

Abstract
We previously showed that autophagy is an important component in human immunodeficiency virus (HIV) replication and in the combined morphine-induced neuroinflammation in human astrocytes and microglia. Here we further studied the consequences of autophagy using glial cells of mice partially lacking the essential autophagy gene Atg6 (Beclin1) exposed to HIV Tat and morphine. Tat is known to cause an inflammatory response, increase calcium release, and possibly interact with autophagy pathway proteins. Following Tat exposure, autophagy-deficient (Becn1+/−) glial cells had significantly and consistently reduced levels in the pro-inflammatory cytokine IL-6 and the chemokines RANTES and MCP-1 when compared to Tat-treated cells from control (C57BL/6J) mice, suggesting an association between the inflammatory effects of Tat and Beclin1. Further, differences in RANTES and MCP-1 secretion between C57BL/6J and Becn1+/− glia treated with Tat and morphine also suggest a role of Beclin1 in the morphine-induced enhancement. Analysis of autophagy maturation by immunoblot suggests that Beclin1 may be necessary for Tat, and to a lesser extent morphine-induced arrest of the pathway as demonstrated by accumulation of the adaptor protein p62/SQSTM1 in C57BL/6J glia. Calcium release induced by Tat alone or in combination with morphine in C57BL/6J glia was significantly reduced in Beclin1+/− glia while minimal interactive effect of Tat with morphine in the production of reactive oxygen or nitrogen species was detected in glia derived from Becn1+/− or C57BL/6J. Overall, the data establish a role of Beclin1 in Tat and morphine-mediated inflammatory responses and calcium release in glial cells and support the notion that autophagy mediates Tat alone and combined morphine-induced neuropathology.
Keywords: Neuroinflammation, HIV Tat, opioid, Beclin1-deficient mouse model
Introduction
HIV neuropathogenesis can be attributed to not only the propagation of infection in the brain, but also to viral proteins being secreted by infected cells that can also cause neurotoxicity. HIV transactivator of transcription (Tat) protein, responsible for regulating the initiation of HIV transcription and elongation, is released from HIV-infected cells and has been found to be neurotoxic independent of virus (Ensoli et al. 1993; Hui et al. 2012). The release of Tat contributes to the microglial-derived release of reactive oxygen species (ROS), nitric oxide (NO), calcium overload, and initiation inflammatory cascades, all of which impede neuronal survival (Haughey et al. 1999; Nath 2002; New et al. 1997; Pocernich et al. 2005; Pulliam et al. 1991). Other proteins within the HIV genome including Nef, gp120, Rev, Vpr and Vif also show varying effects on the brain and influence neurotoxicity through means of oxidative stress, secretion of pro-inflammatory molecules, and induction of apoptosis (Kramer-Hammerle et al. 2005; Nath 2002). Drugs of abuse such as opiates are known to increase risk of exposure to HIV infection and influence the progression to AIDS. Intravenous drug users (IDU) become highly susceptible to HIV not only because of shared contaminated needles but also due to opiate induced immune suppression (Friedman et al. 2006). Of HIV-infected patients that are injection drug users, a population show an accelerated progression of HIV-associated neural pathogenesis characterized by HIV encephalitis (HIVE) (Hauser et al. 2012). HIV viral proteins and drugs of abuse show similar mechanisms of injury such as proinflammatory response, oxidative stress, and induction of apoptosis (Cao et al. 2016b). As such, co-exposure to HIV and opiates metabolites such as morphine have been shown to enhance viral replication and exacerbate the detrimental effects of HIV Tat in the infected brain and promote progression to AIDS dementia (Steele et al. 2003; Zou et al. 2011). Despite studies to understand how the progression of HIV-associated neuronal pathogenesis is exacerbated by opiates, the mechanism is still not fully understood.
