorders (Cullup et al. 2013; Byrne et al. 2016a), congenital midline defects such as cleft lip or palate, thymic aplasia and/or hypospadias (Vici et al. 1988), hydronephrosis, renal dysfunction and renal tubular acidosis (Miyata et al. 2007; Byrne et al. 2016a), thyroid agenesis and dysfunction (Cullup et al. 2013) and idiopathic thrombocytopenic purpura (ITP) (Huenerberg et al. 2016).
The identification of EPG5 encoding a key autophagy regulator (ectopic P-granules autophagy protein 5) as the genetic cause of Vici syndrome (Cullup et al. 2013) has implicated an intriguing link between the autophagy pathway with neurodevelopmental and neurodegenerative features in this paradigm of a human multisystem disorder (Byrne et al. 2016b). Autophagy is an evolutionarily highly conserved intracellular degradative process that delivers cytoplasmic materials to the lysosome for degradation, playing a fundamental role in maintaining cellular homeostasis (Jiang and Mizushima 2014) and defence against infections (Schmid and Munz 2007). Autophagy has also emerged as a key regulator of cardiac and skeletal muscle homeostasis and functional remodelling (Sandri 2010; Portbury et al. 2011). An associated skeletal muscle myopathy as part of the syndrome has been described in detail by McClelland and colleagues in 2010 (McClelland et al. 2010) and subsequently confirmed in other reports (Al-Owain et al. 2010; Finocchi et al. 2012; Said et al. 2012; Byrne et al. 2016b).
We present the diagnostic odyssey in a patient with a multisystem disorder (profound psychomotor retardation, epilepsy, agenesis of corpus callosum, soft dysmorphism, visual impairment, liver dysfunction and microvesicular steatosis) and a predominant neuromuscular phenotype with skeletal muscle myopathy that was suggestive of a mitochondrial respiratory chain disorder.
Materials and Methods
Clinical Summary
The male patient was born to a non-consanguineous couple of English-Australian and Filipino background at 37 weeks of pregnancy with a birth weight of 2.32 kg (3rd–10th percentiles) and head circumference of 32.5 cm (3rd percentile). The antenatal history was unremarkable apart from two episodes of urinary tract infections at 12 and 18 weeks. He was discharged home a few days after birth, however re-presented at 3 weeks with feeding difficulties when he was weaned off breast-feeding to bottle feeds. He subsequently displayed poor weight gain and had occasional episodes of choking on feeds despite various attempts to address the issue including changing formulas, teats and feeding bottles. He was referred to the Princess Margaret Hospital at 4 months because of failure to thrive. He was unable to fix or follow and had marked central hypotonia with diminished deep tendon reflexes. Ophthalmology assessment by video evoked potentials and electroretinogram identified impairment of the visual pathways bilaterally. Newborn hearing screen and formal audiometry with transient evoked otoacoustic emissions and tympanogram at 5 months were all normal. He developed tonic and myoclonic seizures at 4 months which were refractory to treatment with levetiracetam, oxcarbazepine and clonazepam. Griffith’s Mental Development Scale examination performed at 5 months demonstrated global developmental delay, and he showed no evidence of functional vision. He scored the following subquotients: locomotor 15 days, personal and social 12 days, hearing and language 27 days, and eye and hand coordination unscorable and performance 1¾ months. His mother had two older children from a previous relationship, one of whom had a history of unilateral hearing loss.
Significant feeding intolerance and gastroesophageal reflux improved with elemental feeds and anti-reflux medication. He was initially fed through a nasogastric tube and was subsequently changed to gastrostomy feeds. He experienced frequent respiratory infections which were partly caused by his inability to control salivary secretions. He had progressive microcephaly with head circumference at 14 months at 43.5 cm (<3rd percentile), myopathic facies with a tented upper lip and bilateral partial ptosis. Facial dysmorphism included plagiocephaly, a flat nasal bridge, arched eyebrows, high arched plate and mild retrognathia (Fig. 1a, b). His ears were cupped, and the helices were overfolded over the vertical aspect. He had tapered fingers and mild bilateral fifth finger clinodactyly. Skin hypopigmentation was not overtly evident in the first 2–3 years of life; however by 4 years of age, a few macules were observed over his face and back of neck (Fig. 1a, b).
Fig. 1.

