

Abstract
Cancer immunotherapy approaches have emerged as novel treatment regimens against cancer. A particularly interesting avenue is the concept of in situ vaccination, where immunostimulatory agents are introduced into an identified tumor to overcome local immunosuppression and, if successful, mount systemic antitumor immunity. We had previously shown that nanoparticles from cowpea mosaic virus (CPMV) are highly potent in inducing long-lasting antitumor immunity when used as an in situ vaccine in various tumor mouse models. Here we asked whether the nanoparticles from tobacco mosaic virus (TMV) could also be applied as an in situ vaccine and, if so, whether efficacy or mechanism of immune-activation would be affected by the nanoparticle size (300 × 18 nm native TMV vs 50 × 18 nm short TMV nanorods), shape (nanorods vs spherical TMV, termed SNP), or state of assembly (assembled TMV rod vs free coat protein, CP). Our studies indicate that CPMV, but less so TMV, elicits potent antitumor immunity after intratumoral treatment of dermal melanoma (B16F10 using C57BL/6 mice). TMV and TMVshort slowed tumor growth and increased survival time, however, at significantly lower potency compared to that of CPMV. There were no apparent differences between TMV, TMVshort, or the SNP indicating that the aspect ratio does not necessarily play a role in plant viral in situ vaccines. The free CPs did not elicit an antitumor response or immunostimulation, which may indicate that a multivalent assembly is required to trigger an innate immune recognition and activation. Differential potency of CPMV vs TMV can be explained with differences in immune-activation: data indicate that CPMV stimulates an antitumor response through recruitment of monocytes into the tumor microenvironment (TME), establishing signaling through the IFN-γ pathway, which also leads to recruitment of tumor-infiltrated neutrophils (TINs) and natural killer (NK) cells. Furthermore, the priming of the innate immune system also mounts an adaptive response with CD4+ and CD8+ T cell recruitment and establishment of effector memory cells. While the TMV treatment also lead to the recruitment of innate immune cells as well as T cells (although to a lesser degree), key differences were noted in cyto/chemokine profiling with TMV inducing a potent immune response early on characterized by strong pro-inflammatory cytokines, primarily IL-6. Together, data indicate that some plant viral nanotechnology platforms are more suitable for application as in situ vaccines than others; understanding the intricate differences and underlying mechanism of immune-activation may set the stage for clinical development of these technologies.
Keywords: immunotherapy, cancer, in situ vaccination, cowpea mosaic virus, tobacco mosaic virus, melanoma
Graphical Abstract
INTRODUCTION
In the United States, melanoma is the fifth leading cancer in men and sixth in women; and its incidence rates are increasing.1 Of the seven most common cancers in the US, melanoma is the only one whose incidence is increasing. Lifetime risk for a person born in 1935 was estimated at 1 in 1500;2 in stark contrast, for a person born in 2010, risk is estimated at 1 in 39 for Caucasian men and 1 in 58 for Caucasian women.3 Melanoma is also the most common form of cancer for young adults.4 The overall 5 year survival rate for patients whose melanoma is detected early is about 98%. However, when diagnosed late, prognosis is poor: The survival rate falls to 63% when the disease reaches the lymph nodes and 16% when the disease metastasizes to distant organs.2 The decreased survival rate is correlated with the depth of the tumor and thus its ability to metastasize through underlying vasculature. Melanoma is very aggressive and as a result, if given enough time, it will metastasize. Another considerable challenge is the high likelihood of recurrence of this disease.5 Therefore, there is an urgent clinical need to provide novel therapeutic regimes that are effective and broadly applicable in the setting of metastatic disease. The most promising approaches are the cell-mediated immunotherapeutic strategies, which also enable the induction of immune memory to prevent outgrowth and recurrence.6,7
The immune system has evolved to recognize and remove things deemed “foreign” while maintaining harmony with “self”. Cancers produce neoantigens, differentiation antigens, or cancer testis antigens that label tumors as foreign. Under circumstances of normal immune function, these “foreign” signals are used by the immune system to eradicate tumors through the cancer-immunity cycle.8 The cancer-immunity cycle is a sequential set of steps leading to the targeting of tumor cells by the immune system. It progresses through the identification of tumor antigens, presentation of antigens by antigen-presenting cells (APCs), activation and priming of immune mediators, mediator homing and migration to the tumor bed, and immunological destruction of the tumor by cell-mediated mechanisms. With each iteration, the cycle expands until complete eradication of the tumor is achieved. At which point, the immune system uses feedback mechanisms to down regulate immune responses. However, the disruption of any of these steps can bring the cycle to a halt, consequently creating a protumor situation.8 The tumor microenvironment (TME) has been identified as one of the locations where the cancer-immunity cycle is deregulated by functioning as an immune rheostat or “immunostat”.8 The TME can be highly immunosuppressive, due to a variety of factors including the impairment of antigen-presenting machinery, increase of immunosuppressive factors, inhibition of dendritic cell maturation, and up-regulation of regulatory T cells. Interestingly, instead of avoiding interactions with the immune system, tumors actively recruit immunosuppressive immune cells through cytokine signaling.9 In addition, once infiltrated in the TME, signaling further modulates immune responses by switching cells from an antitumor phenotype to a tumor enabling phenotype.7 Not only are these protumor cells tolerant to tumors, they also support tumor development by secreting cytokines and growth factors.10 A promising avenue in cancer therapy is the reprogramming of the TME to restore the cancer-immunity cycle and thus the body’s own defense against tumors.11,12
Cancer immunotherapy approaches generally target one of two mechanisms: blocking immunosuppressive targets (i.e., IL-10, CD73, TIM-3) or activating immune targets (i.e., CD137 and tumor vaccines).13 Over the past three decades, several immunotherapies like immunostimulatory cytokines and recently checkpoint inhibitors (CTLA4 and PD-1/PDL-1) have been developed.14–16 However, the systemic administration of exogenous immune modulators only targets specific steps of the cancer-immunity cycle, are not delivered at physiological ranges, or lack tumor specificity leading to serious immune side effects and suboptimal responses.17,18 To avoid the toxicities, alternative and safer localized strategies are required. The concept of in situ vaccination has emerged. An in situ vaccine, generally an immunostimulatory adjuvant, is administered directly into an identified tumor or metastatic site to reprogram the TME from an immunosurpressive to immunostimulated phenotype, thus restarting the cancer-immunity cycle and, if successful, mobilizing a systemic antitumor immune response. For example, Imlygic (tamilogene laherparepvec or T-VEC) has shown success as an in situ vaccine in the setting of melanoma. T-VEC is a genetically modified oncolytic herpes virus, engineered to express the cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF). While TVEC has shown efficacy in the context of melanoma,19,20 concerns surround the safety aspects of using a human pathogen as a therapeutic. Possible safer alternatives are biologics derived from nonhuman pathogens, such as plant viral-based approaches.
Toward this end, we and others have recently demonstrated that plant viral and viral-like particles from cowpea mosaic virus (CPMV),21 papaya mosaic virus (PapMV),22 and potato virus X (PVX)23 can induce immune-mediated antitumor responses when introduced into the TME as an in situ vaccine. It is hypothesized that the plant virus-based nanoparticles activate the innate immune system locally. This innate immune-activation is thought to overcome the immunosuppressive TME, restarting the cancer-immunity cycle, leading to systemic elimination of cancer cells through the adaptive immune system. While the anti-tumor immunestimulation of PapMV was attributed to its packaged RNA and associated stimulation of toll-like receptors (TLRs),22,24,25 our previous studies with RNA-free “empty” CPMV (termed eCPMV) indicated that the RNA cargo is not required (at least in the case of the CPMV platform).21 Thus, it is likely that innate receptors such as pattern recognition receptors (PRRs) play a key role recognizing the multivalent nature of the plant virus or plant viral-like nanoparticles. The repetitive, multivalent coat protein assemblies are products known as pathogen-associated molecular patterns (PAMPs).26
The potency of the plant viral in situ vaccination strategy is intriguing, and questions remain regarding the underlying immunology as well as the engineering design space, i.e., whether the antitumor effect is a property of any (plant) virus-based nanomaterial or whether shape, size, and molecular composition of the virus relate to its efficacy. Therefore, the present work set out to investigate the following: (1) we asked whether tobacco mosaic virus (TMV) could be used as an in situ vaccine to elicit an antitumor immune response against melanoma and, if so, (2) whether the potency of this response would be influenced by structural parameters. To test this, we compared assembled TMV vs free coat proteins (CPs), full-length 300 × 18 nm TMV vs shorter TMV variants measuring ~50 × 18 nm (TMVshort) vs spherical TMV nanoparticles (termed SNPs). (3) We addressed whether the TMV-mediated antitumor responses against melanoma would match the efficacy observed with CPMV nanoparticles. While filamentous plant viruses, such as PVX and PapMV, were utilized in previous work, TMV provides an interesting alternative because of its robust and well-developed surface chemistry and its shape engineering capabilities; the synthesis of complex shapes such as star-shaped TMV and boomerang-shaped TMV has been described in the literature.27,28 This level of engineer ability has not yet been demonstrated with the filamentous plant virus platforms. Another key difference between the filamentous platforms PVX and PapMV vs TMV is the flexible modulus of the particles: while PVX and PapMV are flexible filaments, TMV is a rigid nanotube. For these reasons, we felt that TMV would be a welcome addition to test as an in situ vaccine.
Figure 1 depicts the library of viral nanoparticles under consideration. Using a mouse model of dermal melanoma, we compared antitumor efficacy of these plant viral nanoparticles side-by-side; temporal changes in cytokine and immune-cell profiles were assessed using Luminex assays, histology, and flow cytometry to gain insights into whether differences in efficacy could be explained merely by differences in magnitude of immune-activation or underlying differences in the mechanism of immune-activation.
Figure 1.

CPMV and TMV nanoparticle library. (A) Schematic showing the different TMV formulations: native TMV measures 300 × 18 nm and consists of 2130 copies of an identical coat protein (CP). Free CP was obtained through TMV disassembly, and TMVshort was obtained through assembly using in vitro transcribed RNA of defined lengths; spherical TMV nanoparticles (SNPs) were obtained through heat-mediated shape-shifting. (B) Depiction of CPMV; CPMV consists of 60 copies each of a small (S) and large (L) CP.
MATERIALS AND METHODS
VLP Production.
TMV was propagated in Nicotiana benthamiana plants and purified as previously reported.29 CPMV was propagated in Vigna unguiculata plants and purified as previously reported.30
TMV disassembly methods to yield purified CPs were as previously described.31,32 In brief, particles were exposed to cold glacial acetic acid in a 1:2 ratio (v/v), vortexed, and allowed to sit on ice for 20 min. Afterward, tubes were centrifuged for 20 min at 20 000g and 4 °C. The supernatant was transferred and diluted in a 1:1 ratio (v/v) with Milli-Q water. The CP were dialyzed against Milli-Q water for at least 48 h at 4 °C, with daily changing of the water. At the end of the dialysis, CP were centrifuged for 20 min at 20 000g and 4 °C. CP were then resuspended in 200 μL of 75 mM sodium phosphate buffer at pH 7.2 and resuspended overnight. The next day, CPs were centrifuged for 20 min at 10 000g at room temperature.