Autophagy is a cellular process in which cytoplasmic contents such as organelles and proteins are encapsulated in a double-layered membrane autophagosome and delivered to the lysosome for degradation and macromolecule recycling (Klionsky et al. 2011). Autophagy can be initiated as a pro-survival response by a variety of stimuli such as environmental and cellular stress such as nutrient starvation, accumulation of aggregated proteins, or for pathogen internalization and degradation in innate immune cells (Feng et al. 2014). This is particularly key in the brain cells since many are post-mitotic and require autophagy for maintaining homeostasis. Moreover, dysregulation in the autophagy pathway contributes to the development of a number of neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases (Kiriyama and Nochi 2015). Patterns of neurodegeneration similar to these diseases during HIV infection have been observed and linked to defects in the autophagy pathway. Though generally cytoprotective, autophagy plays an intricate role in viral replication with respect to infected cell type and can be induced or inhibited in order to bolster virus production at certain stages of the viral life cycle (Espert et al. 2009; Zhou and Spector 2008).
We have previously shown that siRNA mediated silencing of the BECN1 gene can significantly reduce HIV p24 levels and HIV and morphine-induced inflammatory cytokine and chemokine secretion from infected microglia and astrocytes (El-Hage et al. 2015; Rodriguez et al. 2017a; Rodriguez et al. 2017b; Rodriguez et al. 2017c). Beclin1, a component of the phosphatidylinositol 3-kinase nucleation complex, regulates the initiation stages of autophagy preceding the formation of the isolation membrane of the autophagosome. As such, Beclin1 poses an attractive target for future therapeutic intervention and for gaining a better understanding of the interplay between HIV, autophagy, and drugs of abuse. In the present study, we use a mouse model possessing a monoallelic deletion of the Becn1 gene as a potential therapeutic model to provide a direct link between the autophagy protein Beclin1 and HIV Tat and their effects on glial dysfunction. The data also support the role of autophagy in mediating Tat and morphine-induced neuropathology.
Materials and Methods
Animals
C57BL/6J mice (stock # 000664) and Becn1 model (B6.129X1-Becn1tm1Blev/J) were procured from The Jackson Laboratory (Bar Harbor, ME, USA) and bred in the animal facility at Florida International University. The guidelines of the National Institutes of Health Guide for the Care and Use of Laboratory Animals were followed and the Florida International University Institutional Animal Care and Use Committee approved all animal experimental protocols.
Mouse genotyping
Genotyping of tail genomic DNA was performed to detect wild type and Becn1 knockout alleles by PCR amplification. The sense primer 5′-AGCCTCTGAAACTGGACACG-3′and the antisense primer 5′-TGGAAACAGGGTCTCATTCA-3′ were used to detect the wild type beclin1 allele (yielding a PCR product of 380 bp), and the sense primer 5′-CTCCAGACTGCCTTGGGAAAA-3′ and the identical antisense primer were used to detect the knockout beclin1 allele (yielding a PCR product of approximately 400 bp). PCR was performed using AmpliTaq Gold® DNA Polymerase kit (Life Technologies) as per manufacturer’s instructions and the PCR product identified by agarose gel electrophoresis.
Mixed glial murine cell cultures
For primary murine glial culture, post-natal day 4-6 (P4-P6) Becn1+/− mutant mice and C57BL/6J littermates were separated according to phenotypic coat color and sacrificed according to IACUC guidelines. Growth medium contained Dulbecco’s Modified Eagle’s Medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with glucose (2 mg/mL; Sigma-Aldrich), Na2HCO3 (6 mM; Invitrogen), 10% v/v heat inactivated fetal bovine serum (FBS; Hyclone, Logan, UT, USA), and 1% penicillin/streptomycin (100 U/mL/100 μg/mL; Invitrogen) (Gurwell et al. 2001). Striata were isolated and dissociated mechanically and enzymatically with 0.25% trypsin containing DNase (2.5 mg/ml) centrifuged, triturated, and twice filtered through 40 μM nylon mesh cell strainer. Cells were then plated and maintained in plates coated with poly-L-lysine (0.1mg/ml). Cultures were maintained for 5-10 days at 37°C and 5% CO2.
