

Summary
β-Glucan-induced trained immunity in myeloid cells leads to long-term protection against secondary infections. Although previous studies have characterized this phenomenon, strategies to boost trained immunity remain undefined. We found that β-glucan-trained macrophages from mice with a myeloid-specific deletion of the phosphatase SHIP-1 (LysMΔSHIP-1) showed enhanced proinflammatory cytokine production in response to lipopolysaccharide. Following β-glucan training, SHIP-1-deficient macrophages exhibited increased phosphorylation of Akt and mTOR targets, correlating with augmented glycolytic metabolism. Enhanced training in the absence of SHIP-1 relied on histone methylation and acetylation. Trained LysMΔSHIP-1 mice produced increased amounts of proinflammatory cytokines upon rechallenge in vivo and were better protected against Candida albicans infection compared with control littermates. Pharmacological inhibition of SHIP-1 enhanced trained immunity against Candida infection in mouse macrophages and human peripheral blood mononuclear cells. Our data establish proof of concept for improvement of trained immunity and a strategy to achieve it by targeting SHIP-1.
Keywords: trained immunity, innate immune memory, myeloid, glycolytic metabolism, SHIP-1
Graphical Abstract
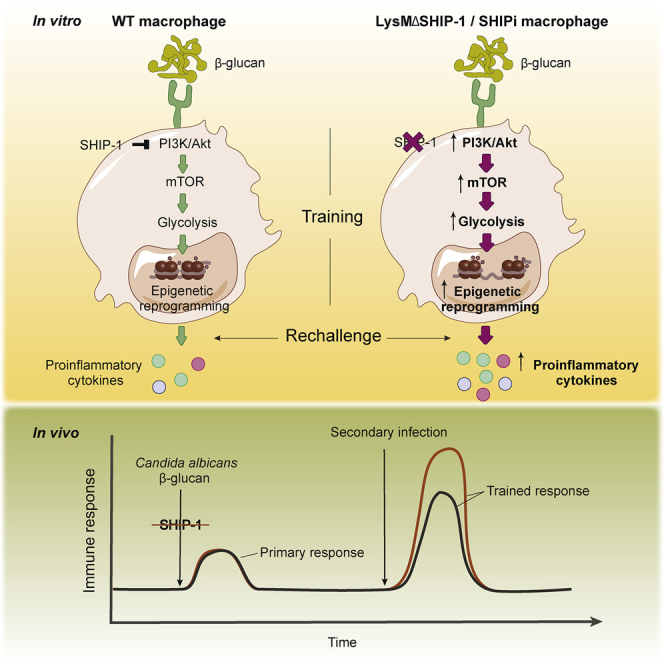
Highlights
-
•
β-glucan-induced trained immunity is enhanced by SHIP-1 deletion in macrophages
-
•
SHIP-1 regulates molecular, metabolic, and epigenetic hallmarks of trained immunity
-
•
Myeloid SHIP-1 deficiency improves protection conferred by trained immunity
-
•
SHIP-1 pharmacological inhibition enhances trained immunity in mice and human cells
Trained immunity leads to long-term protection, but strategies to boost it require further investigation. Saz-Leal et al. show that myeloid SHIP-1 deletion enhances trained immunity, improving the response to pathogen-specific or heterologous challenges. Pharmacological inhibition of SHIP-1 also potentiates this phenomenon, thereby revealing a potential tool to harness trained immunity.
Introduction
Innate immune cells challenged with certain stimuli undergo long-lasting changes that result in improved response to a second challenge by the same or different microbial insult, a process referred to as trained immunity (Quintin et al., 2012). Stimuli driving trained immunity lead to a shift to aerobic glycolysis (Arts et al., 2016b), accompanied by sustained changes in the epigenome, mainly via histone methylation and acetylation (Netea et al., 2016). Among trained immunity inducers, exposure to a low dose of Candida albicans or the fungal cell wall component β-glucan protects mice from secondary lethal systemic candidiasis (Bistoni et al., 1986) or heterologous Staphylococcus aureus septicemia (Di Luzio and Williams, 1978). This acquired resistance does not rely on T/B lymphocytes or natural killer (NK) cells but occurs in a myeloid-dependent manner (Cheng et al., 2014, Quintin et al., 2012).
The C-type lectin receptor Dectin-1 is critical for Candida albicans or β-glucan sensing, leading to immune training of monocytes (Quintin et al., 2012). These primed macrophages show heightened production of proinflammatory cytokines to a wide variety of insults (Ifrim et al., 2013, Quintin et al., 2012). Dectin-1-mediated training relies on activation of the PI3K (phosphoinositide 3-kinase)/mTOR (mammalian target of rapamycin) pathway (Cheng et al., 2014). PI3K-induced Akt signaling is tightly regulated by phosphoinositide phosphatases, which counterbalance PI3K activity (Eramo and Mitchell, 2016). Among those phosphatases, the hematopoietic-restricted SHIP-1 (SH2-containing inositol 5′-phosphatase 1) (Kerr, 2011) is of particular interest, as we showed that it binds to the intracellular tail of Dectin-1 receptor in granulocyte-macrophage colony-stimulating factor (GM-CSF) bone marrow-derived cells (Blanco-Menéndez et al., 2015).
Because Candida albicans-induced trained immunity relies on Dectin-1 and PI3K signaling, and SHIP-1 couples to Dectin-1 and counteracts PI3K function, we postulated that SHIP-1 targeting could modulate trained immunity triggered by Dectin-1. Our results indicate that SHIP-1 has a regulatory role in β-glucan-induced training, affecting all hallmarks involved in that process. Moreover, in vivo SHIP-1 deficiency in the myeloid compartment improves protection conferred by trained immunity. Notably, enhanced proinflammatory cytokine production and better protection was also achieved by pharmacological SHIP-1 inhibition in both mice and human peripheral blood mononuclear cells (PBMCs), providing a potential therapeutic approach to boost trained immunity.