To produce TMVshort, an RNA-templated self-assembly protocol was used.33 In brief, DNA plasmid pIDTblue-TMVshort (Figure S1), containing the first 1177 nt of the TMV genome (6.5 kb) including the origin-of-assembly site (OAS), a T7 promotor, and an ampicillin gene were amplified in E. coli and purified by Qiagen Maxi-Prep. Plasmid pIDTblue-TMVshort was linearized using BamH1 and NgoMIV restriction enzymes (NEB). Following linearization, DNA fragments were purified and transcribed using a T7-based MegaScript kit (Thermo Fisher Scientific), and the resulting RNA was purified with the MEGAclear kit (Thermo Fisher Scientific). To yield TMVshort (measuring 54 nm as defined by the length of the in vitro transcribed RNA template), obtained synthetic RNA transcripts were incubated with purified TMV coat proteins (CPs) at 37 °C in 75 mM sodium phosphate buffer at pH 7.2 overnight (Figure S2).
Spherical TMV nanoparticles (SNPs) were produced through thermal shape-transition as previously reported34,35 (Figure S3).
Agarose Gel Electrophoresis.
To confirm plasmid sizes, restriction digest cuts, and T7 reverse transcription, nucleic acids were loaded with 6× agarose loading dye (Invitrogen) and analyzed in a 0.8% (w/v) agarose gel (in 1× TBE) stained with GelRed nucleic acid stain (BioRad). DNA/RNA samples were separated at 100 V for 30 min. Gels were then visualized under UV light using an Alpha Imager (Alpha Innotech) (Figure S2).
UV-vis Spectroscopy.
The concentration as well as the purity of CPMV, TMV particles, and CPs was determined by UV-vis spectroscopy, using the NanoDrop2000 spectrophotometer (Thermo Fisher Scientific). Protein concentration was determined using the Beer-Lambert law and the protein/assembly specific extinction coefficients (ε); TMV ε = 3.1 mL mg−1 cm−1 at 260 nm), TMV CP ε = 1.2 mL mg−1 cm−1 at 260 nm, and CPMV ε = 8.1 mL mg−1 cm−1 at 260 nm. The RNA/protein (260/280) ratios were used to measure the purity of the CPMV (A260/280 = 1.8), TMV and TMVshort (A260/280 = 1.2), and TMV CPs (A260/280 = 0.65).
Transmission Electron Microscopy (TEM), Scanning Transmission Electron Microscopy (STEM), and Scanning Electron Microscopy (SEM).
TEM and STEM imaging was performed after CPMV and TMV particle purification to confirm their integrity. CPMV and TMV samples (0.1 mg mL−1, in dH2O) were placed on carbon-coated copper grids and negatively stained with 0.2% (w/v) uranyl acetate. For TEM imaging, grids were imaged using a Zeiss Libra 200FE Transmission Electron Microscope, operated at 200 kV. For STEM imaging, samples were prepared as described above and imaged using a FEI Helios NanoLab 650 Field Emission Scanning Electron Microscope with Focused Ion Beam with XEDS, operated at 30 kV and 0.4 nA at 50–100k magnification. For SEM imaging of SNPs, samples were prepared on silica wafers and sputter coated with gold prior to imaging using a FEI Helios NanoLab 650 Field Emission Scanning Electron Microscope with Focused Ion Beam with XEDS, operated at 1 kV at 10k magnification.
Fast Protein Liquid Chromatography (FPLC).
To further confirm purity and structural integrity of TMV particles and CPs, FPLC was performed using an ÄTKA purifier and Superose 6 column and methods as previously reported.30,35,36
Cell Culture.
B16F10 cells (ATCC) were cultured in Dulbecco’s modified Eagle’s media (DMEM, Life Technologies), supplemented with 10% (v/v) fetal bovine serum (FBS, Atlanta Biologicals) and 1% (v/v) penicillin-streptomycin (Life Technologies). Bone-marrow-derived dendritic cells (BMDCs) were harvested from the femurs and tibias of euthanized C57BL/6 mice. The bones were rinsed in 70% ethanol, epiphyses were removed, and the marrow was flushed. Cells were broken up to a single cell suspension and applied to a 40 μm cell strainer. Red blood cells were depleted with ammonium chloride potassium (ACK) lysing buffer, followed by washing with phosphate buffer saline (PBS). The marrow cells were filtered, washed, and resuspended at 1 × 106 cells/mL in T cell media from Roswell Park Memorial Institute RPMI (Corning) supplemented with 10% (v/v) fetal bovine serum (FBS), 1% (v/v) penicillin-streptomycin (Pen/Strep), 1% (v/v) MEM nonessential amino acids (Corning), 1 mM sodium pyruvate, and 50 mM beta-mercaptoethanol (Life Technologies) supplemented with 10 ng/mL mouse interleukin 4 (IL-4) (BioLegend) and 15 ng/mL mouse GM-CSF (BioLegend). Then, 3 mL of cells was plated in each well of a six-well plate and incubated at 37 °C. The media was removed and replaced with fresh T cell media supplemented with IL-4 and GM-CSF on day 3, and an additional 3 mL of fresh T cell media supplemented with IL-4 and GM-CSF was added on day 5. Cells were harvested on day 7.
Animal Studies.
All experiments were conducted in accordance with Case Western Reserve University’s Institutional Animal Care and Use Committee. C57BL/6J male mice (Jackson Laboratory) were used. B16F10 tumors were induced intradermally into the right flank of C57BL/6J mice (1.25 × 105 cells/50 μL of DMEM media). Animals were monitored, and tumor volumes were calculated as V = 0.5 (a × b2); where a is length, and b is width of the tumor.
Treatment Schedule.
Eight days post tumor induction (day 0), mice were randomly assigned to the following groups (n = 8): PBS, TMV, TMVshort, TMV CP, or CPMV. Mice were treated intratumorally (20 μL), every 4 days, with 100 μg of the respective CPMV/TMV particle, CP, or PBS. Mice were sacrificed when tumors reached a volume >1500 mm3 or when tumors exceed 10% body weight.
Luminex Assay.
Intradermal B16F10 melanoma tumors were induced in C57BL/6J male mice (Jackson) as described above. Eight days post tumor induction (day 0), mice were randomly assigned to the following groups (n = 4): PBS, TMV, TMVshort, TMV-CP, or CPMV. Two treatment schedules were analyzed: (1) Mice were treated intratumorally (20 μL) once with 100 μg of the respective CPMV/TMV particle, CP, or PBS, and tumors were harvested at 24 h post treatment. (2) Mice were treated intratumorally (20 μL) on day 0 and day 4 with 100 μg of the respective CPMV/TMV particle, CP, or PBS, and tumors were harvested at 4 h after the second injection (day 4). Millipore Milliplex MAP mouse 32-plex was run by Eve Technologies Corporation (Calgary, Canada).
In vitro Luminex assays were done with BMDCs. Cells were isolated as previously described and treated with PBS (negative control), TMV, TMVshort, TMV CP, CPMV, or lipopolysaccharides (LPS) (positive control). LPS is a known agonist of TLR4. Media was collected 24 h post treatment. Millipore Milliplex MAP mouse 32-plex was run by Eve Technologies Corporation.
Cytokine and Chemokine Pathway Analysis.
Cytokine and chemokines that were significant between treatment groups in the Luminex assay were imported into Crosstalker software (https://sb4j.case.edu) for pathway analysis. The cytokine/chemokines added function as seeds; crosstalker software then develops networks based on published interactions. Seeds were mapped by corresponding gene symbols to human genes using the BioGRID database. Result networks were tested for functional enrichments by hypergeometric test to assess over-representation of gene sets from the selected pathway database. A more comprehensive description can be found.37,38
Flow Cytometry.
Intradermal B16F10 melanomas were induced in C57BL/6J male mice (Jackson) as described above. Eight days post tumor induction (day 0), mice were randomly assigned to the following groups (n = 4): PBS, TMV, or CPMV. Mice were treated intratumorally as described above. Tumors were harvested at 4 and 10 days post initial treatment. Tumors were prepared for flow cytometry as previously described.21 Briefly, single tumor cell suspensions were incubated in anti-CD16/CD32 antibody (BioLegend) for Fc receptors blocking for 15 min in the dark. Cell viability was assessed using Zombie Yellow Fixable Viability Kit (BioLegend) for 30 min incubation in the dark on ice followed by a single wash in 1× PBS. Surface staining was performed in the dark for 30 min at 4 °C in FACS staining buffer (1 mM EDTA, 1% (v/v) FBS, and 25 mM HEPES, pH 7.0 in Ca2+- and Mg2+-free PBS). Cells were then washed twice with staining buffer followed by fixation in 3% (v/v) paraformaldehyde. Fluorophore-labeled monoclonal antibodies were used for surface staining of cell-type-specific markers including CD45, CD11b, CD11c, NK1.1, Ly6C, Ly6G, CD3, CD8, CD4, CD62L, and CD44 (BioLegend). Tumors harvested at day 4 were subjected to the innate cell antibody panel, and the day 10 tumors were tested for the adaptive panel (Table 1). Flow cytometry analyses were performed on a BD LSR II flow cytometer (BD Biosciences), and data was analyzed using the FlowJo software (Tree Star Inc.).
Table 1.
Antibodies Used for Flow Cytometry
| dye | marker | vendor | innate panel catalog no. | clone/host | isotype | concentration |
| pacific blue | CD4S | BioLegend | 103126 | 30-F11 Rat | IgG2b, K | 0.5 mg/mL |
| FITC | CD11b | BioLegend | 10120S | Ml/70 Rat | IgG2b, K | 0.5 mg/mL |
| PE | CD80 | BioLegend | 104707 | 16–10A1A Hamster | IgG | 0.2 mg/mL |
| PE-Cy7 | CD86 | BioLegend | 10S014 | GL-1 Rat | IgG2a, K | 0.2 mg/mL |
| APC | I-A/I-E (=MHCII) | BioLegend | 107613 | MS/114.1S.2 Rat | IgG2b, K | 0.2 mg/mL |
| APC/Cy7 | Ly-6G | BioLegend | 127623 | 1A8 Rat | IgG2a, K | 0.2 mg/mL |
| APC | CD11c | BioLegend | 10120S | N418 A Hamster | 0.2 mg/mL | |
| PE | F4/80 | BioLegend | 123109 | BM8 Rat | IgG2a, K | 0.2 mg/mL |
| PE/Cy7 | Ly6C | BioLegend | 128018 | HK1.4 Rat | IgG2c, K | 0.2 mg/mL |
| BV60S | NK1.1 | BioLegend | 108737 | PK136 | IgG2a, k | 0.2 mg/mL |
| dye | marker | vendor | adaptive panel catalog no. | clone/host | isotype | concentration |
| pacific blue | CD45 | BioLegend | 103126 | 30-F11 Rat | IgG2b, K | 0.5 mg/mL |
| FITC | CD4 | BioLegend | 100405 | GK1.5 Rat | IgG2b, K | 0.5 mg/mL |
| PE | CD44 | BioLegend | 103007 | 1M7 Rat | IgG2b, K | 0.2 mg/mL |
| PE-Cy7 | CD62L (L-selectin) | BioLegend | 104417 | MEL-14 Rat | IgG2a, K | 0.2 mg/mL |
| APC | CD8α | BioLegend | 100712 | S3–6.7 Rat | IgG2a, K | 0.2 mg/mL |
| APC/Cy7 | CD3ε | BioLegend | 100330 | 14S-2C11 A. Hamster | IgG | 0.2 mg/mL |
Histology, Immunostaining, and Immunohistochemistry (IHC).