Immunocytochemistry
Chambers containing cells were washed with PBS, fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked in 0.1% Triton X-100 with 10% milk/0.1% goat serum and immunolabeled. Primary antibodies anti-GFAP (catalog # MAB360, Millipore) and anti-Iba 1 (catalog # 019-19741, Wako Chemicals) were each used at 1:400 and 1:50 dilutions for astrocyte and microglia visualization respectively. Immunoreactivity was visualized with secondary antibodies from Molecular Probes (Carlsbad, CA, USA). Cells were mounted with ProLong® Gold antifade reagent with DAPI (Thermo Fisher Scientific). Images were analyzed using a Zeiss (Germany) inverted fluorescence microscope with a 560 Axiovision camera.
Western blotting
Whole cell lysates were prepared in RIPA buffer supplemented with a mixture of protease and phosphatase inhibitors and separated by SDS-PAGE for western blotting. Primary antibodies: Beclin 1 – 1:500 (Novus Biologicals, NB500-249), LC3-B – 1:1000 (Novus Biologicals, NB600-1384), p62/SQSTM1 – 1:500 (Novus Biologicals, NBP1-48320), JNK - 1:200 (Santa Cruz, sc-571), p38 (α/β/γ) MAPK– 1:1000 (Cell Signaling Technology, #8690), β-actin – 1:200 (Santa Cruz, sc-47778). Following primary antibodies, blots were incubated with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology, 7076S; Sigma-Aldrich, A0545) used at a 1:1000 dilution. Immunoblots were exposed to SuperSignal West Femto Substrate (Thermo Scientific) and visualized using a ChemiDoc imaging system (Bio-Rad, Hercules, California). Protein expression was calculated using ImageJ software (National Institutes of Health (NIH); Bethesda, MD).
LC3 Autophagy Sensor
Levels of autophagy in primary glia derived from C57BL/6J and Becn1+/− mice were assessed using the Premo™ Autophagy Sensor LC3B-GFP kit (Thermo Fisher Scientific, Grant Island, NY, USA) following manufacturer’s instructions. Cells were treated with 10 μL of BacMam reagent containing the LC3B-GFP construct for 16 hours. Rapamycin (Sigma-Aldrich, St. Louis, MO, USA) was used at 2.5 μM as an inducer of autophagy. After incubation, cells were washed with PBS and mounted with ProLong® Gold anti-fade reagent with DAPI (Thermo Fisher Scientific). Images were analyzed for green fluorescence (LC3B positive autophagosomes) using a Zeiss (Germany) inverted fluorescence microscope with a 560 Axiovision camera.
Reagents and treatments
Morphine sulfate (Sigma-Aldrich, St. Louis, MO) and HIV Tat1-86 IIIB (ImmunoDiagnostics, Woburn, MA) were used at concentrations ranging from 100 nM - 1 μM and 10 nM - 100 nM respectively. The HIV Tat is derived from the CXCR4 tropic HIV-1 IIIB strain which was expressed in the E.coli expression system as a recombinant protein. Furthermore, it has been previously shown that the neurotoxic domain of Tat is well conserved across various strains of HIV (Mayne et al. 1998). This concentration of Tat has been shown to not only induce inflammatory cytokine production and other homeostatic dysregulations in glial cells in vitro, but also causes toxicity to neurons. Given that treatments of 100 ng/mL and 1000 ng/mL correspond to approximately 8.3 nM and 83.3 nM concentrations respectively, we deemed the concentration range of 10 nM to 100 nM acceptable. It may also worth noting that both the Kruman and Nath studies utilize the HIV-1 BRU Tat1-72 which may account for differences in extent of effects. In addition, this lab has used these concentrations in previous studies of Tat induced neuroinflammation (El-Hage et al. 2005; Kruman et al. 1998; Nath et al. 1999; Zou et al. 2011). Morphine at a concentration of 500 nM has been previously shown to fully activate MOR as well as enhance HIV Tat mediated neurotoxicity (El-Hage et al. 2005; Gurwell et al. 2001). μ-opioid receptor antagonist naltrexone (Sigma-Aldrich, St. Louis, MO) was used at a concentration of 500 nM and was added 30 minutes prior to morphine exposure. Viral proteins Nef, Rev, and Gag were used at 1 and 5 nM envelope protein gp120 was used at 200 and 500 pM (NIH AIDS reagent program).