Photographs showing facial dysmorphism. (a) Frontal view: flat nasal bridge, arched eyebrows, high arched plate and hypopigmented macule over glabella. (b) Right lateral view: plagiocephaly, a flat nasal bridge, mild retrognathia, cupped ears with overfolded helices over the vertical aspect and hypopigmented macule over posterolateral aspect of his neck
His brain MRI showed complete agenesis of the corpus callosum, colpocephaly with a high riding 3rd ventricle and disproportionate prominence of the occipital horns of the lateral ventricles, periventricular leukomalacia and delayed myelination as evidenced by little internal capsule myelination (Fig. 2a, b). He had decreased muscle tone and deep tendon reflexes. He was commenced on oral coenzyme Q 10, vitamin C, riboflavin, thiamine and carnitine. Liver histopathology and electron microscopy examination at 6 months showed prominent microvesicular steatosis within hepatocytes with fine cytoplasmic vacuolation and PAS-positive, diastase-sensitive glycogen demonstrated within the hepatocytes, in amounts compatible with the patient’s age. Muscle biopsy from the right vastus lateralis muscle showed features consistent with a myopathic process with increased variability in fibre size, marked type 1 fibre predominance and small type 2 fibres. Staining for utrophin was positive in the muscle fibres. Dystrophin, sarcoglycans, alpha- and beta-dystroglycan, dysferlin, merosin, emerin, collagen VI and caveolin-3 immunohistology were normal. Muscle enzyme histochemistry for NADH- tetrazolium reductase (NADH-TR), succinate dehydrogenase and cytochrome oxidase was normal. Acid phosphatase stain showed patchy sarcoplasmic positivity in several fibres, and the spectrin stain displayed occasional vacuoles in few fibres. There was granular sarcoplasmic positivity for p62 in many fibres. Immunohistochemistry for LC3 was not performed as this stain was not available in our laboratory.Electron microscopy showed increased variability in fibre size with a few atrophic fibres showing redundant basal lamina. Increased intermyofibrillar lipid was observed in many fibres, some of which were associated with mild to moderately pleomorphic mitochondria. Several fibres showed aggregates of autophagic vacuoles (Fig. 3). A mitochondrial disorder was considered a possible differential diagnosis. Liver and muscle respiratory chain enzymology (RCE) were normal (Table 1). A summary of muscle morphology in our patient and in other previously reported cases is summarised in Table 2.
Fig. 2.

MRI of the brain at 4 months of age. (a) Axial view: complete agenesis of the corpus callosum, colpocephaly with a high riding 3rd ventricle and disproportionate prominence of the occipital horns of the lateral ventricles, periventricular leukomalacia and delayed internal capsule myelination. (b) Sagittal view: complete agenesis of the corpus callosum and high riding 3rd ventricle
Fig. 3.
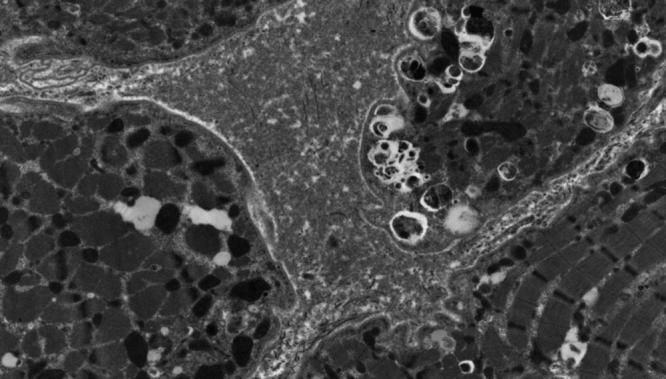
Muscle electron microscopy. Aggregates of autophagic vacuoles in a muscle fibre
Table 1.
Respiratory chain enzyme activities for patient 1
| Assay | Enzyme activity | CS ratio (%) | ||
|---|---|---|---|---|
| Muscle | Liver | Muscle | Liver | |
| Complex I (nmol/min/mg) | 84 (19–72) | 26 (7.8–11.2) | 200 | 202 |
| Complex II (nmol/min/mg) | 112 (26–63) | 93 (54–73) | 249 | 109 |
| Complex II + III (nmol/min/mg) | 46 (30–76) | 100 | ||
| Complex III (min/mg) | 46.1 (12.8–50.9) | 15.5 (5.2–10.3) | 158 | 146 |
| Complex IV (min/mg) | 8.06 (3.3–9.1) | 0.56 (0.45–0.87) | 122 | 59 |
| Citrate synthase (nmol/min/mg) | 227 (85–179) | 38 (26–31) | 176 | – |
Expressed relative to protein and as % residual activity relative to citrate synthase (CS)
Values outside the normal range are shown in bold. Reference ranges are shown in brackets
Table 2.