Intradermal B16F10 melanomas were induced in C57BL/6J male mice (Jackson) as described above. Eight days post tumor induction (day 0), mice were randomly assigned to the following groups (n = 4): PBS, TMV, or CPMV. Mice were treated intratumorally as described above, and tumors where harvested at 4, 6, or 10 days post initial treatment. Tumors were then fixed in 4% (v/v) paraformaldehyde in PBS for 5–7 days and then transferred to 1% (v/v) paraformaldehyde in PBS and taken to the Case Western Reserve University Pathology core for sectioning and staining. Tumors for frozen IHC were flash frozen and also delivered to the Pathology core for processing.
Paraformaldehyde tumors were treated serially in 10% (v/v) neutral buffered formalin (twice), 70% (v/v) ethanol, and 80% (v/v) ethanol for 90 min at 45 °C each. Subsequently, tumors were serially treated for 2 h at 45 °C in 95 and 100% (v/v) ethanol (three times). Next tumors were placed in xylene for 90 min at 45 °C (this step was carried out twice, i.e., slides were moved to a solution of fresh xylene after the first 90 min incubation step). Finally, tumors were kept in paraffin for 2 h at 45 °C (this step was carried out three times). Then, samples were kept at 60 °C until they were embedded in paraffin. Paraffin blocks were soaked in cold dH2O followed by sectioning of 5 μM thin tissue sections using a microtome. Then, the sections were placed in 45 °C dH2O, placed on a charged slide, and allowed to dry overnight at room temperature.
For H&E staining, slides from paraffin embedded tissues were baked at 60 °C for 90 min and then deparaffinized in xylene for 2 min. Slides were then rehydrated through a series of graded ethanol washes for 2 min each: 100, 95, and 70% (v/v) followed by a rinse in dH2O. Slides were stained by placing them for 15 min in Harris Modified Hematoxylin (Fisher Scientific) followed by rinsing in water five times (10 dips each). Slides were differentiated with eight dips in 1% (v/v) acid alcohol (hydrochloric acid in 70% (v/v) ethanol) followed by five dips in saturated lithium chloride (1% (v/v) in dH2O); then, slides were washed by rinsing in tap water (10 dips, five times). Slides were placed in 70 and then 95% (v/v) ethanol for 2 min each prior to counterstaining with Esosin Y (Fisher Scientific) for 3 min. Slides were subsequently dehydrated in 100% ethanol for 2 min (five times) and cleared in xylene for 2 min (twice). Slides were mounted with resinous mounting medium (Mercedes Medical, OpticMount) and allowed to dry overnight.
IHC slides from paraffin embedded tissues were baked at 60 °C for 75 min and then deparaffinized in xylene twice for 7 min. Slides were then rehydrated through a series of graded ethanols: 100% (twice for 2 min each), 95% (twice for 2 min each), and 70% for 2 min followed by a rinse in dH2O. Antigen retrieval was achieved using 0.01 M citrate buffer at pH 6.0 (Vector Laboratories, H3300) and by placing slides in a pressure cooker for 30 s at 123 °C followed by cooling at room temperature for 20 min, and followed by a rinse in dH2O for 2 min. The endogenous peroxidase activity was then blocked in Peroxidazed (PX968M, BioCare Medical) for 8 min, followed by rinsing in dH2O. Following peroxidase block, endogenous mouse IgG was blocked using Rodent Block (RBM961, BioCare Medical) for 20 min, followed by a rinse using Tris-buffered saline (TBS) with Tween-20 (TBST). The slides were then immunostained with the corresponding primary antibodies (Table 2) or without antibodies for the negative controls (Protein Block Serum-Free [Antibody Diluent], X0909, DAKO Cytomation) for 1 h at room temperature, followed by TBST rinse. Slides were then treated with Rabbit-on-Rodent HRP Polymer (RMR622H, BioCare Medical) for 30 min and rinsed with TBST. The slides were incubated with the Betazoid DABKit (BDB2004L, Betazoid DAB Chromogen, BioCare Medical) mix for 5 min in the dark followed by rinsing with dH2O. This was followed by counterstaining with CAT Hematoxylin (CATHE-M, BioCare Medical).
Table 2.
Antibodies Used for Immunohistochemistry
| marker | vendor | catalog no. | clone/host |
|---|---|---|---|
| CD45 | Abcam | ab10558 | Rabbit |
| CD3 | Abcam | abl6669 | Rabbit |
After processing, all slides were scanned digitally using the Leica SCN400 Slide Scanner (Leica Biosystems, Wetzlar, Germany) for digital image storage and analysis.
RESULTS
CPMV and TMV Library Production.
In this study, we set out to address (i) whether TMV would be effective as an in situ vaccine against melanoma and whether the shape (TMV rod vs SNP), aspect ratio (AR17 for native TMV measuring 300 × 18 nm and AR3 for TMVshort measuring 54 × 18 nm), or state of assembly (multivalent TMV particle vs soluble CP) of TMV would impact its efficacy as an in situ vaccine. The icosahedron CPMV, measuring 30 nm in diameter, was used as a standard, because we previously reported its potent efficacy when used as an in situ vaccine against melanoma among other metastatic tumors.21
The viral nanoparticle library was obtained using a combination of molecular farming and self-assembly methods (Figure 2). CPMV, a 30 nm-sized icosahedron with P3 symmetry consisting of 60 copies each of a small (S) and large (L) coat protein, was produced and isolated from Vigna unguiculata (black-eyed peas or cowpea plants No. 5, Figure 2A). The following TMV con-figurations were obtained: native TMV measuring 300 × 18 nm was produced in and isolated from Nicotiana benthamiana (Australian tobacco, Figure 2B). To obtain TMV CP and TMVshort, established dis and reassembly methods were employed.31,33 CP was obtained through disassembly of native TMV, and purified CP was assembled on synthetic TMV transcripts containing an origin-of-assembly site (OAS) to initiate the self-assembly reaction; TMV transcripts containing 1177 nt of the 6.5 kb TMV genome (Figure 2) were used as a template yielding TMVshort particles measuring 54 nm in length. The width is constant at 18 nm (Figure 2C,D). Lastly, SNP measuring ~250 nm in diameter were obtained through previously described heat-transformation of TMV to SNP (Figure 2E).
Figure 2.

Production of viral nanoparticle library. Schematic showing the molecular farming of CPMV (A) and TMV (B). (C) Disassembly of TMV into its CPs (and genomic RNA [gRNA]). (D) Assembly TMVshort via RNA-templated assembly. (E) Thermal transition of TMV into SNP.
The CPMV and TMV particles and CPs thereof were characterized by UV-vis spectroscopy, FPLC, and S/TEM to confirm concentration, purity, and structural integrity prior to use in animal studies (Figure 3).
Figure 3.

Characterization of viral nanoparticle libraries. (A) UV-vis spectroscopy of CPMV, TMV, TMVshort (TMV-S), and TMV-CP. (B) Fast protein liquid chromatography (FPLC) of CPMV, TMV, TMVshort (TMV-S), and TMV-CP. (C) TEM of CPMV and TMV and STEM of TMVshort; all samples were negatively stained with 0.2% (w/v) uranyl acetate.
The concentration as well as the purity of CPMV and TMV particles and CPs was determined by UV-vis spectroscopy. The RNA:protein (260/280) ratios indicated that pure and intact CPMV (A260/280 = 1.8), TMV and TMVshort (A260/280 = 1.2), and TMV CPs (A260/280 = 0.65) were produced (Figure 3A). FPLC using ÄTKApurifier and a Superose6 column showed the typical elution profiles for intact CPMV at 12–15 mL, for intact TMV and TMVshort at ~8 mL (due to the high molecular weights of either assembly, 5.6 × 106 and 40 × 106 kDa for TMVshort and TMV, respectively; differences in elution profiles are not apparent due to the limitations of the pore size resolution of the Superose6 column), and for soluble CPs at 16–17 mL (Figure 3B). TEM and STEM analysis of negatively stained samples further confirmed structural integrity and the expected sizes of the viral nanoparticles (Figure 3C) with CPMV nanoparticles appearing spherical in shape with a diameter of 30 nm, and native TMV forms high aspect ratio soft-matter nanorods measuring 300 × 18 nm. A total of 240 TMVshort particles were analyzed, and their length was measured (Figure 3C); the mean length was determined at 50 ± 20 nm (with the 57 nm in the 75%); the minimum length was 13 nm (likely incomplete assemblies), and the longest length was measured at 155 nm (either non-RNA-templated assemblies or end-to-end assemblies of multiple TMVshort or fragments thereof). SNPs were also characterized by STEM, and the averaged diameter was determined at ~250 nm (Figure S3).
In Vivo Efficacy of TMV vs CPMV in Situ Vaccines against Dermal Melanoma.
To test whether TMV, TMVshort, its CP, or SNPs would stimulate an antitumor response when used as an in situ vaccine, we used the B16F10 melanoma model. B16F10 is a highly aggressive and poorly immunogenic tumor model used extensively for immunotherapy studies; it also has served as a model for the evaluation of the immunotherapeutic potential of virus-based therapies; for example, we and others have previously shown the efficacy of icosahedral CPMV as well as filamentous PapMV and PVX.21–23 Its low immunogenicity makes it an attractive platform to investigate new immunostimulatory therapies. B16F10 isografts were induced intradermally on the right flank of C57BL/6J mice. Eight days post induction (tumor starting volume < 100 mm3), mice were randomized (n = 8) and treated every 4 days intratumorally with PBS or 100 μg of CPMV, TMV, TMVshort, TMV CP, or SNP. Tumor volumes were measured daily, and mice were sacrificed when tumors reached >1500 mm3 or 10% body weight.