ELISA
Mouse glial cell culture supernatants were used to measure the levels of cytokines and chemokines, which have been shown to be released upon HIV Tat treatment (El-Hage et al. 2005). These included interleukin (IL) -6, monocyte chemotactic protein-1 (MCP-1), regulated upon activation normal T-cell expressed and secreted (RANTES), and tumor necrosis factor alpha (TNF-α) which were assayed by ELISA (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. The optical density was read at A450 with wavelength correction at A570 on a Synergy HTX plate reader (Biotek, Winooski, VT)
Quantitative Reverse Transcription-Polymerase Chain Reaction
Total RNA was isolated from glia following 24-hour treatments using the miRNeasy Mini Kit (Qiagen, Valencia, CA). Purity was assessed by microspot RNA reader (Synergy HT Multi-Mode Microplate Reader from BioTek) and RNA preparations with an OD260 nm/OD280 nm absorbance ratio of at least 2.0 were used for cDNA synthesis. 1 μg of RNA was used for cDNA synthesis using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems; Cat #: 4368814) as per the manufacturer’s protocol. Relative abundance of mRNA was assessed by SsoAdvanced Universal SYBR Green Supermix (BioRad, Cat #: 172-5271) in 20 μL real-time PCR reactions with gene specific primers using a BioRad CFX96 real time system. All data were normalized to β-actin mRNA and presented as 2^−ΔΔCt for fold-change values. Primers used for amplification were mouse MOR, 5′-TGGAAACTTCCTGGTCATGT-3′ and 5′- AGATCACGATCTTGCAGAGG-3′ (182bp); and mouse β-actin, 5′-AAGAGCTATGAGCTGCCTGA-3′ and 5′-TACGGATGTCAACGTCACAC-3′ (160bp).
Cell viability
Toxicity of HIV Tat and morphine in mixed glial cultures was assessed using 12 mM MTT (3-(4, 5-Dimethylthiazol-2-yl)-2, 5-Diphenyltetrazolium Bromide) for cell survival and proliferation (Millipore, Temecula, CA). Cells were plated in poly-L-lysine coated 96 well plates at a density of 3 × 104 cells/well from T75 flasks and cultured overnight followed by treatment with 100 nM HIV Tat and increasing concentrations of morphine ranging from 100 nM to 1 μM. After 24 hours, cells were refed with 100 μL fresh media and 10 μL of 12 mM MTT was added to each well except for control and incubated for 2 hours to allow the formation of formazan crystals. Media was removed from wells and formazan crystals dissolved using DMSO. Absorbance was read at 570 nm with reference at 630 nm on a Synergy HTX plate reader (Biotek, Winooski, VT).
Reactive oxygen species (ROS)
Levels of intracellular ROS production were measured using dichlorodihydrofluorescein diacetate (CM-H2 DCFDA, Invitrogen, Carlsbad, CA), which is de-acetylated to dichlorofluorescein (DCF). Mixed glial cells were incubated with 10 μM CM-H2 DCFDA in warm PBS for 1 hour according to the manufacturer’s protocol, washed then treated. N-acetylcysteine, a precursor to the antioxidant glutathione, was used as a control at 10μM and pre-treated for 1 hour. H2O2 (0.001% v/v) was added at time of treatment as a positive indicator of oxyradical species. Dichlorofluorescein (DCF) fluorescence was measured at an excitation wavelength (λex) of 485 nm and an emission wavelength (λem) of 520 nm over 24 hours using a Synergy HTX plate reader (Biotek, Winooski, VT). Relative DCF fluorescence estimated the levels of ROS present and were reported as DCF mean fluorescence intensity (MFI).
Nitric oxide (NO) production
Measurement of nitric oxide (NO) was assessed from the supernatant of control and HIV Tat ± morphine treated cells after 0, 1, 6, and 24 hours. Collected supernatant was measured for the concentration of nitrite (the oxidized metabolite of NO) and evaluated using a nitric oxide assay kit (BioVision, Milpitas, CA) according to manufacturer’s instructions. Absorbance was measured at 540 nm using a Synergy HTX plate reader (Biotek, Winooski, VT). The concentration of NO in samples was calculated based on the standard curve using known concentrations of nitrite.