Summary of muscle morphology in our patient and in other previously reported cases
| Our patient | del Campo et al. (1999) – Case 4 | Chiyonobu et al. (2002) | Miyata et al. (2007) | McClelland et al. (2010) | Al-Owain et al. (2010) | Rogers et al. (2011) | Finocchi et al. (2012) | Said et al. (2012) | Ozkale et al. (2012) | Ehmke et al. (2014) | Byrne et al. (2016a, b) | Hedberg-Oldfors et al. (2017) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sibling 1 | Sibling 2 | Sibling 1 | Sibling 2 | Sibling 1 | Sibling 2 | |||||||||||
| Global delay | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Hypotonia | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Elevated CK | + | NA | + | + | + | + | CK normal | NA | NA | NA | + | + | + | + | + | + |
|
Summary of muscle pathology
Increased fibre size variability |
+ | + | ND | ND | ND | ND | + | + | − | ND | + | + | ND | ND | 16/17 patients | + |
| Type 1 fibre predominance | + | NA | NA | + | − | + | + | 5/17 patients | NA | |||||||
| Internalised/central nuclei | + | − | + | + | − | + | + | 12/17 patients | + | |||||||
| Vacuoles | + | + | − | + | − | − | − | 12/17 patients | + | |||||||
| Increased glycogen content | + | − | + | + | − | − | + | 7/17 patients | + | |||||||
| Mitochondrial pathology | + | + | + | + | − | + | + | 9/17 patients | + | |||||||
| Increased number of autophagosomes | − | − | − | − | − | − | − | 1/17 patients | + | |||||||
| Exocytosis | + | − | + | − | − | − | − | 1/17 patients | − | |||||||
| Autophagic vacuoles with sarcolemmal features | + | 2/17 patients | − | |||||||||||||
| p62-positive inclusions | + | ND | ND | ND | ND | ND | ND | 2 patients | + | |||||||
| High expression of foetal and embryonic MyHC | − | ND | ND | ND | ND | ND | ND | ND | + | |||||||
| Respiratory chain enzyme studies | N | N | N | ND | ND | ND | N | 3/8 patients: reduced complexes I, III and IV 2 patients, reduced complex IV 1 patient |
ATPase activity – 259 nmol/min/mg protein (N > 415) | |||||||
N normal, ND not done, NA not available
Laboratory investigations performed showed elevated creatine kinase 1,020 U/L (ref range 25–220), blood lactate 7.3 mmol/L (ref range < 2.1), pyruvate 0.15 mmol/L (ref range < 0.12), lactate/pyruvate ratio 48 (ref range 14–28), liver enzymes (ALT 134 U/L (ref < 60), ALT 74 U/L (ref < 35)) and plasma alanine 582 umol/L (ref range 143–439). Alphafetoprotein, CSF lactate 1.3 mmol/L (ref range 0.7–1.8) and pyruvate 0.08 mmol/L (ref range 0.03–0.10), ammonia, plasma acylcarnitine profile, urine amino and organic acid screens, serum transferrin, apolipoprotein C III isoforms, serum uric acid, plasma very-long-chain fatty acids, plasma phytanic acids, white blood cell lysosomal enzymes, serum 7-dehydrocholesterol, urine succinylpurines, purines, pyrimidines, polyols and oligosaccharides, CSF lactate, pyruvate, glucose and glycine and fibroblast pyruvate dehydrogenase enzymology were all normal. Immune workup including immunoglobulin levels, white cell counts and lymphocyte subsets were normal, suggesting normal humoral and cellular immune functions. Echocardiography performed at 4 years of age was normal. Genetic studies including chromosomal microarray, mitochondrial DNA sequencing in muscle and fibroblasts, POLG gene mutation analysis, targeted exome sequencing using an absent speech multigene panel which investigates overlapping conditions presenting with global developmental delay predominantly affecting the speech (ANKRD11, ARX, ATRX, CDKL5, CNTNAP2, DYRK1A, EEF1A2, EHMT1, FOLR1, FOXG1, FRAXA, FRAXE, GRIN2A, HERC2, L1CAM, MBD5, MECP2, MEF2C, NRXN1, OPHN1, PCDH19, PNKP, SETBP1, SLC9A6, TCF4, UBE3A and ZEB2) and whole exome sequencing results were non-contributory.