As previously shown, CPMV particles elicited a potent antitumor response in this mouse model and significantly slowed the tumor growth rate and extended the survival time compared to PBS (p < 0.005, Mantel Cox log-rank, Figure 4A-D). Disassembled TMV CP had no effect on B16F10 tumor growth and progression. Interestingly, TMV and TMVshort slowed tumor growth and increased survival time (p < 0.05, Mantel Cox log-rank, Figure 4A-D), however, at significantly lower potency compared to that of CPMV. There were no apparent differences between TMV and TMVshort indicating that the aspect ratio (and rate of phagocytosis33) does not necessarily play a role in plant viral in situ vaccines.
Figure 4.

Efficacy study. Tumors were induced with an intradermal injection of 125 000 cells/mouse. Mice (n = 8) were treated with 100 μg of particle or PBS control once every 4 days, starting 8 days post induction. Arrows indicate in situ vaccination schedule. Time course tumor growth curves with overall growth (A), by treatment group (B), and compared at individual time points (C). Survival curve of mice post first in situ vaccination (time 0) (D). SNP tumor growth curve [left] and survival curve [right] (E). TMV dosing experiment tumor growth curve (100 vs 500 μg) (F).
To address whether nanoparticle shape would impact the effectiveness of the TMV vaccine, TMVshort vs SNP were tested side-by-side using the dermal B16F10 model: the SNP in situ vaccination approach also reduced tumor growth rates and thus increased survival time (Figure 4E); nevertheless, the tumor growth curves and survival curves between the SNP and TMVshort groups indicated matched efficacy (it should be noted that the SNPs tested here were ~250 nm in size–we cannot rule out that SNPs of smaller diameters may trigger more potent responses and that this question could be addressed in future work).
Lastly, to test whether increased dosing would allow an increase of the potency of the TMV in situ vaccination approach, TMV was administered every 4 days, 8 days post B16F10 tumor establishment at a dosage of 100 vs 500 μg (higher dosing is technically challenging to achieve, due to the small intratumoral injection volume of <20 μL): both dosing schedules led to slowed tumor growth rates vs the PBS group, but there were no apparent differences between the treatment groups, indicating that higher doses do not increase efficacy (Figure 4F).
In summary, while in situ vaccination with TMV, TMVshort, and SNP resulted in delayed tumor growth and increased survival time in mice with B16F10 tumors, no efficacy was observed when animals were treated with soluble CPs, thus indicating that mutivalency and particle assembly is a requirement. Nevertheless, neither shape nor aspect ratio were indicated as parameters influencing outcomes. While delayed tumor growth was observed after in situ vaccination with TMV, TMVshort, and SNP, the significantly lower rate of efficacy compared to CPMV may indicate distinct mechanism of action. To further investigate this, immune cells and chemo/cytokine profiles were assessed in temporal studies as described below.
Cytokine Profiling of BMDCs and Tumor Cell Suspensions from Tumor-Bearing Animals Treated with CPMV vs TMV-Based in Situ Vaccine.
While immune-cell profiling (next section) provides information on which cell types infiltrate and become activated within the TME in response to the viral in situ vaccine, cytokine and chemokine profiling provides clues about how this cellular response is orchestrated (this section). Therefore, we first measured cyto/chemokine expression levels in vitro and ex vivo using either isolated BMDCs or homogenized tumor lysates. Furthermore, we performed a pathway analysis to gain insights into how these identified cyto/chemokines are interconnected and to gain clues about the key pathways that are being activated.
BMDCs were stimulated in vitro with PBS (negative control), TMV, CP, TMVshort, and LPS (positive control, TLR4 agonist), and cytokine/chemokine panels were assessed using Luminex 32-plex panels. Data indicate that all plant virus-based samples, including the free CPs, are immune-stimulatory with no apparent differences between any of the formulations under the conditions tested (Figures 4A and S4). Nevertheless, it is difficult to draw conclusions from in vitro studies that only consider a single cell type in 2D culture; therefore, in vivo studies were performed.
To determine whether CPMV vs TMV particles elicit different cytokine profiles in vivo, B16F10 isografts were induced intradermally on the right flank of C57BL/6J mice. Eight days post induction (tumor starting volume < 100 mm3), mice were randomized (n = 4) and treated with PBS or 100 μg of CPMV, TMV, TMVshort, or TMV CP. Tumors were harvested at 24 h post initial injection, homogenized, and prepared for Luminex assay. At 24 h post treatment, data indicate that the TMV formulation elicited more profound cytokine/chemokine release within the TME compared to CPMV (Figures 4B, S5). In comparison of the various TMV formulations, it appears that while both TMV and TMVshort prime immunostimulation within the TME, intratumoral treatment of B16F10 melanomas with free CP did not stimulate cytokine/chemokine release within the TME, which is in line with the lack of efficacy of free CP (Figure 5B).
Figure 5.

Cytokine release profiles. (A) Heat maps depicting cytokine and chemokine levels of BMDCs after exposure to PBS, TMV, TMV-CP, TMV-S (TMVshort), CPMV, or LPS. Data were obtained using a 32-plex Luminex assay; all samples were analyzed in duplicates. Averaged values were analyzed, and % sum of the means are plotted (green signal indicates an increase, and red indicates a decrease in signal). (B,C) 32-Plex Luminex assay of homogenized B16F10 dermal tumors at 24 h post initial in situ vaccination (B) and 4 days post initial treatment (C; animals received two treatments, the first on day 1 and the second on day 4; tumors were harvested 4 h post the second treatment). Results are normalized to PBS levels and by the sum of each column. Yellow and green arrows highlight critical chemo/cytokines (see Discussion). See also Figures S4 and S5 for additional data presentations. The asterisks (*) indicate statistical significance over PBS with p < 0.05 being statistical significant.
While somewhat similar, different trends could be observed comparing the cytokine/chemokines released upon TMV vs TMVshort treatments, with the following cytokines and chemokines showing statistical significant differences between TMV vs TMVshort: G-CSF, IL-1α, IL-3, IL-6, IL-17 were elevated for TMV but not for TMVshort (or CPMV), while IP-10 and RANTES were elevated for the TMVshort vs TMV (or CPMV). Overall, it was apparent that in particular proinflammatory cytokines were upregulated in the TME post TMV treatment: IL-6 vs PBS (p = 0.006, fold change [fc] = 13.4), IL-1β vs PBS (p = 0.0006, fc = 8.9), IL-17 vs PBS (p = 0.002, fc = 7.8), and TNF-α vs PBS (p = 0.005, fc = 10.6). Furthermore, macrophage inflammatory proteins MIP-1β vs PBS (p = 0.006, fc 23.2) and MIP-2 vs PBS (p = 0.0002, fc = 58) were up-regulated. Both chemokines are produced by macrophages, and they activate granulocytes, such as neutrophils; as such, these chemokines are also key players in inducing pro-inflammatory responses toward infection and inflammation.39 This may also be consistent with the observation that the cytokine G-CSF appears to be released upon TMV treatment with G-CSF vs PBS (p = 0.0001, fc = 170) showing significant elevation in the TME (Figure 5B).
At 24 h, CPMV did not elicit a strong response for most of the cytokines/chemokines tested. Nevertheless, CPMV did have a statistical significant increase in the expression of IFN-γ (p = 0.009, fc = 2.4) and the downstream IFN-γ-regulated genes IP-10 (p = 0.001, fc = 4.8), MCP-1 (p < 0.0001, fc = 4.5), RANTES (p < 0.0001, fc = 5.8), and MIG (p = 0.0044, fc = 3.1) (Figure 5B).
Together, the cytokine/chemokine profiles indicate that at early time points (i.e., 24 h post treatment), TMV and CPMV-based viral nanoparticle in situ vaccines both activate the immune system with TMV having a more profound effect, and cytokine/chemokine profiles appearing to be related to distinguishable pathways (see Discussion below).
To gain insights into how the initial tumor cytokine/chemokine profile changes would maintain or change over time, we repeated the above Luminex experiments at 4 days post initial treatments on day 1 and day 4. This time point was chosen, because at day 4 post first treatment, one can appreciate the antitumor effect based on the tumor growth curves that bifurcate for the various treatment arms (see Figure 4). In these experiments (and studies going forward), we simplified our experimental set up and only considered PBS vs CPMV and TMV; this was done because (i) there were no apparent differences in efficacy comparing TMV vs TMVshort, and (ii) there were no stark differences in the early cytokine profile.
At first glance, the Luminex results indicate that at 4 days, CPMV instead of TMV had the pre-eminent profile or, in other words, that CPMV at this time point established a more robust cytokine/chemokine secretion within the TME (Figures 4C and S5). Another considerable difference is that expression levels of cytokines and chemokines were significantly higher at 4 days compared to levels measured at the 24 h time point (Figure S5).
Noticeably, the characteristic IFN-γ CPMV profile that was seen at 24 h was maintained (Figure 5C, yellow arrows; see also pathway analysis and Discussion below). Moreover, another subset of cytokines and chemokines became apparent: this set includes Eotaxin (p = 0.0003, fc = 2.1), G-CSF (p < 0.0001, fc = 7.5), GM-CSF (p < 0.0001, fc = 11.3), IL-12 (p = 0.02, fc = 2.7), KC (p < 0.0001, fc = 8.2), MIP2 (p < 0.0001, fc = 14.5), and TNF-α (p < 0.0001, fc = 8.4) (Figure 5C; green arrows highlight cytokines that were specific to CPMV. In other words, only CPMV showed statistically significant elevation of these cytokines).
Together, data indicate that while TMV elicits a pro-inflammatory response upon injection, it does not establish a long-lasting response. In stark contrast, the onset of the CPMV-induced cytokine/chemokine profile is delayed and distinct, likely due to initial recruitment and/or reprogramming of immune cells, which in turn mount more potent antitumor immunity. While data indicate differences in immune-activation, the underlying mechanism for these differences are yet to be identified: it is possible that CPMV and TMV are recognized by different receptors, but it is also possible that different pathways are triggered due to differences in metabolic processing of CPMV vs TMV. Answering these particular questions however was beyond the present study.
Immune Networks and Pathway Analysis Comparing CPMV vs TMV in Situ Vaccination.
To gain a deeper understanding of the differential cyto/chemokine profiles measured within the TME upon in situ treatment of B16F10 dermal tumors with CPMV vs TMV, immune network and pathway analysis was conducted. Cytokine pathway analysis is a powerful tool to gain insights into the global interplay of the immune mediators. Case Western Reserve University’s Crosstalker software (https://sb4j.case.edu) was used for pathway analysis. Cyto/chemokines with significant expression levels (compared to PBS controls) and/or cyto/chemokines expressed differentially (with statistical significance) between the treatment arms CPMV and TMV were used as “seeds” for the computation. The Crosstalker software considers these “seeds” or entry points globally based on established networks–the output is a map of the identified signaling pathway highlighting how the different cyto/chemokine players are interconnected and how this may differ between the treatment arms.