Results
SHIP-1 Deletion Boosts β-Glucan-Induced Trained Immunity in Macrophages
Dectin-1 sensing of β-glucan induces trained immunity in myeloid cells, including PBMCs (Ifrim et al., 2013) or bone marrow-derived macrophages (BMDMs) (Walachowski et al., 2017). We initially stimulated BMDMs with purified particulate β-glucan from S. cerevisiae, a well-known ligand for Dectin-1 (Rosas et al., 2008). SHIP-1 basal expression in BMDMs was further induced after 1 day of β-glucan stimulation (Figure 1A). To study the potential involvement of SHIP-1 in Dectin-1-triggered trained immunity, we generated BMDMs from wild-type (WT) mice or mice bearing a myeloid-specific deletion of SHIP-1 in the myeloid compartment (LysMΔSHIP-1) (Collazo et al., 2012) (Figure 1B). Next, we adapted the proposed in vitro long-term scheme of trained immunity (Quintin et al., 2012) to IFN-γ-primed BMDMs, evaluating whether training with β-glucan boosts cytokine production in response to lipopolysaccharide (LPS) (Figure 1C). Of note, as previously described (Mosser and Zhang, 2008), IFN-γ priming was required to detect LPS-induced cytokines in BMDMs (Figure S1A), regardless of the induction of training. Surface expression of the receptors involved in β-glucan (Dectin-1; Figures 1D and S1B) and LPS (TLR4; Figures 1E and S1C) recognition were comparable between WT and LysMΔSHIP-1 BMDMs. We found that β-glucan-induced training resulted in increased cell viability in WT BMDMs (Figure S2), concurring with previous results (Bekkering et al., 2016, Garcia-Valtanen et al., 2017). Non-trained SHIP-1-deficient BMDMs showed higher viability than their WT counterparts, but the relative cell number after β-glucan training was similar between genotypes (Figure S2). To ensure the analysis of cell-intrinsic responses as described (Bekkering et al., 2016), cytokine production was normalized to the relative cell number present in each treatment.
Figure 1.

SHIP-1 Deletion Boosts β-Glucan-Induced Trained Immunity in Macrophages
(A) SHIP-1 expression by WB, normalized to β-actin, in bone marrow-derived macrophages (BMDMs) exposed (+) or not (−) to β-glucan (whole glucan particles) for the indicated time. Representative experiment of three performed.
(B) SHIP-1 protein expression in BMDMs. Representative experiment of six performed.
(C) Trained immunity in vitro model in mouse BMDMs. See also Figure S1A.
(D and E) Dectin-1 expression in BMDMs before β-glucan stimulation (D) or TLR4 expression both under non-trained (left) or β-glucan-primed (right) conditions, just before LPS stimulation (E), according to Figure 1C. FACS histograms representative of four independent experiments. See also Figures S1B and S1C.
(F) BMDMs were stimulated (+) or not (−) with β-glucan or LPS, and IL-1β (left), IL-6 (middle), and TNFα production (right) was analyzed in supernatants according to Figure 1C.
See also Figure S2. Independent experiments (N = 4 or 5) are shown. ∗p < 0.05 and ∗∗p < 0.01, paired Student’s t test comparing wild-type (WT) and LysMΔSHIP-1. #p < 0.05, paired Student’s t test comparing stimulated or not with β-glucan within the same genotype.
Pre-incubation of WT BMDMs with β-glucan prompted a greater production of IL-1β and TNFα in response to LPS (Figures 1F and S1A), reproducing trained immunity (Quintin et al., 2012). Notably, β-glucan-trained LysMΔSHIP-1 BMDMs showed an increased production of these trained immunity-associated cytokines compared with trained WT BMDMs (Figure 1F). Conversely, IL-6 was not induced following training or in the absence of SHIP-1 in this setting (Figure 1F). Of note, SHIP-1 deletion did not affect any of these inflammatory responses under non-trained conditions. These data indicate that SHIP-1 modulates the extent of LPS-induced proinflammatory cytokine production specifically during β-glucan training.
SHIP-1 Regulates Molecular and Metabolic Hallmarks of Trained Immunity
We tested whether the boost in β-glucan training in the absence of SHIP-1 was accompanied by regulation of key hallmarks involved in the process. Regarding the molecular pathway, Akt was phosphorylated in response to β-glucan in a time-dependent manner in WT BMDMs (Figure 2A, left, and Figure S3A), concurring with previous results (Cheng et al., 2014). Notably, LysMΔSHIP-1 BMDMs showed significantly increased and sustained Akt phosphorylation upon β-glucan stimulation (Figure 2A, left, and Figure S3A). The analysis of mTOR targets, S6 and 4EBP1, also revealed a maintained and significantly increased phosphorylation during the treatment with β-glucan in SHIP-1-deficient BMDMs (Figure 2A, right, and Figure S3B). Of note, a basal activation of the Akt pathway occurs in LysMΔSHIP-1 BMDMs. This is consistent with previous results in which the absence of SHIP-1 was associated with Akt overactivation and survival advantage (Antignano et al., 2010, Wang et al., 2002), which would concur with our results in non-trained BMDMs (Figure S2).
Figure 2.

SHIP-1 Regulates Molecular and Metabolic Hallmarks of Trained Immunity
(A) BMDMs were exposed to β-glucan for the indicated time, and phospho-Akt, Akt, phospho-S6, phospho-4EBP1, and β-actin were analyzed by WB. Representative experiment of five performed. See also Figure S3.