At 4 years, he remains profoundly delayed, hypotonic, non-ambulatory with only partial head control and non-verbal. He has significant failure to thrive, progressive microcephaly and refractory seizures. His electroencephalogram (EEG) at 4 years of age has remained abnormal with frequent epileptiform discharges, electroclinical seizures, asynchronous background with high amplitude slow waves and superimposed slightly faster frequencies at times. His electroencephalogram (EEG) at 4 years of age has remained abnormal with frequent epileptiform discharges, which continue to be maximal in the parietal regions. Sharp waves are often asynchronous, whilst the background at times is also asynchronous with high amplitude slow waves and superimposed slightly faster frequencies at times which may occur over either hemisphere. Epileptiform activity was seen throughout the recording. Brief relative decrements are seen of less than 1-s duration. Electroclinical seizures were also noted, and the video recording of the first demonstrated a brisk extensor-type movement of the upper limbs. This was associated with a reduction in the background amplitude and an admixture of frequencies. Compared to the interictal background, there was an increase in the amount of faster frequencies. A second event of slightly longer duration was associated with video showing movement with shaking of the head followed by extension of the upper limb and then movement of the head from side to side. The EEG correlate is of decrement with an increase in faster beta activity of 2–3 s duration followed by a posteriorly dominant rhythm of around 8 Hz with greater amplitude in the right hemisphere with an admixture of faster frequencies lasting for a further 3–4 s. Some nonspecific movement for 1 s was followed by a further brief relative decrement before the recording returns to the interictal state. The asynchronous EEG recorded on several occasions over the first 4 years of life is in keeping with absence of the corpus callosum. Repeat ophthalmology assessment at 4.5 years revealed impaired vision with absence of defensive blink and failure to react to visual stimuli, which was attributed to a cortical cause. His right optic disc was noted to be pale. No cataracts or ocular albinism were observed.
Respiratory Chain Enzyme Activities
Respiratory chain enzyme activities were assayed in patient skeletal muscle and liver as previously described (Frazier and Thorburn 2012).
Whole Genome Sequencing
Whole genome sequencing was performed at the Kinghorn Centre for Clinical Genomics (Garvan Institute, Sydney) on genomic DNA extracted from blood collected from the patient and his parents as previously described (Riley et al. 2017).
cDNA Synthesis Studies
RNA was obtained from white blood cell that had been cultured for 3–5 days with RMPI 1640, foetal calf serum, heparin, and glutamine together with phytohaemagglutinin (PHA). At the end of the culture period, cycloheximide was added, for 4–6 h before harvesting to inhibit nonsense-mediated decay. RNA was extracted using a QIAGEN RNeasy kit and kept at −80°C until cDNA was synthesised.
cDNA was made from 300 ng total RNA with poly dT and SuperScript III. PCR reactions were carried out with specific exonic primers designed to exclude known SNVs.
Results
Whole genome sequencing was undertaken on the patient and his parents to provide a genetic diagnosis. We identified compound heterozygous variants in EPG5 Chr18(GRCh37): g.43456196C>T, c.4327C>T, p.Gln1443* and g.43487925G>A, c.6049+5G>A. Neither variant was present in ExAC. EPG5 encodes ectopic P-granules autophagy protein 5 homologue (C. elegans), a large (2,579 amino acids) coiled coil domain containing protein that functions in autophagy. Variants in EPG5 have been associated with Vici syndrome. The c.4327C>T variant in exon 24 introduces a premature stop codon, p.Gln1443*. The mRNA produced is most likely targeted for nonsense-mediated decay as the variant occurs in exon 24 of 44 of EPG5 (Popp and Maquat 2016). The c.6049+5G>A variant is predicted to affect splicing, reducing the strength of the donor site by ~25% (14–52% across five prediction programmes, Alamut Visual). No candidates were identified within genes in the MitoCarta2.0 database (Calvo et al. 2015) or the mitochondrial genome. cDNA studies (Fig. 4) have confirmed that the c.6049+5G>A variant results in an out of frame deletion (exon 35 spliced out) that is expected to result in a truncated protein or nonsense-mediated RNA decay (Fig. 4).
Fig. 4.

cDNA sequencing studies. Exon 35 is missing in both forward and reverse directions, confirming that the c.6049+5G>A variant results in an out of frame deletion that is expected to result in a truncated protein or nonsense-mediated RNA decay
Discussion
The EPG5 gene, on chromosome 18q12.3, encodes the key autophagy regulator ectopic P-granules autophagy protein 5 (EPG5), a protein of 2,579 amino acids, and is predominantly expressed in the CNS, skeletal and cardiac muscle, thymus, immune cells, lungs and kidneys (Halama et al. 2007), tissues which are all involved in Vici syndrome. EPG5 was initially identified amongst a group of genes found to be mutated in breast cancer tissue (Sjoblom et al. 2006) before Cullup and colleagues elucidated its causative role in Vici syndrome (Cullup et al. 2013).
Autophagy is an evolutionarily conserved lysosomal pathway that degrades intracellular constituents. It begins with the initial formation of isolation membranes or phagophores which subsequently envelop the portion of cytoplasm to be removed, forming autophagosomes that subsequently fuse with lysosomes resulting in autolysosomes. The EPG5 protein has been implicated in the late step of autophagy in human cells involving autophagosome-lysosome fusion (Cullup et al. 2013) and, ultimately, impaired cargo delivery to the lysosome (Byrne et al. 2016b).