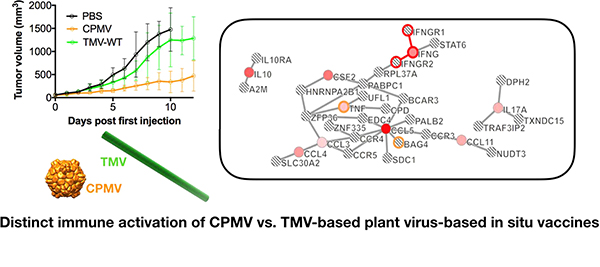
At 24 h compared to PBS, CPMV produced seven “seeds” that were significantly expressed: MCP-1, RANTES, IFN-γ, IL-5, MIG, IP-10, and M-CSF (Figure 6B, seeds are shown in red). Computed protein networks and pathways are shown in Figure 6; this analysis showed that CPMV-treated tumors were preferentially signaling through regulation of the IFN-γ pathway (Figure 6B, highlighted in green and aqua). At the same time point, when comparing PBS and TMV, 18 “seeds” that were significantly expressed were imported for analysis, of which the following 16 mapped to the software-predicted networks: IL-6, IL-3, M-CSF, LIF, MCP-1, and IL-1β, IL-4, IL-10, and MIP-1β, MIP-1α, TNF-α, IL-15, IL-17, KC, IL-7, and IL-1α (the following two “seeds” did not produce any hits with the predicted networks: IL-12 and MIP-2). The main pathways identified were IL-6 and IL-3 signaling pathways (Figure 6C, highlighted in aqua and brown, respectively). Lastly, TMV vs CPMV “seeds” were loaded into the Crosstalker software; and data further support that TMV and CPMV treatment activates distinct, nonover-lapping pathways: for TMV signaling occurs through IL-6 and IL-3 (aqua and blue), CPMV signaling occurs through IFN-γ (orange), and there was no apparent crosstalk between the involved immune mediators (Figure 6D). These results corroborate that the different cyto/chemokine profiles measured at 24 h are indeed a coordinated profile with distinct signaling pathways at play.
Figure 6.

Cytokine pathway analyses. Cytokine/chemokine release data were used to develop pathways based on p-value and fold changes in expression. (A) Table showing cytokine/chemokine names along with gene symbol [used in pathways]. Cytokine pathways comparing (B) CPMV vs PBS, (C) TMV vs PBS, and (D) TMV vs CPMV at 24 h post in situ vaccination. Pathways at 4 days post in situ vaccination compare (E) CPMV vs PBS and (F) CPMV vs TMV. Colored circles represent input cytokines whose expression was significant compared to reference group (p < 0.05). Intensity of color indicates degree of fold change with brighter colors having the largest difference (red is a positive fold change, while blue is a negative fold change). Lined circles indicate other genes that were mapped based on interactions with input genes. Outlines of circles and connections indicate the enriched pathway associated with the specific color.
At 4 days post injection, as predicted by the Luminex data (see Figure 5), there was a significantly higher level of immune-activation in tumors from CPMV-treated animals; 21 significant “seeds” were identified with 17 mapped. Chemokine signaling was most apparent: IFN-γ and GM-CSF, KC, TNF-α, MIP-1β, MIP-1α, RANTES, MCP-1, Eotaxin, and IL-12p40, IL-17p70, and IL-10, IL-1β, and MIP-2, IL-6, M-CSF, LIX (Figure 6E). TMV vs PBS only resulted in 12 “seeds,” and data analysis indicated that TMV also preferentially enriched for chemokine intersignaling (data not shown). Comparing CPMV vs TMV showed 14 significant “seeds” and 9 could be mapped to IFN-γ signaling and regulation as well as TNF-α signaling (Figure 6F, highlighted in red and orange): IL-10 and MIP-1β, MIP-1α, RANTES, TNF-α, GM-CSF, IFN-γ, Eotaxin, and IL-17 (Figure 6F). Throughout the time points considered (24 h and 4 days), IFN-γ signaling remained a key differentiator for CPMV. In addition, several cytokines emerged for the CPMV-treatment arm at 4 days. These include GM-CSF (CSF2) and Eotaxin (CCL11), both of which are interconnected to the main network, therefore further indicating manifestation of an IFN-γ-mediated antitumor response.
Overall, Luminex and pathway analysis are in agreement and support that CPMV vs TMV intratumoral injection leads to immune-activation; however, distinct cyto/chemokine profiles were observed, and data point toward distinct pathways of immune-activation and signaling.
Tumor Histology and Immunohistochemistry Comparing CPMV vs TMV in Situ Vaccination.
Tumor growth curves and cyto/chemokine analyses indicate differential efficacy of CPMV vs TMV-based in situ vaccines based on inherent differences in immune-activation. Therefore, histology and flow cytometry (see next section) were performed to gain insights into the immune cells at play. For histology and immunohistochemistry (IHC), dermal B16F10 tumor-bearing animals were treated with PBS, TMV vs CPMV as described above; tumors were harvested and analyzed 6 days post first treatment (i.e., these animals received two treatments, the first on day 1 and the second on day 4). First, tumors were stained with H&E to assess the gross tumor morphology. In sections, tumors were circumscribed, which was attributed to proper placement in the dermis (Figure 7A-C). Due to the fast growth rate of the B16F10 tumors, there was a noticeable central necrosis (Figure 7A-C, middle row); central necrosis was apparent in all treatment groups, with a higher degree of necrosis being noticed in tumors from animals receiving the PBS sham treatment. B16F10 tumor morphology is characterized by sheets of cells that have diffuse nuclei (Figure 7A-C, top inset). In areas of tumor death, the tumor tissue appears darker with nuclear condensation and reduction of cytoplasmic volume (Figure 7B-C, bottom inset). Second, sections were stained for total leukocytes (CD45). At 6 days, PBS tumors showed minimal presence of leukocytes shown by the absence of red signal throughout the tumor (Figure 7D). TMV-treated tumors showed a presence of leukocytes that were concentrated pericentrally (Figure 7E), while CPMV was littered with CD45+ cells throughout the sections (Figure 7F). The diffuse presence of CD45+ correlated with the diffuse alteration of tumor morphology on H&E. This was the case at all time points (4 and 10 days [data not shown]).
Figure 7.

Histological analyses. Paraffin embedded B16F10 tumors. (A-C) H&E stained tumors 6 days post in situ vaccination with PBS, TMV vs CPMV. Inset row one (orange boxes) showing normal tumor morphology. Inset row two (black boxes) with altered tumor morphology. Insets at 20× magnification. (D-F) CD45 stained tumors 6 days post in situ vaccination with PBS, TMV vs CPMV. Inset row at 20× magnification.
Flow Cytometric Analysis of B16F10 Tumors Receiving CPMV vs TMV in Situ Treatment.
Histology and IHC indicates differences in tumor morphology and increased infiltration of leukocytes into tumors of animals receiving the CPMV in situ vaccine (see Figure 7). Flow cytometry analysis was used to gain further insights into the differences in immune-cell profiles within the TME comparing PBS, CPMV vs TMV treatment groups. Dermal B16F10 tumors were studied and treated as described above; tumor tissue was harvested at day 4 or 10 days post first treatment. Day 4 was considered for innate panel analysis: 4 h after the day 4 treatment (animals received two treatments; one on day 1 and one on day 4), tumor tissue was collected and analyzed by FACS to characterize innate immune-cell infiltration. The numbers of live leukocytes (CD45+) in tumors were increased in TMV (p = 0.0192) and CPMV (ns)-treated mice compared to the PBS control group (Figure 8A). However, among the elevated infiltrating leukocytes, CPMV treatment induced slightly more monocytic cells (Figure 8E, p = 0.0289), while TMV treatment induced more granulocytic cells (Figure 8D, p = 0.0056) in tumors, in particular, granulocytic myeloid-derived suppressor cells (G-MDSCs, CD11b+LY6GhiLY6Clo) (Figure 8G, p = 0.0005 vs PBS and p = 0.0013 vs CPMV). While MDSCs have been considered to promote tumor growth and induce CD8+ T cell tolerance,40 G-MDSCs can also be converted to neutrophils in the presence of GM-CSF.41 Significant increase of GM-CSF levels were found in the TME in response to CPMV and TMV treatment (see Figure 5). Therefore, these changes could lead to G-MDSC activation and enhanced phagocytic function leading toward a better prognosis. In line with this discussion, both treatments induced recruitment of tumor-infiltrating neutrophils (TINs, CD11b+LY6GhiF4/80+); however, only the CPMV group showed statistical significance (p = 0.0227). Lastly, CPMV-treated tumors showed high levels of proportions of NK cells (Figure 8I, p = 0.0034 vs PBS and p = 0.0108 vs TMV).
Figure 8.

CPMV and TMV administration induces tumor immune-cell infiltration and activation. C57BL/6 mice bearing dermal B16F10 tumors were treated intratumorally with 100 μg of CPMV, TMV, or PBS on days 8 and 12 post implantation. For the innate panel, 4 h following the second treatment, tumor cells were harvested to quantify immune-cell infiltration by flow cytometry. Proportions of total live CD45+ cells. (A) Proportions of tumor-infiltrated dendritic cells (B) and macrophages (C), granulocytic cells (D), monocytic cells (E), M-MDSCs (F), G-MDSCs (G), and tumor-infiltrated neutrophils (TINs) (H) within the live CD45+ cells. (I) Proportions of NK1.1+ in M-MDSCs. For the adaptive panel, animals were sacrificed and tumor harvested 10 days post the first treatment (which occurred on day 8). Analysis of the percentage of CD4+ T cell (J), and CD8+ T cell (K) infiltration is depicted as a percentage of CD45+ cells. Analysis of percentage of effector memory cells (CD44+CD62L−) in CD4+ T cells (L) and CD8+ T cells (M). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We also evaluated the population of infiltrated monocytic MDSCs (M-MDSCs, CD11b+LY6G−LY6C+) among different treatment groups (Figure 8F) and found that CPMV treatment resulted in a higher cell population of M-MDSCs compared to TMV or PBS. M-MDSCs have the ability to differentiate to macrophage and DCs under appropriate cytokines/chemokine stimulation (e.g., GM-CSF).41 Different trends in tumor-infiltrated dendritic cells (CD11b+CD11c+) and macrophage (CD11b+F4/80+) populations were also observed comparing CPMV vs TMV: the percentages of both DCs (p = 0.0143) and macrophages (p = 0.0023) significantly increased in CPMV-treated mice; in contrast, these cell populations decreased in TMV-treated mice (p = 0.0008 for DCs; p < 0.0001 for macrophages) (Figure 8B,C).