(B–E) BMDMs were left untreated (dashed lines) or treated for 1 day with β-glucan (solid lines), washed, rested for 3 days, and re-plated in equal numbers for determination of extracellular acidification rate (ECAR) in a glycolysis stress test upon sequential addition of glucose, oligomycin, and 2-deoxyglucose (2DG) as indicated (B). Analysis of basal glycolysis (C), maximal glycolysis (D), and glycolytic reserve (E). See also Figure S4. Mean ± SEM (B) or individual data (C–E) of five independent cultures are shown.
(F) BMDMs were trained (+) or not (−) with β-glucan for 1 day, washed, and rested for 4 days and chromatin immunoprecipitation (ChIP) against H3K4me3 was performed in which enrichment on the TNFα promoter was analyzed using qPCR. Mean ± SEM of five independent experiments is shown.
(G and H) BMDMs were incubated (+) or not (−) with the methyltransferase inhibitor 5′-deoxy-5′-(methylthio)adenosine (MTA), the histone demethylase inhibitor pargyline (G), the histone deacetylase activator resveratrol, or the histone acetyltransferase inhibitor EGCG (H) for 30 min before β-glucan training and after wash-out. TNFα production was analyzed in supernatants after LPS stimulation according to Figure 1C. Individual data corresponding to three (G) or four (H) independent experiments are shown.
In (C)–(H), ∗p < 0.05 and ∗∗p < 0.01, paired Student’s t test comparing WT and LysMΔSHIP-1. In (C)–(F), #p < 0.05, paired Student’s t test comparing stimulated or not with β-glucan within the same genotype.
Next, we measured the extracellular acidification rate (ECAR) in β-glucan-trained BMDMs in a glycolysis stress test prior to LPS stimulation (Figure 2B). Training with β-glucan for 5 days increased ECAR in WT BMDMs, a metabolic shift that was significantly boosted in trained SHIP-1-deficient BMDMs (Figure 2B), as reflected by enhanced basal (Figure 2C) and maximal (Figure 2D) glycolysis, together with a higher glycolytic reserve (Figure 2E). Increase in glycolytic reserve is the first metabolic signature associated with SHIP-1-deficient BMDMs upon β-glucan training (Figure S4). Consistent with data on signaling pathway activation (Figures 2A and S3), basal enhanced glycolysis was found in LysMΔSHIP-1 BMDMs (Figures 2C–2E and S4), although it did not result in higher cytokine production unless β-glucan-induced training was established (Figure 1F). These results suggest that SHIP-1 controls the extent of the glycolytic switch. Therefore, SHIP-1 deficiency may promote a pro-glycolytic state that could boost inflammatory response upon β-glucan-trained conditions.
To assess whether SHIP-1 could affect epigenetic reprogramming induced by β-glucan, we tested the presence of the activating histone 3 Lys 4 trimethylation (H3K4me3) (Cheng et al., 2014, Quintin et al., 2012). Chromatin immunoprecipitation (ChIP) analysis showed that H3K4me3 was specifically enriched by β-glucan training at TNFα promoter in WT BMDMs and further augmented in trained SHIP-1-deficient macrophages (Figure 2F), concurring with final enhanced TNFα production (Figure 1F). Moreover, inhibition of histone methyltransferases using 5′-deoxy-5′-(methylthio)adenosine (MTA) (Quintin et al., 2012) abolished TNFα overproduction in the absence of SHIP-1 upon training, whereas the histone demethylase inhibitor pargyline (Quintin et al., 2012) did not have any effect (Figure 2G). Considering that β-glucan-induced training relies also on histone acetylation, training in the presence of the histone deacetylase activator resveratrol (Cheng et al., 2014) or the histone acetyltransferase inhibitor EGCG (Ifrim et al., 2014) inhibited the enhanced TNFα produced by trained SHIP-1-deficient macrophages (Figure 2H). These results highlight SHIP-1 as a regulator of trained immunity by dampening the Akt/mTOR molecular pathway and the glycolytic switch and relying on the epigenetic reprogramming induced by β-glucan.
Myeloid-Specific Deletion of SHIP-1 Improves Trained Immunity In Vivo
The generation of trained immunity in vivo leads to cross-protection against diverse secondary infections (Netea et al., 2016). Signaling through PI3K is the canonical molecular pathway implicated in the development of these trained responses (Arts et al., 2016a, Cheng et al., 2014). To test the role of myeloid SHIP-1 in cytokine production under β-glucan training in vivo, WT and LysMΔSHIP-1 mice were challenged with LPS after training with β-glucan (Cheng et al., 2014), and serum cytokines were measured (Figure 3A). LPS-induced levels of IL-6 and TNFα were increased in sera from WT mice upon β-glucan pre-treatment (Figure 3B), indicative of the generation of a trained response (Quintin et al., 2012). Notably, serum levels of IL-1β, IL-6, and TNFα were further increased in LysMΔSHIP-1 trained mice compared with trained WT mice (Figure 3B), supporting the regulatory role of SHIP-1 upon β-glucan training also in vivo.
Figure 3.

Myeloid-Specific Deletion of SHIP-1 Improves Trained Immunity In Vivo
(A) In vivo model of training by two intraperitoneal (i.p.) β-glucan injections and secondary i.p. LPS challenge for measuring serum cytokines.
(B) Mice were treated according to Figure 3A. Serum was collected after 60 min (TNFα) or 90 min (IL-1β and IL-6) of LPS challenge, and cytokines were analyzed.
(C) In vivo model of training as in (A) but with secondary Candida albicans lethal infection.
(D) Survival curve according to Figure 3C.
(E) In vivo model of training by a systemic infection with a low dose of C. albicans followed by a secondary lethal challenge with the same pathogen.
(F) Survival curve according to Figure 3E.