Intracellular calcium [Ca2+]i measurements
Levels of [Ca2+]i release in glial cultures were measured using the fluorescent marker Fura-2-AM (Invitrogen, Carlsbad, CA). Cells were loaded with 5 μM fura-2-AM for 30 minutes at 37°C, 5% CO2. After three washes, the cells were incubated in neuronal medium for an additional 30 minutes to complete de-esterification of the AM group of the fluorophores. Cells were pre-treated with 10μM of the cell permeant calcium chelator BAPTA (Tocris Bioscience) as a control prior to addition of fura-2-AM. Baseline measurements were recorded, followed by exposure to Tat and/or morphine. Fura-2 ratio at 340/380 nm excitation measurements were taken every 10 seconds for 30 minutes. Data is presented as percent of control values ± SEM from 3 separate experiments (15-25 cells).
Statistical analysis
Results are reported as mean ± SEM of 3-6 independent experiments. Data were analyzed using analysis of variance (ANOVA) techniques, followed by the appropriate post hoc test for multiple comparisons (GraphPad Prism 7 software, La Jolla, CA). A value of p < 0.05 was considered significant.
Results
Molecular characterization of Beclin1 using primary glial cell cultures derived from C57BL/6J and autophagy deficient mouse
Beclin1-mediated autophagy is of key importance in maintaining brain homeostasis, with dual deletion of the Becn1 allele (Becn1−/−) being embryonically lethal and conditional knockout causing neurodegeneration in mice (McKnight et al. 2014; Yue et al. 2003). In order to study the role of host autophagy in the context of HIV viral protein effects, we instead make use of a monoallelic deletion mutant of Beclin1 (Becn1+/−). B6.129X1-Becn1tm1Blev/J mice are heterozygous for the Becn1 allele (Becn1+/−) and have a brown (agouti) coat color which is believed to be a result of the effect of the Becn1 mutation on melanogenesis and can thus be distinguished from wild-type littermates phenotypically (JAX Website). This study makes use of primary glia derived from C57BL/6J and Becn1+/− pups according to defined breeding scheme (Fig. 1a). We first characterized Beclin1 expression within the primary mixed glia derived from the mutant Becn1+/− mice. Mice initially separated by coat color 4-6 days after birth were genotyped to confirm the BECN1 single allele mutation using DNA isolated from mouse tails and amplified by PCR. Amplification of the wild type sequence (14946) yielded a PCR product of 380bp in both black and agouti pups as expected, whereas the mutant primer (oIMR8619) yields a product of 400bp only in agouti pups, confirming the mutation (Fig. 1b–c). Knockdown of Beclin1 protein in murine mixed glia was confirmed by western blot analysis and indicated a 60% reduction in expression levels (Fig. 1d). To determine whether a single allele mutation in the BECN1 gene is sufficient to considerably impair the autophagy pathway, we compared induction of autophagy in C57BL/6J and Becn1+/− derived glia using a fluorescent autophagy marker LC3-GFP assay. Upon induction of autophagy, LC3 localizes to pre-autophagosomal and autophagosomal membranes which can be indicated by the punctate appearance of GFP staining (Kabeya et al. 2000). We observed that untreated Becn1+/− glia show a slight but insignificant decrease in LC3-GFP puncta as compared to untreated C57BL/6J glia (Fig. 1f–h); however, when treated with the mTOR inhibitor and autophagy inducer rapamycin, C57BL/6J glia showed a 64% increase in LC3-GFP puncta whereas there was no corresponding increase in Becn1+/− glia. These findings further demonstrate that a 60% reduction in protein levels due to the heterozygous deletion of the Becn1 allele correlates to a reduction of autophagic activity in glial cells upon autophagy induction. Morphology and glial cell components were also assessed by immunofluorescent labeling with the astrocyte marker, GFAP, and the microglia marker, Iba1 (Fig. 1e) and showed no apparent cellular morphological differences between the different strains.
Fig. 1.