The ubiquitous expression of EPG5 during embryonic development and physiologically enhanced autophagy in neurons and muscle likely explains the prominent central nervous system and neuromuscular involvement in Vici syndrome (Byrne et al. 2016a). The prominent feature of agenesis of the corpus callosum, often associated with colpocephaly and other brain malformations including neuronal migration and myelination defects, cerebellar vermis dysplasia, pontine hypoplasia and abnormalities of the septum pellucidum in patients with Vici syndrome, suggests that aberrant autophagy during foetal development adversely affects CNS development (Cullup et al. 2013). This suggestion is supported by the demonstration of increased neuronal proliferation and severe neural tube defects in mice with null mutations in AMBRA1, a positive autophagy regulator with predominant expression in neural tissues (Fimia et al. 2007; Cullup et al. 2013). Interestingly, the progressive loss of skills and profound acquired microcephaly in children surviving beyond infancy suggest a neurodegenerative component in addition to the prominent neurodevelopmental defects (Ebrahimi-Fakhari et al. 2016). Supporting this idea is the neurodegenerative phenotype in Epg5-deficient mice, with selective neurodegeneration of cortical and spinal cord motor neurons resembling amyotrophic lateral sclerosis (Zhao et al. 2013) and the neurodegenerative phenotype in Epg5-deficient Drosophila melanogaster (Byrne et al. 2016b). These dual effects of defective autophagy on cellular development and maintenance (Byrne et al. 2016b) place EPG5-related Vici syndrome within a novel subclass of inborn errors of metabolism termed ‘congenital disorders of autophagy’, wherein dysregulated autophagy results in early-onset neurodevelopmental and neurodegenerative disorders (Ebrahimi-Fakhari et al. 2016).
Skeletal muscle myopathy, an important, less documented feature of Vici syndrome, had previously been suggested by the presence of profound hypotonia, generalised weakness, paucity of movements and variable elevations of creatine kinase in early case reports (Chiyonobu et al. 2002; Miyata et al. 2007) and morphological abnormalities in muscle biopsy in one reported patient that were suggestive of a mitochondrial cytopathy but with normal respiratory chain enzyme analysis (del Campo et al. 1999). Histopathologically, the associated myopathic features were subsequently documented in detail in an infant with a neuromuscular phenotype, characterised by increased variability in fibre size with type 1 fibre hypotrophy and normally sized type 2 fibres (potentially fulfilling the criteria for fibre type disproportion), prominent centralised nuclei in atrophic fibres, increased glycogen storage and variable vacuoles on light microscopy. Electron microscopy revealed redundant basal lamina and some small areas of debris between duplicated basal lamina around several small fibres suggesting exocytosis, enlarged mitochondria with abnormal cristae and areas of membrane glycogen which may relate to degenerating mitochondria. Additional findings of increased Gomori trichrome and abnormal oxidative stains in some small fibres, increased lipid content and the suggestive electron microscopy also raised the possibility of a mitochondrial cytopathy; however, this was not supported by CSF and plasma lactate levels which were normal, as were respiratory chain enzyme activities (McClelland et al. 2010). In a separate study, skeletal myopathy was confirmed in 17 patients on pre-mortem muscle biopsy which was corroborated by evidence of myopathy on EMG/nerve conduction studies in six patients (Byrne et al. 2016a).
Morphologically, considerable overlap exists with primary neuromuscular disorders that have been linked to defects in the autophagic pathway, in particular centronuclear myopathies (Jungbluth and Gautel 2014) and vacuolar myopathies including Pompe disease, Danon disease (Malicdan and Nishino 2012) and X-linked myopathy with excessive autophagy (XMEA) (Ramachandran et al. 2013), glycogen storage disorders and mitochondrial myopathies (Byrne et al. 2016b). Whilst EPG5-related skeletal muscle myopathy has been defined as a primary vacuolar myopathy based on the presence of vacuoles visible on both light and electron microscopy, vacuoles can be absent or inconspicuous (Byrne et al. 2016a) as observed in our patient. Other prominent morphological features of myopathy in Vici syndrome include marked variability in fibre size with type 1 predominance and atrophy, increase in internal and centralised nuclei and increased glycogen storage on light microscopy (Byrne et al. 2016a). Ultrastructural examination by electron microscopy reveals altered mitochondrial morphology and variable degrees of subsarcolemmal accumulation of autophagic vacuoles, glycogen and mitochondria (Byrne et al. 2016a; Hedberg-Oldfors et al. 2017). Additional findings include reddish Gomori trichrome staining of atrophic fibres suggesting mitochondrial abnormalities and variably increased acid phosphatase staining (Byrne et al. 2016a). Immunofluorescence studies in skeletal muscle tissue showed upregulation of sarcomere-associated autophagy proteins (p62/SQSTM1), lysosome-associated membrane protein (LAMP2) (Byrne et al. 2016a; Hedberg-Oldfors et al. 2017) and NBR1 with numerous LC3-positive autophagosomes, potentially reflecting impaired autophagic flux (Cullup et al. 2013). The observation of variable respiratory chain enzyme abnormalities (Byrne et al. 2016a; Hedberg-Oldfors et al. 2017), in addition to abnormalities of mitochondrial internal structure and positioning reported in earlier studies (McClelland et al. 2010; Al-Owain et al. 2010; Finocchi et al. 2012), supports secondary mitochondrial dysfunction as a possible downstream effect of defective autophagy (Byrne et al. 2016a, b) and likely reflects the evolutionarily conserved role of the autophagy pathway in maintaining mitochondrial quality and function (Zhang et al. 2007). Defective autophagy has also been implicated in secondary mitochondrial dysfunction in various neuromuscular disorders (Katsetos et al. 2013).