(G and H) Renal cytokines on day 2 post-infection (p.i.) (G) and kidney fungal burden (H) at indicated time points p.i. were evaluated in trained mice, following model in Figure 3E.
In (B), (G), and (H), single dots correspond to individual mice. Mean ± SEM of two (B and H) or three (G) pooled experiments is shown, including at least 5 mice per condition. ∗p < 0.05 and ∗∗p < 0.01, unpaired Student’s t test comparing WT and LysMΔSHIP-1. #p < 0.05, unpaired Student’s t test comparing the same genotype stimulated or not with β-glucan. In (D) and (F), a pool of two experiments is shown, including between 6 and 16 mice per group as indicated. ∗∗p < 0.01, log rank test between WT and LysMΔSHIP-1 mice. #p < 0.05, log rank test comparing trained or not with β-glucan (D) or C. albicans (F) within the same genotype.
Protective response against lethal systemic Candida albicans infection by trained immunity relies on monocytes and macrophages (Quintin et al., 2012). After training with β-glucan, WT and LysMΔSHIP-1 mice were intravenously infected with a lethal dose of the clinical isolate C. albicans SC5314 (Figure 3C). Both WT and LysMΔSHIP-1 non-trained mice rapidly succumbed upon these infectious conditions (Figure 3D, dashed lines), indicating that SHIP-1 expression in the myeloid compartment is redundant for the primary response to lethal candidiasis. The protective effect of β-glucan administration against a lethal C. albicans infection was significantly improved in LysMΔSHIP-1 mice compared with WT littermates (Figure 3D, solid lines).
As trained immunity can be defined as a protection mechanism from secondary lethal C. albicans infection induced by a nonlethal encounter with the same pathogen (Quintin et al., 2012), we trained mice with a low dose of C. albicans followed by a lethal dose of the fungus (Figure 3E). Again, the training stimulus enlarged the survival time of WT mice and LysMΔSHIP-1 trained mice were more resistant than WT to lethal systemic candidiasis (Figure 3F, solid lines). Notably, we observed enhanced production of IL-1β and TNFα in Candida lethally infected kidneys from LysMΔSHIP-1 trained mice (Figure 3G), together with a decreased renal fungal burden (Figure 3H). These data indicate that SHIP-1 deficiency in myeloid cells enhances β-glucan- and Candida-induced trained immunity in vivo, improving the response to pathogen-specific or heterologous challenges.
Pharmacological Inhibition of SHIP-1 Enhances Trained Immunity
The relevance of the PI3K pathway in distinct pathologies has promoted the development of chemical SHIP-1 phosphatase inhibitors such as 3α-aminocholestane (3AC; SHIPi) (Brooks et al., 2010). We thus tested 3AC as a potential tool to boost trained immunity. BMDMs were trained with β-glucan in presence of different doses of 3AC (half maximal inhibitory concentration [IC50] = 13.5 μM; Brooks et al., 2015), and LPS-induced TNFα was measured (Figure 4A). Upon β-glucan training, SHIP-1 inhibition boosted TNFα production in a dose-dependent manner (Figure 4B).
Figure 4.

Pharmacological Inhibition of SHIP-1 Enhances Trained Immunity
(A) In vitro experimental model applied to mouse BMDMs, indicating when the SHIP-1 inhibitor (SHIPi) 3α-aminocholestane (3AC) was added.
(B) Mouse BMDMs were incubated with the SHIPi at the indicated concentrations. TNFα production was analyzed in supernatants of β-glucan-trained cells after LPS stimulation according to Figure 4A. Mean + SEM of four independent experiments is shown. ∗∗p < 0.01, paired Student’s t test between SHIPi-treated and non-treated cells.
(C) In vivo model of training by a systemic infection with a low dose of Candida albicans in the presence of SHIPi followed by a second lethal challenge with the same pathogen. When indicated, the inhibitor was administered intraperitoneally.
(D) Survival curve of 0.3% hydroxypropylcellulose (control) or SHIPi-treated mice according to Figure 4C. A pool of two experiments is shown, including between 10 and 19 mice per group as indicated. ∗∗p < 0.01, log rank test between trained control and SHIPi-treated. #p < 0.05, log rank test comparing trained or not with C. albicans within the same treatment.
(E) In vitro experimental model applied to human peripheral blood mononuclear cells (PBMCs) indicating when SHIPi was added.
(F) IL-1β, IL-6, and TNFα production was analyzed in supernatants of β-glucan-trained human PBMCs after LPS stimulation according to Figure 4E. Samples from seven independent donors are shown. ∗p < 0.05, paired Student’s t test.
To analyze the effect of 3AC SHIPi under in vivo infectious conditions, mice were administered SHIPi twice on consecutive days following the published regimen (Gumbleton et al., 2017), and coincident with the second day of 3AC administration, mice were trained with a low dose of C. albicans. Seven days later, mice were lethally infected with the same fungus (Figure 4C). Inhibition of SHIP-1 did not affect the survival of non-trained mice (Figure 4D, dashed lines) but improved the survival of Candida-trained mice (Figure 4D, solid lines).
To further explore the potential relevance of the use of 3AC SHIPi, we trained human PBMCs in the presence of SHIPi, rested and stimulated with LPS, and cytokine production was measured (Figure 4E). This scheme mirrors the stimulation pattern proposed for human PBMCs elsewhere (Quintin et al., 2012). Importantly, SHIP-1 inhibition boosted IL-1β, IL-6, and TNFα production in these β-glucan-trained human PBMCs (Figure 4F). Thus, our data indicate that SHIP-1 can be targeted with pharmacological inhibitors in both mice and human cells to boost trained immunity.