Next, tumors were collected at later time points, i.e., 10 days post first treatment to investigate whether adaptive immune responses are mounted by either treatment. CPMV treatment led to significant recruitment of both CD4+ (p < 0.0001) and CD8+ (p = 0.0040) T cells. TMV treatment only enhanced the CD4+ T cells infiltration (p = 0.0023) (Figure 8J,K). To assess whether memory T cells were generated in tumor tissues after treatments, the levels of effector memory T cells (EMT, CD44+CD62L−) were quantified. A higher proportion of effector memory CD4+ (p = 0.0001) and CD8+ (p = 0.0040) T cells were found in CPMV-treated mice (Figure 8L), whereas the TMV treatment only accumulated a higher level of CD4+ EMT (p = 0.0287) (Figure 8M).
Together, these results indicate that CPMV and TMV in situ vaccination of B16F10 tumors accelerates the recruitment of innate and adaptive immune cells in TME, with TMV and CPMV leading to the recruitment of differential immune-cell profiles; these differences alongside with the observed distinct cyto/chemokine profiles explain the observed differential potency of CPMV vs TMV applied as an in situ vaccine.
DISCUSSION
In this work, we set out to gain understanding in how structural differences between CPMV and TMV-based in situ vaccine formulations may enhance or hinder effective immune-activation leading to a TME conducive for an antitumor response. We found that in the setting of dermal melanoma in mice: (1) tobacco mosaic virus (TMV) used as an in situ vaccine elicits a weak anti-tumor immune response against melanoma showing a trend of reduced tumor burden and prolonging survival–however, the efficacy of CPMV could not be matched; (2) there were no apparent differences comparing the efficacy of native 300 × 18 nm-sized TMV vs TMVshort measuring ~50 × 18 nm or spherical TMV, SNP; however, free CPs did not elicit an antitumor response or immune-stimulation (as measured per cytokine/chemokine levels). The latter may be attributed to the fact that a multivalent assembly is required to trigger activation through innate receptors such as PRRs. Another consideration is the stability or possibly the lack of stability of free CP vs TMV/TMVshort in their assembled state; instability and degradation of the CP through proteases within the TME may also explain the lack of immune-stimulation in vivo (this hypothesis may be supported by the observation that in in vitro BMDC stimulation assays, the free CP did indeed induce immune-stimulation). Furthermore, it should also be noted that besides the state of assembly, TMV/TMVshort contain RNA, and the CP is devoid of RNA. Therefore, it cannot be ruled out that besides multivalency, the encapsidated RNA may be a contributing factor. For example, in studies with PapMV, the antitumor immune-stimulation was attributed to its packaged RNA and resulting stimulation of TLRs.22,24,25 On the contrary, our previous studies with RNA-free eCPMV indicated that the RNA cargo is not a requirement. Also, in the present study, we demonstrated that TMVshort and SNPs have matched efficacy, and SNPs are devoid of RNA.34 Therefore, while RNA may contribute to immune-stimulation, it is unlikely the key requirement. In fact, further systematic studies would be required to elucidate the role of the RNA cargo; CPMV may be a suitable model system, because we demonstrated that both RNA-free eCPMV21 and RNA-containing CPMV (e.g., this work) have efficacy against tumors. The RNA cargo may affect the type of immune response induced; RNA is an immune-stimulatory PAMP known to stimulate innate immune responses through TLRs (TLR7/8), RIG-I-like receptors (RLRs), and STING (stimulator of interferon genes).24,42,43 It remains unknown what the receptor for eCPMV is, i.e., what the initial events of immune recognition are. It cannot be ruled out that RNA-containing CPMV and nucleic-acid-free eCPMV have distinct potencies based on differences in immune-activation. Our laboratory is investigating this question, but this is beyond the present study.
When comparing the TMV vs TMVshort formulation, there were no apparent differences in antitumor efficacy. Cyto/chemokine analysis indicated subtle differences; however, the overall trend was similar, indicating that TMV and TMVshort administration into dermal B16F10 tumors lead to secretion of cyto/chemokine profiles associated with inflammation (as determined by 32-plex Luminex 24 h post treatment). Prominent cytokines were IL-6, IL-1β, IL-17, TNF-α, and G-CSF as well as macrophage inflammatory chemokines MIP-1β and MIP-2. These cytokines and chemokines are also key players in inducing pro-inflammatory responses toward infection and inflammation.39 It is interesting to note that while our previous work indicated that lower aspect ratio TMV particles have higher rates of phagocytosis,33 this did not correlate with increased immune-activation comparing TMVshort vs TMV. Both TMVshort and TMV are phagocytosed by immune cells, and it is possible that the dose used exceeded a threshold for immune-activation thus negating the role of cell uptake rates. Furthermore, phagocytosis plays a role for intracellular TLR activation; it is possible that signaling occurs on the cell surface, not requiring cell uptake.
When comparing CPMV vs TMV, TMVshort, or SNPs, it is clear that CPMV outperforms any of the TMV-based formulations in terms of efficacy against dermal melanoma. We investigated whether higher dosing of TMV could achieve matched efficacy, but this was not the case indicating the underlying differences in immune-activation (as opposed to magnitude of immune-activation) are the underlying cause for the observed differences in potency. Indeed, cytokine/chemokine as well as immune cell profiling indicate distinct differences comparing CPMV vs TMV. While TMV stimulates an early inflammatory response with pro-inflammatory cytokine/chemokine elevation at early time points, CPMV has a delayed onset of immune-activation with IFN-γ signaling being identified as a key signaling pathway, and this is consistent with our previous studies.21 In CPMV-treated animals, changes within the TME appear delayed but result in a sustained antitumor response.
The benefits of IFN-γ signaling in the context of developing an antitumor immune response through recruitment of cells and steering of their maturation is well-known.44,45 Here, we show that CPMV stimulates the IFN-γ pathway as early as 24 h post intratumoral treatment and that this response is maintained over at least 4 days (later time points were not considered in our Luminex studies). In fact, the CPMV-induced IFN-γ predominant response increases and expands over time to include inflammatory cytokines like Eotaxin, GM-CSF, and IL-12 (these players are specific to CPMV, and elevation was not observed for any of the TMV formulations). GM-CSF and IL-12 have been identified as promising cytokines for cancer immunotherapy, including treatment of melanoma; these cytokines activate antitumor immune cells and expand IFN-γ signaling through a positive-feedback loop.46–49
In stark contrast, TMV induces a potent immune response early on, which is characterized by strong pro-inflammatory cytokines, primarily IL-6. IL-6 signaling has been associated with protumorigenic attributes including the ability to modulate polarization of macrophages and neutrophils to their protumorigenic phenotypes, M2 and N2 specifically.50–53 This inflamma-tory early response could cause the tumors to “burn out” early, which might explain the high numbers of G-MDSCs observed in the innate panel.
While the TME is dominated by granulocytes, in particular, G-MDSCs, in response to TMV treatment, the TME of CPMV-treated tumors was dominated by monocytes, including M-MDSCs, DCs, and macrophages. Furthermore, TNF-α, which is upregulated within the TME, has also been shown to be able to drive differentiation to M1 vs M2.54 M1 macrophages are able to kill tumor cells, present antigens, and activate type I T cell responses by producing high levels of IL-12/IL-23 and toxic intermediates (i.e., NO, reactive oxygen intermediates, and TNF). Alternatively, M2 macrophages are characterized by protumor phenotype.55 CPMV treatment shows potential to initiate M1 macrophage polarization, therefore, promoting local antitumor immune response. Conceivably, these upregulated peripheral cells can be attracted to the tumors by the immune profile induced by CPMV and be the source of the increased CD45+ cells apparent in histology and IHC as well as flow cytometry. Overall, the striking differences in immune-cell profiles indicate that TMV and CPMV activate distinct immune pathways.
It appears that both TMV and CPMV intratumoral treatments lead to the recruitment of TINs. This is also consistent with our previous study,21 in which we showed that neutrophils are the initial and primary responders to eCPMV, and that these TINs have the appearance of antigen-presenting cells with high Class II and CD86 expression.21 TINs produce substantial quantities of MCP-1, MIP-1α, IL6, and IL-8 and are able to stimulate T cell proliferation and IFN-γ release in human lung cancer.56 The increased levels of MCP-1, MIP-1α, IL6 in TME of both treatment groups might be secreted by CPMV/TMV-induced TINs, and the cross-talk between TINs and T cells may boost the production of IFN-γ by T cells. IFN-γ, together with other cyto/chemokines, GM-CSF, MIP-2, KC, and TNF-α, might attract or activate more neutrophils into TME.57 This positive-feedback loop indicates that TINs might play the essential role in CPMV treatment to convert immunosuppressive TME into immunostimulatory and further enhance T cell proliferation and activation.
It is also of note that CPMV treatment leads to the recruitment of NK cells. While the role of NK cells within the TME is not fully understood, the antitumor potential of NK cells has been recognized.58,59 Either through their direct action or their capacity to induce DC maturation, which may amplify T cell responses.60 And indeed, for CPMV-treated tumors, significant recruitment of CD4+ and CD8+ positive cells and effector memory cells was apparent. The presence of T cells, and in particular the presence of effector memory cells, is in agreement with our previous studies demonstrating that CPMV treatment not only elicits a potent antitumor response at the primary site of treatment but also at distinct metastatic sites; most importantly, our previous work indicates that CPMV in situ vaccination mounts immune memory protecting animals from rechallenge with the same tumor.21
CONCLUSIONS
While the potential of mammalian viruses or other microbes in medicine has long been recognized, the application of plant virus-based materials, in particular, their use as in situ vaccines is a novel and emerging field. Recent data indicate that plant viral nanoparticles and virus-like particles thereof can stimulate an antitumor response when administered as an in situ vaccine.21–23 While plant viruses are neither oncolytic or do they replicate or infect mammalian cells, it is thought, that plant virus-based nanoparticles activate the innate immune system locally through interaction with PRRs, thereby reprogramming the immunosuppressive TME toward an immune-activated state. This in turn restarts the cancer-immunity cycle leading to systemic elimination of cancer cells through the adaptive immune system. Previously, it has been reported that nanoparticles from CPMV (a 30 nm-sized icosahedron) and filamentous platforms PapMV and PVX can induce antitumor responses upon intratumoral application.21–23 The high aspect ratio filamentous particles were able to significantly decrease the growth of the tumors and increase survival upon intratumoral administration. Furthermore, these particles were able to modulate intratumoral cytokine and chemokine production. Nonetheless, neither was able to eliminate tumors as was seen with the nanoparticles from CPMV. Here we demonstrate that CPMV, and to a lesser degree TMV, elicits antitumor immunity after intratumoral treatment in a mouse model of dermal melanoma. Comparing the various TMV-based formulations: native 300 × 18 nm-sized TMV vs TMVshort measuring ~50 × 18 nm vs SNP, there was no difference in efficacy or underlying immune-stimulation–each formulation elicited a pro-inflammatory response that resulted in moderately slowed tumor growth. The free CPs did not elicit an antitumor response or immune-stimulation, which may indicate that a multivalent assembly is required to trigger innate immune recognition and activation. Differential potency of CPMV is explained with differences in immune-activation. While the initial receptor recognizing CPMV has not been identified yet, data indicate that CPMV stimulates an antitumor response through recruitment of monocytes into the TME, establishing signaling through the IFN-γ pathway, which also leads to recruitment of TINs and NKs. Data indicate that initial priming of the innate immune system mounts also an adaptive response with CD4+ and CD8+ T cell recruitment and establishment of effector memory cells. While the TMV treatment also lead to the recruitment of innate immune cells as well as T cells (although to a lesser degree), key differences were noted in cyto/chemokine profiling with TMV inducing a potent immune response early on characterized by strong pro-inflammatory cytokines, primarily IL-6. CPMV had more modest responses early, but those responses were much higher by day 4 compared to those of TMV and that would make a clear biological difference. Additionally, and perhaps surprisingly, only CPMV made high levels of IFN-γ early or late, and this cytokine is commonly associated with potent T cell-mediated antitumor immunity.