In our patient, the compilation of histopathological features, including patchy sarcoplasmic positivity on acid phosphatase staining, electron microscopic findings of aggregates of sarcoplasmic autophagic vacuoles in several fibres and the increase in autophagy marker protein p62 in many fibres, is consistent with perturbed autophagy. Characterisation of autophagy in Vici syndrome revealed a wide spectrum of abnormalities, making it a paradigm for a human multisystem disorder associated with defective autophagy (Cullup et al. 2013; Ebrahimi-Fakhari et al. 2016). To further delineate if the increased p62/SQSTM1 and NBR1 levels are reflective of enhanced upstream induction or downstream clearance in autophagic flux, patient-derived and healthy control fibroblasts were treated with autophagy inducer rapamycin and inhibitor bafilomycin which suppresses the autolysosomal H+ ATPase required for acidification and subsequent degradation of lysosomal contents. The results precluded defects in the early steps in autophagy, including autophagosome formation and processing of LC3-I to LC3-II. Autophagosomal clearance was however impaired as demonstrated by decreased colocalisation of LC3 and NBR1 with LAMP1, indicative of reduced fusion of autophagosomes with lysosomes. This was further corroborated by accumulation of lysine-63-polyubiquitinated proteins, which are usually destined for autolysosomal degradation (Cullup et al. 2013). Deranged transcriptional regulation was observed with altered phosphorylation in the AKT-mTOR protein kinase pathway, a major pathway controlling the expression of autophagy proteins (Cullup et al. 2013). In retrospect, these histopathological features are consistent with previous studies suggestive of defective autophagy (Al-Owain et al. 2010; McClelland et al. 2010; Cullup et al. 2013), as demonstrated by the prominence of autophagic vacuoles, storage of abnormal material, secondary mitochondrial abnormalities in skeletal muscle and multisystem defects in the heart, immune system, skin pigmentation and central nervous system, implicating autophagy in a wide range of cellular processes (Cullup et al. 2013).
Our patient had histopathological and electron microscopic findings in muscle consistent with a myopathic process, with associated mitochondrial pleomorphism and aggregates of autophagosomes, making mitochondrial cytopathy a likely differential. In conjunction with his multisystemic clinical presentation with a predominant neuromuscular phenotype, biochemical parameters of lactic acidemia, elevated plasma alanine and raised lactate/pyruvate ratio, he was classified as being in the “probable” category of a mitochondrial respiratory chain defect based on the modified Nijmegen criteria (http://mitochondrialdiseases.org/fmm/docs/CLINICAL%20CRITERIA.pdf) after exclusion of other differential diagnoses. In retrospect, several features of Vici syndrome were manifested in the patient; however due to the absence of a few cardinal findings including cardiomyopathy, cataracts and combined immunodeficiency, Vici syndrome had not been considered as a potential differential. This case highlights the utility of next-generation sequencing technologies including whole genome sequencing in providing a genetic diagnosis for complex rare diseases, particularly when the clinical phenotype does not conform in entirety to our current knowledge of a specific disorder. The emergence of rapid and more widely available next-generation DNA sequencing technology has not only led to the identification of novel diseases but will potentially contribute to the expansion of the phenotypic spectrum of known rare genetic disorders.