Discussion
Herein, we define SHIP-1 in myeloid cells as a target to improve trained immunity. Additionally, we provide a pharmacological approach, the SHIP-1 inhibitor 3AC, improving training-induced resistance to Candida infection and trained immunity in human PBMCs. Because modulation of myeloid progenitors in the bone marrow is an integral component of trained immunity (Mitroulis et al., 2018), and 3AC administration expands the hematopoietic stem cell compartment (Brooks et al., 2015), SHIP-1 inhibition could also influence this compartment. Although Candida-induced training and the primary response to the fungus are T/B cell independent (Bär et al., 2014, Quintin et al., 2012), systemic inhibition of SHIP-1 can also influence NK and T cells (Gumbleton et al., 2017), and we cannot rule out indirect effects on non-myeloid cells.
We show that SHIP-1 inhibition potentiates the canonical PI3K/Akt activation pathway, which also mediates trained immunity in response to other stimuli such as the bacillus Calmette-Guérin (BCG) vaccine (Arts et al., 2016a). SHIP-1 inhibition could represent a broad strategy to boost trained immunity. Indeed, SHIP-1 displays an inhibitory function in NOD2 signaling (Condé et al., 2012), the BCG-mediated trained immunity pathway (Kleinnijenhuis et al., 2012). Considering that BCG vaccination confers cross-protection to human viral infections (Arts et al., 2018b), SHIP-1 inhibitor could improve the protective effect of BCG.
Enhanced trained immunity could raise as an important host defense mechanism against infections or sepsis (Netea et al., 2016). However, because diverse endogenous danger signals from injured tissues can trigger innate immune memory hallmarks (Crișan et al., 2016b), caution is needed regarding the potential deleterious function of boosting trained immunity in diseases characterized by excessive inflammation. This notion could apply to atherosclerosis (Leentjens et al., 2018), cardiovascular events (Hoogeveen et al., 2017), gout (Crișan et al., 2016a), and a variety of autoimmune diseases and autoinflammatory disorders such as rheumatoid arthritis, systemic lupus erythematosus, and hyper-IgD syndrome (Arts et al., 2018a), in which monocytes and macrophages share a detrimental trained immunity-like phenotype. Similarly, boosting microglia-dependent training (Wendeln et al., 2018) through SHIP-1 inhibition could be detrimental for neurological disorders and stroke. Under these settings, SHIP-1 activators such as AQX-1125 (Stenton et al., 2013) could be potentially used to ameliorate an excessive and detrimental activation of trained immunity.
In conclusion, although studies on trained immunity have focused on the characterization of this phenomenon, strategies to enhance trained immunity deserve further investigation. Our data indicate that the trained immunity process can be boosted by targeting SHIP-1 in myeloid cells. Moreover, SHIP-1 inhibitors could be proposed as potential pharmacological tools to improve trained immunity in clinical settings in which enhancement of inflammatory responses is beneficial.
Experimental Procedures
Mice and Human Samples
Mice were bred at Centro Nacional de Investigaciones Cardiovasculares (CNIC) under specific pathogen-free conditions. WT C57BL/6J mice were used for SHIP-1 inhibition experiments. LysM+/+SHIP-1flox/flox (WT) and LysMCre/+SHIP-1flox/flox (LysMΔSHIP-1) (Collazo et al., 2012) were kept as littermates. Experiments were conducted with 8- to 12-week-old age-matched mice (regardless of gender). Experiments were approved by the animal ethics committee at CNIC and conformed to Spanish law under Real Decreto 1201/2005. Animal procedures were also performed in accordance to European Union (EU) Directive 2010/63EU and Recommendation 2007/526/EC.
Buffy coats from healthy volunteers were obtained from the Andalusian Biobank after ethical approval was obtained from the local Instituto de Salud Carlos III (ISCIII) Research Ethics Committee (PI 36_2017).
Trained Immunity In Vitro Models
BMDMs
BMDMs (105) were plated in 96-well plates (200 μL final volume; Corning) and stimulated with R10 or 100 μg/mL β-glucan (whole glucan particles, Biothera) for 24 hr. Then, cells were washed and rested 3 days in culture medium. On day 4, unless indicated, BMDMs were washed again and primed with 25 ng/mL IFN-γ (BD Biosciences) for 24 hr. On day 5, a final wash was performed, and cells were stimulated with R10 or 1 μg/mL standard Escherichia coli LPS (EK; Invivogen). To measure IL-1β production, following 4 hr of LPS challenge, cells were further stimulated for 2 hr with 5 mM ATP (Sigma-Aldrich), needed for inflammasome activation and pro-IL-1β processing (Schroder and Tschopp, 2010), and supernatants were harvested for ELISA assay. For TNFα and IL-6, after 24 hr of LPS stimulation, supernatants were collected for ELISA.
When required, BMDMs were pre-incubated for 30 min prior to β-glucan stimulation with 500 μM 5′-deoxy-5′-(methylthio)adenosine (MTA), 6 μM pargyline, 50 μM resveratrol, and 50 μM (-)-epigallocatechin-3-gallate (EGCG) (all from Sigma-Aldrich). SHIP-1 inhibitor (SHIPi; 3AC; Calbiochem) was also used at the indicated doses on β-glucan-trained BMDMs (toxic for non-trained cells). Inhibitors were also added in the first wash-out, before the resting period.
To assess receptor expression and cell viability, 6 × 105 BMDMs were plated in non-treated 24-well plates (1,200 μL final volume; Corning) and followed the training scheme described above. Dectin-1 expression was evaluated on day 0 prior to β-glucan addition. Cell viability and TLR4 expression were assessed on day 5 before LPS stimulation. At indicated times, cells were collected in PBS/EDTA and stained on ice-cold fluorescence-activated cell sorting (FACS) buffer (PBS/EDTA plus 3% fetal bovine serum [FBS]) for flow cytometry analysis.