It is clear that further research is required to fully elucidate the mechanism of plant viral in situ vaccines. Our data indicate that some plant viral nanotechnology platforms are more potent than others, the enhanced efficacy of CPMV vs TMV-based nanoparticles could be explained with underlying differences in immune-activation. The question as to how or why CPMV in particular is more effective requires further investigation, i.e., a critical question to answer would be what is the molecular receptor that recognizes CPMV and initiates and orchestrates the potent antitumor immune response. Once this question is answered, one may test this hypothesis by re-engineering TMV or other platforms to target the identified receptor and/or pathway. Understanding the intricate differences and underlying mechanism of immune-activation of different (plant) virus-based platforms may set the stage for clinical development of these technologies.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge use of the Leica SCN slide scanner in the Light Microscopy Imaging Facility at Case Western Reserve University made available through the Office of Research Infrastructure (NIH-ORIP) Shared Instrumentation Grant S10RR031845. Amy M. Wen is thanked for assistance with TEM imaging, and Sourabh Shukla is thanked for assistance with BMDC isolation. This work was funded in part by a grant from the National Institute of Health U01-CA218292 to N.F.S. and training grants from the National Institutes of Health, T32 GM007250 to A.A.M. and TL1 TR000441 to A.A.M.
ABBREVIATIONS
- ACK
ammonium chloride potassium
- APCs
antigen-presenting cells
- BMDCs
bone-marrow-derived dendritic cells
- CPMV
cowpea mosaic virus
- CPs
coat proteins
- CTLA4
cytotoxic T-lymphocyte-associated protein 4
- eCPMV
“empty” CPMV
- FBS
fetal bovine serum
- FPLC
fast protein liquid chromatography
- G-MDSC
granulocytic myeloid-derived suppressor cells
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- IFN
interferon
- IHC
immunohistochemistry
- IL
interleukin
- LPS
lipopolysaccharides
- M-MDSC
monocytic MDSC
- NK
natural killer
- NLRs
NOD-like receptors
- OAS
origin-of-assembly site
- PAMP
pathogen-associated molecular patterns
- PapMV
papaya mosaic virus
- PBS
phosphate buffer saline
- PD-1
programmed cell death protein 1
- PDL-1
programmed death-ligand 1
- Pen/Strep
penicillin/streptomycin
- PRRs
pattern recognition receptors
- RLR
RIG-I-like receptors
- SDS PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEM
scanning electron microscopy
- SNPs
spherical TMV nanoparticles
- STEM
scanning transmission electron microscopy
- STING
stimulator of interferon genes
- T-VEC
tamilogene laherparepvec
- TBS
Tris-buffered saline
- TBST
TBS with Tween-20
- TEM
transmission electron microscopy
- TIN
tumor-infiltrated neutrophil
- TLR
toll-like receptors
- TME
tumor microenvironment
- TMV-CP
TMV coat proteins
- TMVS
TMV shortend
- TMV
tobacco mosaic virus
- VLPs
virus-like nanoparticles
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A Cancer Statistics, 2018. Ca-Cancer J. Clin. 2018, 68 (1), 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Rigel DS; Russak J; Friedman R The Evolution of Melanoma Diagnosis: 25 Years Beyond the ABCDs. Ca-Cancer J. Clin 2010, 60 (5), 301–316. [DOI] [PubMed] [Google Scholar]
- (3).Guy GP Jr; Thomas CC; Thompson T; Watson M; Massetti GM; Richardson LC Vital Signs: Melanoma Incidence and Mortality Trends and Projections - United States, 1982–2030. MMWR Morb. Mortal. Wkly. Rep 2015, 64 (21), 591–596. [PMC free article] [PubMed] [Google Scholar]
- (4).Altekruse SF; Kosary CL; Krapcho M; Cancer, N. N. M. A. C. J.; SEER Cancer Statistics Review, 1975–2007 National Cancer Institute, Bethesda, MD; 2010. [Google Scholar]
- (5).Weir HK; Marrett LD; Cokkinides V; Barnholtz-Sloan J; Patel P; Tai E; Jemal A; Li J; Kim J; Ekwueme DU Melanoma in Adolescents and Young Adults (Ages 15–39 Years): United States, 1999–2006. J. Am. Acad. Dermatol 2011, 65 (5 SUPPL. 1), S38.e1–S38.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rubinstein N; Alvarez M; Zwirner NW; Toscano MA; Ilarregui JM; Bravo A; Mordoh J; Fainboim L; Podhajcer OL; Rabinovich GA Targeted Inhibition of Galectin-1 Gene Expression in Tumor Cells Results in Heightened T Cell-Mediated Rejection. Cancer Cell 2004, 5 (3), 241–251. [DOI] [PubMed] [Google Scholar]
- (7).Zou W Regulatory T Cells, Tumour Immunity and Immunotherapy. Nat. Rev. Immunol 2006, 6 (4), 295–307. [DOI] [PubMed] [Google Scholar]
- (8).Penn I Malignant Melanoma in Organ Allograft Recipients. Transplantation 1996, 61 (2), 274–278. [DOI] [PubMed] [Google Scholar]
- (9).Chen DS; Mellman I Oncology Meets Immunology: the Cancer-Immunity Cycle. Immunity 2013, 39 (1), 1–10. [DOI] [PubMed] [Google Scholar]
- (10).Curiel TJ; Coukos G; Zou L; Alvarez X; Cheng P; Mottram P; Evdemon-Hogan M; Conejo-Garcia JR; Zhang L; Burow M; Zhu Y; Wei S; Kryczek I; Daniel B; Gordon A; Myers L; Lackner A; Disis ML; Knutson KL; Chen L; Zou W Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med 2004, 10 (9), 942–949. [DOI] [PubMed] [Google Scholar]
- (11).Beavis PA; Slaney CY; Kershaw MH; Gyorki D; Neeson PJ; Darcy PK Reprogramming the Tumor Microenvironment to Enhance Adoptive Cellular Therapy. Semin. Immunol 2016, 28 (1), 64–72. [DOI] [PubMed] [Google Scholar]
- (12).Allison KE; Coomber BL; Bridle BW Metabolic Reprogramming in the Tumour Microenvironment: a Hallmark Shared by Cancer Cells and T Lymphocytes. Immunology 2017, 152 (2), 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lanca T; Silva-Santos B The Split Nature of Tumor-Infiltrating Leukocytes. OncoImmunology 2012, 1 (5), 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Topalian SL; Sznol M; McDermott DF; Kluger HM; Carvajal RD; Sharfman WH; Brahmer JR; Lawrence DP; Atkins MB; Powderly JD; Leming PD; Lipson EJ; Puzanov I; Smith DC; Taube JM; Wigginton JM; Kollia GD; Gupta A; Pardoll DM; Sosman JA; Hodi FS Survival, Durable Tumor Remission, and Long-Term Safety in Patients with Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol 2014, 32 (10), 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tumeh PC; Harview CL; Yearley JH; Shintaku IP; Taylor EJM; Robert L; Chmielowski B; Spasic M; Henry G; Ciobanu V; West AN; Carmona M; Kivork C; Seja E; Cherry G; Gutierrez AJ; Grogan TR; Mateus C; Tomasic G; Glaspy JA; Emerson RO; Robins H; Pierce RH; Elashoff DA; Robert C; Ribas A PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515 (7528), 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Curran MA; Montalvo W; Yagita H; Allison JP PD-1 and CTLA-4 Combination Blockade Expands Infiltrating T Cells and Reduces Regulatory T and Myeloid Cells Within B16 Melanoma Tumors. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (9), 4275–4280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Postow MA Managing Immune Checkpoint-Blocking Antibody Side Effects. American Society of Clinical Oncology Educational Book 2015, 35, 76–83. [DOI] [PubMed] [Google Scholar]
- (18).Pintova S; Sidhu H; Friedlander PA; Holcombe RF Sweet’s Syndrome in a Patient with Metastatic Melanoma After Ipilimumab Therapy. Melanoma Res 2013, 23 (6), 498–501. [DOI] [PubMed] [Google Scholar]
- (19).Johnson DB; Puzanov I; Kelley MC Talimogene Laherparepvec (T-VEC) for the Treatment of Advanced Melanoma. Immunotherapy 2015, 7 (6), 611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Andtbacka RHI; Kaufman HL; Collichio F; Amatruda T; Senzer N; Chesney J; Delman KA; Spitler LE; Puzanov I; Agarwala SS; Milhem M; Cranmer L; Curti B; Lewis K; Ross M; Guthrie T; Linette GP; Daniels GA; Harrington K; Middleton MR; Miller WH; Zager JS; Ye Y; Yao B; Li A; Doleman S; VanderWalde A; Gansert J; Coffin RS Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol 2015, 33 (25), 2780–2788. [DOI] [PubMed] [Google Scholar]
- (21).Lizotte PH; Wen AM; Sheen MR; Fields J; Rojanasopondist P; Steinmetz NF; Fiering S In Situ Vaccination with Cowpea Mosaic Virus Nanoparticles Suppresses Metastatic Cancer. Nat. Nanotechnol 2016, 11, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lebel M-È; Chartrand K; Tarrab E; Savard P; Leclerc D; Lamarre A Potentiating Cancer Immunotherapy Using Papaya Mosaic Virus-Derived Nanoparticles. Nano Lett 2016, 16 (3), 1826–1832. [DOI] [PubMed] [Google Scholar]
- (23).Lee KL; Murray AA; Le DHT; Sheen MR; Shukla S; Commandeur U; Fiering S; Steinmetz NF Combination of Plant Virus Nanoparticle-Based in Situ Vaccination with Chemotherapy Potentiates Antitumor Response. Nano Lett 2017, 17 (7), 4019–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Carignan D; Herblot S; Laliberté-Gagné M-È; Bolduc M; Duval M; Savard P; Leclerc D Activation of Innate Immunity in Primary Human Cells Using a Plant Virus Derived Nanoparticle TLR7/8 Agonist Nanomedicine 2017; in press.10.1016/j.nano; 2017.10.015 [DOI] [PubMed] [Google Scholar]