In conclusion, EPG5-related Vici syndrome is a paradigm of an emerging class of neurometabolic disorders termed ‘congenital disorders of autophagy’ as it highlights the consequences of dysfunctional autophagy and indicates an intriguing link between neurodevelopment and neurodegeneration. Substantial overlap exists with other multisystem disorders including mitochondrial disease, congenital disorders of glycosylation and lysosomal and glycogen storage disorders (Byrne et al. 2016b). Further characterisation of the phenotypic spectrum of Vici syndrome will enhance genetic diagnosis of the disorder which is imperative in facilitating genetic counselling and prenatal diagnosis. Similarly, exploring the associated secondary mitochondrial dysfunction observed in Vici syndrome could further facilitate our insights into the role of autophagy in maintaining mitochondrial quality and function (Ebrahimi-Fakhari et al. 2016).
Acknowledgements
This research was supported by a New South Wales Office of Health and Medical Research Council Sydney Genomics Collaborative grant (CS and JC). We also gratefully acknowledge donations to JC by the Crane and Perkins families as well as the participation of the research subjects.
Key Messages
A male patient presented with clinical features resembling a mitochondrial disorder including profound developmental delay, visual impairment, epilepsy and skeletal myopathy
Two novel EPG5 variants identified on WGS
Muscle morphological studies showed increased variability of fibre size, rare internalised nuclei, mitochondrial abnormalities and aggregates of autophagosomes
Considerable overlap exists between EPG5-related Vici syndrome and mitochondrial disorders
Details of the Contributions of Individual Authors
SB drafted the manuscript and was involved in the clinical management and diagnostic workup of the patient with AV, CK-B and PR. LR, MC, VG, CS, CE, EE and JC performed/supervised/interpreted molecular workup. All authors have read/critically revised the manuscript.
Compliance with Ethical Standards
Conflict of Interest
SB, LR, AV, MC, VG, TR, CS, RJ, CK and PR declare they have no conflict of interest. JC is a communicating editor of the Journal of Inherited Metabolic Disease.
Ethics
All procedures followed in this study were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, and its later amendments, and this project was approved by the Sydney Children’s Hospitals Network Human Research Ethics Committee (reference number 10/CHW/113). Informed consent was obtained for all participants included in the study.
Footnotes
Lisa G. Riley and Anand Vasudevan contributed equally to this work.
Contributor Information
Shanti Balasubramaniam, Email: saras329@hotmail.com.
Collaborators: Matthias Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Al-Owain M, Al-Hashem A, Al-Muhaizea M, et al. Vici syndrome associated with unilateral lung hypoplasia and myopathy. Am J Med Genet A. 2010;152A(7):1849–1853. doi: 10.1002/ajmg.a.33421. [DOI] [PubMed] [Google Scholar]
- Byrne S, Jansen L, U-King-Im JM, et al. EPG-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain. 2016;139(3):765–781. doi: 10.1093/brain/awv393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne S, Vici CD, Smith L, et al. Vici syndrome: a review. Orphanet J Rare Dis. 2016;11:21. doi: 10.1186/s13023-016-0399-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo S, Clauser K, Mootha V. MitoCarta2.0: an updated inventory of mammalian proteins. Nucleic Acids Res. 2015;44:D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiyonobu T, Yoshihara T, Fukushima Y. Sister and brother with Vici syndrome: agenesis of the corpus callosum, albinism, and recurrent infections. Am J Med Genet. 2002;109(1):61–66. doi: 10.1002/ajmg.10298. [DOI] [PubMed] [Google Scholar]
- Cullup T, Kho AL, Dionisi-Vici C, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat Genet. 2013;45(1):83–87. doi: 10.1038/ng.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Campo M, Hall BD, Aeby A, et al. Albinism and agenesis of the corpus callosum with profound developmental delay: Vici syndrome, evidence for autosomal recessive inheritance. Am J Med Genet. 1999;85(5):479–485. doi: 10.1002/(SICI)1096-8628(19990827)85:5<479::AID-AJMG9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Saffari A, Wahlster L, et al. Congenital disorders of autophagy: an emerging novel class of inborn errors of neuro-metabolism. Brain. 2016;139(Pt 2):317–337. doi: 10.1093/brain/awv371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehmke N, Parvaneh N, Krawitz P, et al. First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. Am J Med Genet A. 2014;164A(12):3170–3175. doi: 10.1002/ajmg.a.36772. [DOI] [PubMed] [Google Scholar]