For western blotting (WB) assays and ChIP analysis, 3 × 106 BMDMs were plated in six-well plates (3 mL final volume; Corning) and stimulated with R10 or 200 μg/mL β-glucan (to maintain mass/cell ratio) for given times. ChIP was performed on day 5, without IFN-γ priming, as detailed below.
To address metabolic status, 3 × 106 BMDMs were plated in non-treated 6-well plates (3 mL final volume; Corning) and followed the training scheme described above but with 200 μg/mL β-glucan (to maintain mass/cell ratio). On day 4, without IFN-γ priming, cells were detached in PBS/EDTA, plated at 105 cells/well, and rested overnight in R10 prior to the Seahorse XF glycolysis stress test (Agilent Technologies). When glycolytic metabolism was evaluated after overnight stimulation with β-glucan, BMDMs (105) were directly plated in 96-well Seahorse cell culture plates (200 μL final volume; Agilent Technologies) and stimulated with R10 or 100 μg/mL β-glucan the following day.
PBMCs
Total PBMCs (5 × 105) were plated in 96-well plates (200 μL final volume) and stimulated with 100 μg/mL β-glucan for 24 hr. Then cells were washed and rested for 6 days in culture medium. On day 7, PBMCs were stimulated with 1 μg/mL LPS (EK). After 24 hr, supernatants were collected for IL-1β, IL-6, and TNFα measurement by ELISA. When required, β-glucan-trained PBMCs were pre-incubated for 30 min with 10 μM 3AC (toxic for non-trained cells). Inhibitor was also added together with the first wash-out, before the resting period.
To assess cell viability, 3 × 106 total PBMCs were plated in 24-well plates (1,200 μL final volume; Corning) and followed the training scheme described here. On day 7, prior to LPS stimulation, cells were collected in PBS/EDTA and stained on ice-cold FACS buffer for flow cytometry analysis.
For normalization of cytokine production, the fold cell number in each condition was calculated as follows: live cell number/live cell number in average non-trained WT, according to Figure S2. In case of SHIP-1 inhibition experiments, non-treated cells were used as reference. Thus, cytokine production was normalized per cell number as (absolute cytokine value/fold cell number) in each condition.
In Vivo Models
Mice were trained with either two intraperitoneal (i.p.) injections of 1 mg β-glucan particles on days −7 and −4 or 2 × 104 Candida albicans intravenously (i.v.) on day −7. Sterile PBS was used as control. One week later, mice were challenged with 5 μg E. coli LPS (serotype O55:B5; Sigma-Aldrich) i.p., and blood was collected 60 min later to assess serum TNFα (Mouse TNFα DuoSet; R&D Systems) or 90 min later to evaluate serum IL-1β and IL-6. Alternatively, mice were lethally infected with 2 × 106 C. albicans i.v. and monitored daily for weight, general health, and survival, following institutional guidance. For qPCR analysis of renal cytokines, RNA was purified from whole kidneys on day 2 post-secondary infection (p.i.). Kidney fungal burden at indicated time points p.i. was determined by plating organ homogenates obtained mechanically over 70 μm cell strainers (BD Biosciences) after slicing the tissue, in serial dilutions on YPD agar plates; colony-forming units (CFUs) were counted after growth at 30°C for 48 hr, and data are shown as CFUs in total kidney. When required, mice were i.p. treated with 0.11 mg 3AC on days −8 and −7. 3AC was diluted in PBS 0.3% hydroxypropylcellulose (Sigma-Aldrich), used as control.
Quantification and Statistical Analysis
The statistical analysis was performed using Prism (GraphPad Software). Unless specified, statistical significance for comparison between two sample groups with a normal distribution (Shapiro-Wilk test for normality) was determined using two-tailed paired or unpaired Student’s t test. When groups were too small to estimate normality, a Gaussian distribution was assumed. Comparison of survival curves was carried out using the log rank (Mantel-Cox) test. Outliers were identified by means of Tukey’s range test. Differences were considered significant at p < 0.05 as indicated. Except when specified, only significant differences are shown. As indicated in figure legends, either a representative experiment or a pool is shown, and the number of repetitions of each experiment and number of experimental units (either cultures or mice) is indicated. In vitro experiments are shown as a pool of experiments, in which linked WT-LysMΔSHIP-1 dots represent independent cultures that were processed within the same experiment. In this way, an internal comparison between genotypes can be visually done. Different conditions within the same genotype in a particular experiment, although not connected by a matter of clarity, were also pair-analyzed, and statistically significant differences are indicated by pound signs (#).
Acknowledgments
We thank the members of the Immunobiology Lab for useful discussions. We thank the CNIC facilities and personnel, particularly Santiago Rodríguez and Ruben Mota, for their support. P.S.-L. is funded by grant BES-2015-072699 (“Ayudas para Contratos Predoctorales para la Formación de Doctores 2015”) from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO). C.d.F. is supported by the Asociación Española Contra el Cáncer (AECC) Foundation as a recipient of an “Ayuda Fundación Científica AECC a Personal Investigador en Cancer” grant. Work in the Sancho laboratory is funded by CNIC and grant SAF2016-79040-R from MINECO, Agencia Estatal de Investigación, and FEDER (European Fund for Regional Development); grant B2017/BMD-3733 Immunothercan-CM from Comunidad de Madrid; grant RD16/0015/0018-REEM from FIS-Instituto de Salud Carlos III, MINECO, and FEDER; Foundation Acteria; a Constantes y Vitales prize (Atresmedia); Foundation La Marató de TV3 (grant 201723); the European Commission (grant 635122-PROCROP H2020); and the European Research Council (ERC-2016-Consolidator Grant 725091). CNIC is supported by MINECO and the Pro-CNIC Foundation and is a Severo Ochoa Center of Excellence (MINECO award SEV-2015-0505). W.G.K. is an Empire Scholar of the State of New York, the Murphy Family Professor of Children’s Oncology Research, and is supported by funds from the Paige Arnold Butterfly Run.