- (25).Lebel M-È; Daudelin J-F; Chartrand K; Tarrab E; Kalinke U; Savard P; Labrecque N; Leclerc D; Lamarre A Nanoparticle Adjuvant Sensing by TLR7 Enhances CD8+ T Cell-Mediated Protection From Listeria Monocytogenes Infection. J. Immunol 2014, 192 (3), 1071–1078. [DOI] [PubMed] [Google Scholar]
- (26).Mogensen TH Pathogen Recognition and Inflammatory Signaling in Innate Immune Defenses. Clin. Microbiol. Rev 2009, 22 (2), 240–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Eber FJ; Eiben S; Jeske H; Wege C Bottom-Up-Assembled Nanostar Colloids of Gold Cores and Tubes Derived From Tobacco Mosaic Virus. Angew. Chem., Int. Ed 2013, 52 (28), 7203–7207. [DOI] [PubMed] [Google Scholar]
- (28).Eber FJ; Eiben S; Jeske H; Wege C RNA-Controlled Assembly of Tobacco Mosaic Virus-Derived Complex Structures: From Nanoboomerangs to Tetrapods. Nanoscale 2015, 7 (1), 344–355. [DOI] [PubMed] [Google Scholar]
- (29).Bruckman MA; Steinmetz NF Chemical Modification of the Inner and Outer Surfaces of Tobacco Mosaic Virus (TMV) In Recombinant and In Vitro RNA Synthesis; Conn GL, Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2013; Vol. 1108, pp 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wen AM; Shukla S; Saxena P; Aljabali AAA; Yildiz I; Dey S; Mealy JE; Yang AC; Evans DJ; Lomonossoff GP; Steinmetz NF Interior Engineering of a Viral Nanoparticle and Its Tumor Homing Properties. Biomacromolecules 2012, 13 (12), 3990–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).fraenkel-conrat H Degradation of Tobacco Mosaic Virus with Acetic Acid. Virology 1957, 4 (1), 1–4. [DOI] [PubMed] [Google Scholar]
- (32).Maharaj P; Mallajosyula J; Lee G; Thi P; Zhou Y; Kearney C; McCormick A Nanoparticle Encapsidation of Flock House Virus by Auto Assembly of Tobacco Mosaic Virus Coat Protein. Int. J. Mol. Sci 2014, 15 (10), 18540–18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shukla S; Eber FJ; Nagarajan AS; DiFranco NA; Schmidt N; Wen AM; Eiben S; Twyman RM; Wege C; Steinmetz NF The Impact of Aspect Ratio on the Biodistribution and Tumor Homing of Rigid Soft-Matter Nanorods. Adv. Healthcare Mater 2015, 4 (6), 874–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Atabekov J; Nikitin N; Arkhipenko M; Chirkov S; Karpova O Thermal Transition of Native Tobacco Mosaic Virus and RNA-Free Viral Proteins Into Spherical Nanoparticles. J. Gen. Virol 2011, 92 (2), 453–456. [DOI] [PubMed] [Google Scholar]
- (35).Bruckman MA; Hern S; Jiang K; Flask CA; Yu X; Steinmetz NF Tobacco Mosaic Virus Rods and Spheres as Supramolecular High-Relaxivity MRI Contrast Agents. J. Mater. Chem. B 2013, 1 (10), 1482–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Steinmetz NF; Cho C-F; Ablack A; Lewis JD; Manchester M Cowpea Mosaic Virus Nanoparticles Target Surface Vimentin on Cancer Cells. Nanomedicine 2011, 6 (2), 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Karim AF; Sande OJ; Tomechko SE; Ding X; Li M; Maxwell S; Ewing RM; Harding CV; Rojas RE; Chance MR; Boom WH Proteomics and Network Analyses Reveal Inhibition of Akt-mTOR Signaling in CD4 + T Cells by Mycobacterium tuber-culosisMannose-Capped Lipoarabinomannan. Proteomics 2017, 17 (22), 1700233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Maxwell S; Chance MR; Koyutürk M Linearity of Network Proximity Measures: Implications for Set-Based Queries and Significance Testing. Bioinformatics 2016, btw733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Driscoll KE Macrophage Inflammatory Proteins: Biology and Role in Pulmonary Inflammation. Exp. Lung Res 1994, 20 (6), 473–490. [DOI] [PubMed] [Google Scholar]
- (40).Cuenca A; Cheng F; Wang H; Brayer J; Horna P; Gu L; Bien H; Borrello IM; Levitsky HI; Sotomayor EM Extra-Lymphatic Solid Tumor Growth Is Not Immunologically Ignored and Results in Early Induction of Antigen-Specific T-Cell Anergy: Dominant Role of Cross-Tolerance to Tumor Antigens. Cancer Res 2003, 63 (24), 9007–9015. [PubMed] [Google Scholar]
- (41).Youn J-I; Collazo M; Shalova IN; Biswas SK; Gabrilovich DI Characterization of the Nature of Granulocytic Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. J. Leukocyte Biol 2012, 91 (1), 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Yoneyama M; Onomoto K; Jogi M; Akaboshi T; Fujita T ScienceDirect Viral RNA Detection by RIG-I-Like Receptors. Curr. Opin. Immunol 2015, 32, 48–53. [DOI] [PubMed] [Google Scholar]
- (43).Liu Y; Goulet M-L; Sze A; Hadj SB; Belgnaoui SM; Lababidi RR; Zheng C; Fritz JH; Olagnier D; Lin R RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection . J. Virol 2016, 90 (20), 9406–9419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kursunel MA; Esendagli G The Untold Story of IFN-Γ in Cancer Biology. Cytokine Growth Factor Rev 2016, 31, 73–81. [DOI] [PubMed] [Google Scholar]
- (45).Yoshimura T; Takahashi M IFN-Gamma-Mediated Survival Enables Human Neutrophils to Produce MCP-1/CCL2 in Response to Activation by TLR Ligands. J. Immunol 2007, 179 (3), 1942–1949. [DOI] [PubMed] [Google Scholar]
- (46).García Paz F; Madrid Marina V; Morales Ortega A; Santander González A; Peralta Zaragoza O; Burguete García A; Torres Poveda K; Moreno JX; Alcocer González J; Hernandez Marquez E; Bermúdez Morales V The Relationship Between the Antitumor Effect of the IL-12 Gene Therapy and the Expression of Th1 Cytokines in an HPV16-Positive Murine Tumor Model. Mediators Inflammation 2014, 2014 (20), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Elzaouk L; Moelling K; Pavlovic J Anti-Tumor Activity of Mesenchymal Stem Cells Producing IL-12 in a Mouse Melanoma Model. Exp. Dermatol 2006, 15 (11), 865–874. [DOI] [PubMed] [Google Scholar]
- (48).Seo SH; Kim KS; Park SH; Suh YS; Kim SJ; Jeun S-S; Sung YC The Effects of Mesenchymal Stem Cells Injected via Different Routes on Modified IL-12-Mediated Antitumor Activity. Gene Ther 2011, 18 (5), 488–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Kaufman HL; Ruby CE; Hughes T; Slingluff CL Current Status of Granulocyte-Macrophage Colony-Stimulating Factor in the Immunotherapy of Melanoma. J. Immunother Cancer 2014, 2 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sanmarco LM; Ponce NE; Visconti LM; Eberhardt N; Theumer MG; Minguez ÁR; Aoki MP IL-6 Promotes M2Macrophage Polarization by Modulating Purinergic Signaling and Regulates the Lethal Release of Nitric Oxide During Trypanosoma Cruzi Infection. Biochim. Biophys. Acta, Mol. Basis Dis 2017, 1863 (4), 857–869. [DOI] [PubMed] [Google Scholar]
- (51).Fisher DT; Appenheimer MM; Evans SS The Two Faces of IL-6 in the Tumor Microenvironment. Semin. Immunol 2014, 26 (1), 38–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jiang M; Chen J; Zhang W; Zhang R; Ye Y; Liu P; Yu W; Wei F; Ren X; Yu J Interleukin-6Trans-Signaling Pathway Promotes Immunosuppressive Myeloid-Derived Suppressor CellsviaSuppression of Suppressor of Cytokine Signaling 3 in Breast Cancer. Front. Immunol 2017, 8, 1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Zou J-M; Qin J; Li Y-C; Wang Y; Li D; Shu Y; Luo C; Wang S-S; Chi G; Guo F; Zhang G-M; Feng Z-H IL-35 Induces N2 Phenotype of Neutrophils to Promote Tumor Growth. Oncotarget 2017, 8 (20), 33501–33514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Kratochvill F; Neale G; Haverkamp JM; Van de Velde L-A; Smith AM; Kawauchi D; McEvoy J; Roussel MF; Dyer MA; Qualls JE; Murray PJ TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep 2015, 12 (11), 1902–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Balkwill F; Charles KA; Mantovani A Smoldering and Polarized Inflammation in the Initiation and Promotion of Malignant Disease. Cancer Cell 2005, 7 (3), 211–217. [DOI] [PubMed] [Google Scholar]
- (56).Eruslanov EB; Bhojnagarwala PS; Quatromoni JG; Stephen TL; Ranganathan A; Deshpande C; Akimova T; Vachani A; Litzky L; Hancock WW; Conejo-Garcia JR; Feldman M; Albelda SM; Singhal S Tumor-Associated Neutrophils Stimulate T Cell Responses in Early-Stage Human Lung Cancer. J. Clin. Invest 2014, 124 (12), 5466–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Gao JL; Wynn TA; Chang Y; Lee EJ; Broxmeyer HE; Cooper S; Tiffany HL; Westphal H; Kwon-Chung J; Murphy PM Impaired Host Defense, Hematopoiesis, Granulomatous Inflammation and Type 1-Type 2 Cytokine Balance in Mice Lacking CC Chemokine Receptor 1. J. Exp. Med 1997, 185 (11), 1959–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Mgrditchian T; Arakelian T; Paggetti J; Noman MZ; Viry E; Moussay E; Van Moer K; Kreis S; Guerin C; Buart S; Robert C; Borg C; Vielh P; Chouaib S; Berchem G; Janji B Targeting Autophagy Inhibits Melanoma Growth by Enhancing NK Cells Infiltration in a CCL5-Dependent Manner. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (44), E9271–E9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Guillerey C; Smyth MJ NK Cells and Cancer Immunoediting. Curr. Top. Microbiol. Immunol 2015, 395, 115. [DOI] [PubMed] [Google Scholar]
- (60).Moretta A; Marcenaro E; Sivori S; Chiesa MD; Vitale M; Moretta L Early Liaisons Between Cells of the Innate Immune System in Inflamed Peripheral Tissues. Trends Immunol 2005, 26 (12), 668–675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.