- Fimia GM, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- Finocchi A, Angelino G, Cantarutti N, et al. Immunodeficiency in Vici syndrome: a heterogeneous phenotype. Am J Med Genet A. 2012;158A(2):434–439. doi: 10.1002/ajmg.a.34244. [DOI] [PubMed] [Google Scholar]
- Frazier A, Thorburn D. Biochemical analyses of the electron transport chain complexes by spectrophotometry. Methods Mol Biol. 2012;837:49–62. doi: 10.1007/978-1-61779-504-6_4. [DOI] [PubMed] [Google Scholar]
- Halama N, Grauling-Halama SA, Beder A, et al. Comparative integromics on the breast cancer associated gene KIAA1632: clues to a cancer antigen domain. Int J Oncol. 2007;31:205–210. [PubMed] [Google Scholar]
- Hedberg-Oldfors C, Darin N, Oldfors A. Muscle pathology in Vici syndrome – a case study with a novel mutation in EPG5 and a summary of the literature. Neuromuscul Disord. 2017;27(8):771–776. doi: 10.1016/j.nmd.2017.05.005. [DOI] [PubMed] [Google Scholar]
- Huenerberg K, Hudspeth M, Bergmann S, et al. Two cases of Vici syndrome associated with Idiopathic Thrombocytopenic Purpura (ITP) with a review of the literature. Am J Med Genet A. 2016;170A(5):1343–1346. doi: 10.1002/ajmg.a.37589. [DOI] [PubMed] [Google Scholar]
- Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. doi: 10.1038/cr.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungbluth H, Gautel M. Pathogenic mechanisms in centronuclear myopathies. Front Aging Neurosci. 2014;6:339. doi: 10.3389/fnagi.2014.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsetos CD, Koutzaki S, Melvin JJ. Mitochondrial dysfunction in neuromuscular disorders. Semin Pediatr Neurol. 2013;20:202–215. doi: 10.1016/j.spen.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Malicdan MC, Nishino I. Autophagy in lysosomal myopathies. Brain Pathol. 2012;22:82–88. doi: 10.1111/j.1750-3639.2011.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland V, Cullup T, Bodi I, et al. Vici syndrome associated with sensorineural hearing loss and evidence of neuromuscular involvement on muscle biopsy. Am J Med Genet A. 2010;152A(3):741–747. doi: 10.1002/ajmg.a.33296. [DOI] [PubMed] [Google Scholar]
- Miyata R, Hayashi M, Sato H, et al. Sibling cases of Vici syndrome: sleep abnormalities and complications of renal tubular acidosis. Am J Med Genet A. 2007;143(2):189–194. doi: 10.1002/ajmg.a.31584. [DOI] [PubMed] [Google Scholar]
- Ozkale M, Erol I, Gumus A, Ozkale Y, Alehan F. Vici syndrome associated with sensorineural hearing loss and laryngomalacia. Pediatr Neurol. 2012;47(5):375–378. doi: 10.1016/j.pediatrneurol.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Popp MW, Maquat LE. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell. 2016;165(6):1319–1322. doi: 10.1016/j.cell.2016.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portbury AL, Willis MS, Patterson C. Tearin’ up my heart: proteolysis in the cardiac sarcomere. J Biol Chem. 2011;286:9929–9934. doi: 10.1074/jbc.R110.170571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran N, Munteanu I, Wang P, et al. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol. 2013;125:439–457. doi: 10.1007/s00401-012-1073-6. [DOI] [PubMed] [Google Scholar]
- Riley LG, Cowley MJ, Gayevskiy V, et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. 2017;40(2):261–269. doi: 10.1007/s10545-016-0010-6. [DOI] [PubMed] [Google Scholar]
- Rogers CR, Aufmuth B, Monesson S. Vici syndrome: a rare autosomal recessive syndrome with brain anomalies, cardiomyopathy, and severe intellectual disability. Case Rep Genet. 2011;2011:1. doi: 10.1155/2011/421582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said E, Soler D, Sewry C. Vici syndrome-a rapidly progressive neurodegenerative disorder with hypopigmentation, immunodeficiency and myopathic changes on muscle biopsy. Am J Med Genet A. 2012;158A(2):440–444. doi: 10.1002/ajmg.a.34273. [DOI] [PubMed] [Google Scholar]
- Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- Schmid D, Munz C. Innate and adaptive immunity through autophagy. Immunity. 2007;27:11–21. doi: 10.1016/j.immuni.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Vici CD, Sabetta G, Gambarara M, et al. Agenesis of the corpus callosum, combined immunodeficiency, bilateral cataract, and hypopigmentation in two brothers. Am J Med Genet. 1988;29(1):1–8. doi: 10.1002/ajmg.1320290102. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Qi H, Taylor R, et al. The role of autophagy in mitochondria maintenance: characterization of mitochondrial functions in autophagy-deficient S. cerevisiae strains. Autophagy. 2007;3:337–346. doi: 10.4161/auto.4127. [DOI] [PubMed] [Google Scholar]
- Zhao H, Zhao YG, Wang X, Xu L, et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol. 2013;200:731–741. doi: 10.1083/jcb.201211014. [DOI] [PMC free article] [PubMed] [Google Scholar]