Author Contributions
P.S.-L., C.d.F., P.B., and S.M.-C. performed the experiments. W.G.K. shared reagents. O.M.D. and J.D.C. prepared 3AC for the pharmacological targeting to SHIP-1. P.S.-L., C.d.F., and D.S. conceived and designed experiments, analyzed data, and wrote the manuscript. All authors discussed the results and the manuscript.
Declaration of Interests
C.d.F., P.S.-L., J.D.C., W.G.K., and D.S. have patents pending and issued on the use of SHIPi in modulating immune function. W.G.K. is chief scientific officer at Alterna Therapeutics, which is devoted to developing SHIPi for therapeutic purposes. J.D.C. serves on the Scientific Advisory Board of Alterna Therapeutics. Both J.D.C. and W.G.K. hold equity in Alterna Therapeutics. All other authors declare no competing interests.
Published: October 30, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.092.
Supplemental Information
References
- Antignano F., Ibaraki M., Ruschmann J., Jagdeo J., Krystal G. SHIP negatively regulates Flt3L-derived dendritic cell generation and positively regulates MyD88-independent TLR-induced maturation. J. Leukoc. Biol. 2010;88:925–935. doi: 10.1189/jlb.1209825. [DOI] [PubMed] [Google Scholar]
- Arts R.J.W., Carvalho A., La Rocca C., Palma C., Rodrigues F., Silvestre R., Kleinnijenhuis J., Lachmandas E., Gonçalves L.G., Belinha A. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 2016;17:2562–2571. doi: 10.1016/j.celrep.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts R.J., Joosten L.A., Netea M.G. Immunometabolic circuits in trained immunity. Semin. Immunol. 2016;28:425–430. doi: 10.1016/j.smim.2016.09.002. [DOI] [PubMed] [Google Scholar]
- Arts R.J.W., Joosten L.A.B., Netea M.G. The potential role of trained immunity in autoimmune and autoinflammatory disorders. Front. Immunol. 2018;9:298. doi: 10.3389/fimmu.2018.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts R.J.W., Moorlag S., Novakovic B., Li Y., Wang S.Y., Oosting M., Kumar V., Xavier R.J., Wijmenga C., Joosten L.A.B. BCG vaccination protects against experimental viral infection in humans through the induction of cytokines associated with trained immunity. Cell Host Microbe. 2018;23:89–100.e5. doi: 10.1016/j.chom.2017.12.010. [DOI] [PubMed] [Google Scholar]
- Bär E., Whitney P.G., Moor K., Reis e Sousa C., LeibundGut-Landmann S. IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity. 2014;40:117–127. doi: 10.1016/j.immuni.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Bekkering S., Blok B.A., Joosten L.A., Riksen N.P., van Crevel R., Netea M.G. In vitro experimental model of trained innate immunity in human primary monocytes. Clin. Vaccine Immunol. 2016;23:926–933. doi: 10.1128/CVI.00349-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bistoni F., Vecchiarelli A., Cenci E., Puccetti P., Marconi P., Cassone A. Evidence for macrophage-mediated protection against lethal Candida albicans infection. Infect. Immun. 1986;51:668–674. doi: 10.1128/iai.51.2.668-674.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Menéndez N., Del Fresno C., Fernandes S., Calvo E., Conde-Garrosa R., Kerr W.G., Sancho D. SHIP-1 couples to the Dectin-1 hemITAM and selectively modulates reactive oxygen species production in dendritic cells in response to Candida albicans. J. Immunol. 2015;195:4466–4478. doi: 10.4049/jimmunol.1402874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks R., Fuhler G.M., Iyer S., Smith M.J., Park M.Y., Paraiso K.H., Engelman R.W., Kerr W.G. SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J. Immunol. 2010;184:3582–3589. doi: 10.4049/jimmunol.0902844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks R., Iyer S., Akada H., Neelam S., Russo C.M., Chisholm J.D., Kerr W.G. Coordinate expansion of murine hematopoietic and mesenchymal stem cell compartments by SHIPi. Stem Cells. 2015;33:848–858. doi: 10.1002/stem.1902. [DOI] [PubMed] [Google Scholar]
- Cheng S.C., Quintin J., Cramer R.A., Shepardson K.M., Saeed S., Kumar V., Giamarellos-Bourboulis E.J., Martens J.H., Rao N.A., Aghajanirefah A. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collazo M.M., Paraiso K.H.T., Park M.-Y., Hazen A.L., Kerr W.G. Lineage extrinsic and intrinsic control of immunoregulatory cell numbers by SHIP. Eur. J. Immunol. 2012;42:1785–1795. doi: 10.1002/eji.201142092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condé C., Rambout X., Lebrun M., Lecat A., Di Valentin E., Dequiedt F., Piette J., Gloire G., Legrand S. The inositol phosphatase SHIP-1 inhibits NOD2-induced NF-κB activation by disturbing the interaction of XIAP with RIP2. PLoS ONE. 2012;7:e41005. doi: 10.1371/journal.pone.0041005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crișan T.O., Cleophas M.C.P., Oosting M., Lemmers H., Toenhake-Dijkstra H., Netea M.G., Jansen T.L., Joosten L.A.B. Soluble uric acid primes TLR-induced proinflammatory cytokine production by human primary cells via inhibition of IL-1Ra. Ann. Rheum. Dis. 2016;75:755–762. doi: 10.1136/annrheumdis-2014-206564. [DOI] [PubMed] [Google Scholar]
- Crișan T.O., Netea M.G., Joosten L.A. Innate immune memory: Implications for host responses to damage-associated molecular patterns. Eur. J. Immunol. 2016;46:817–828. doi: 10.1002/eji.201545497. [DOI] [PubMed] [Google Scholar]
- Di Luzio N.R., Williams D.L. Protective effect of glucan against systemic Staphylococcus aureus septicemia in normal and leukemic mice. Infect. Immun. 1978;20:804–810. doi: 10.1128/iai.20.3.804-810.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eramo M.J., Mitchell C.A. Regulation of PtdIns(3,4,5)P3/Akt signalling by inositol polyphosphate 5-phosphatases. Biochem. Soc. Trans. 2016;44:240–252. doi: 10.1042/BST20150214. [DOI] [PubMed] [Google Scholar]
- Garcia-Valtanen P., Guzman-Genuino R.M., Williams D.L., Hayball J.D., Diener K.R. Evaluation of trained immunity by β-1, 3 (d)-glucan on murine monocytes in vitro and duration of response in vivo. Immunol. Cell Biol. 2017;95:601–610. doi: 10.1038/icb.2017.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbleton M., Sudan R., Fernandes S., Engelman R.W., Russo C.M., Chisholm J.D., Kerr W.G. Dual enhancement of T and NK cell function by pulsatile inhibition of SHIP1 improves antitumor immunity and survival. Sci. Signal. 2017;10:eaam5353. doi: 10.1126/scisignal.aam5353. [DOI] [PubMed] [Google Scholar]
- Hoogeveen R.M., Nahrendorf M., Riksen N.P., Netea M.G., de Winther M.P.J., Lutgens E., Nordestgaard B., Neidhart M., Stroes E.S.G., Catapano A.L., Bekkering S. Monocyte and haematopoietic progenitor reprogramming as common mechanism underlying chronic inflammatory and cardiovascular diseases. Eur. Heart J. 2017 doi: 10.1093/eurheartj/ehx581. Published online October 24, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifrim D.C., Joosten L.A., Kullberg B.J., Jacobs L., Jansen T., Williams D.L., Gow N.A., van der Meer J.W., Netea M.G., Quintin J. Candida albicans primes TLR cytokine responses through a Dectin-1/Raf-1-mediated pathway. J. Immunol. 2013;190:4129–4135. doi: 10.4049/jimmunol.1202611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifrim D.C., Quintin J., Joosten L.A., Jacobs C., Jansen T., Jacobs L., Gow N.A., Williams D.L., van der Meer J.W., Netea M.G. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin. Vaccine Immunol. 2014;21:534–545. doi: 10.1128/CVI.00688-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr W.G. Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann. N Y Acad. Sci. 2011;1217:1–17. doi: 10.1111/j.1749-6632.2010.05869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinnijenhuis J., Quintin J., Preijers F., Joosten L.A., Ifrim D.C., Saeed S., Jacobs C., van Loenhout J., de Jong D., Stunnenberg H.G. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. U S A. 2012;109:17537–17542. doi: 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leentjens J., Bekkering S., Joosten L.A.B., Netea M.G., Burgner D.P., Riksen N.P. Trained innate immunity as a novel mechanism linking infection and the development of atherosclerosis. Circ. Res. 2018;122:664–669. doi: 10.1161/CIRCRESAHA.117.312465. [DOI] [PubMed] [Google Scholar]
- Mitroulis I., Ruppova K., Wang B., Chen L.S., Grzybek M., Grinenko T., Eugster A., Troullinaki M., Palladini A., Kourtzelis I. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. 2018;172:147–161 e12. doi: 10.1016/j.cell.2017.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser D.M., Zhang X. Activation of murine macrophages. Curr. Protoc. Immunol. 2008;Chapter 14:Unit 14.12. doi: 10.1002/0471142735.im1402s83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea M.G., Joosten L.A., Latz E., Mills K.H., Natoli G., Stunnenberg H.G., O’Neill L.A., Xavier R.J. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintin J., Saeed S., Martens J.H.A., Giamarellos-Bourboulis E.J., Ifrim D.C., Logie C., Jacobs L., Jansen T., Kullberg B.J., Wijmenga C. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas M., Liddiard K., Kimberg M., Faro-Trindade I., McDonald J.U., Williams D.L., Brown G.D., Taylor P.R. The induction of inflammation by dectin-1 in vivo is dependent on myeloid cell programming and the progression of phagocytosis. J. Immunol. 2008;181:3549–3557. doi: 10.4049/jimmunol.181.5.3549. [DOI] [PubMed] [Google Scholar]
- Schroder K., Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Stenton G.R., Mackenzie L.F., Tam P., Cross J.L., Harwig C., Raymond J., Toews J., Chernoff D., MacRury T., Szabo C. Characterization of AQX-1125, a small-molecule SHIP1 activator: part 2. Efficacy studies in allergic and pulmonary inflammation models in vivo. Br. J. Pharmacol. 2013;168:1519–1529. doi: 10.1111/bph.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walachowski S., Tabouret G., Fabre M., Foucras G. Molecular analysis of a short-term model of β-glucans-trained immunity highlights the accessory contribution of GM-CSF in priming mouse macrophages response. Front. Immunol. 2017;8:1089. doi: 10.3389/fimmu.2017.01089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.W., Howson J.M., Ghansah T., Desponts C., Ninos J.M., May S.L., Nguyen K.H., Toyama-Sorimachi N., Kerr W.G. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science. 2002;295:2094–2097. doi: 10.1126/science.1068438. [DOI] [PubMed] [Google Scholar]
- Wendeln A.-C., Degenhardt K., Kaurani L., Gertig M., Ulas T., Jain G., Wagner J., Häsler L.M., Wild K., Skodras A. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–338. doi: 10.1038/s41586-018-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.