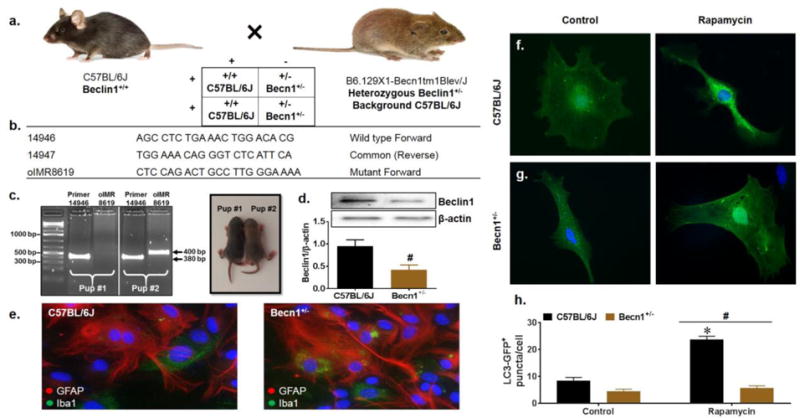
Characterization of Beclin1-deficient mixed glia. C57BL/6J (Beclin1+/+) and Becn1+/− mutant breeding scheme (a). Primers corresponding to the wild type and mutant beclin1 DNA sequence used for genotyping (b). Pups at P4-P6 were distinguished by coat color and mutation of the beclin1 gene was confirmed at the DNA and protein levels by PCR genotyping (c) and western blot (d). Lanes are grouped according to sample number (pup #1 vs pup #2) and labeled according to the primer sequence used for amplification. Wild type sequence generates a PCR product of 380 bp and mutant sequence generates a PCR product of 400bp (indicated by black arrows). Beclin1 protein expression from isolated primary glia was normalized to β-actin. Data is presented as the mean ± SEM of 4 independent experiments. Immunocytochemistry for glial cell presence identifying GFAP+ astrocytes in red and Iba1+ microglia in green (e). Representative images were acquired at 63X magnification. Representative images assessing basal autophagy induction by LC3-GFP puncta quantification upon treatment with the autophagy inducer rapamycin. Data was quantified from 3 independent experiments, averaging puncta per cell for 5 fields of view presented as mean ± SEM. P < 0.05 * vs. Control; # vs. C57BL/6J
Beclin1+/− glia show reduced inflammatory molecule induction upon exposure to the viral protein Tat
We have recently shown that HIV replication and viral-induced neuroinflammation are regulated via a Beclin1-dependent pathway (El-Hage et al. 2015; Rodriguez et al. 2017a; Rodriguez et al. 2017c). Given the established role of Tat as a mediator of neuroinflammation, we sought to determine whether the underlying mechanism is through Beclin1. Mouse models, including the use of glial cells, have been widely used in the studies of HIV-induced neurotoxicity. While true that murine cells are restricted for HIV infection, exposure to viral proteins such as Tat, Nef, and gp120 elicit glial cell activation, inflammatory molecule secretion, macrophage infiltration into the brain, metabolic dysfunction, and signaling pathway alterations which are also seen in primary human glia, despite lacking infectious capability, and reflect levels seen pathophysiologically (Conant et al. 1998; El-Hage et al. 2008; El-Hage et al. 2005; Jones et al. 1998; Philippon et al. 1994). Glial cells derived from C57BL/6J and Beclin1 (Becn1+/−) deficient mice were treated with increasing concentrations of the viral protein Tat for 8 and 24 hours for assessment of cytokine and chemokine secretion by ELISA (Fig. 2a–b). After 8 hours post-treatment, Tat caused a significant concentration-dependent increase in the release of RANTES, MCP-1, and IL-6 in C57BL/6J glia where Becn1+/− glia showed significantly less secretion by comparison (Fig.2a; red, blue, and green arrows). These decreases were maintained at 24 hours for RANTES and IL-6 secretion. At 100 nM Tat treatment, RANTES, MCP-1, and IL-6 secretion in Becn1+/− glia were reduced by 1.6, 2.7, and 1.3-fold at 8 hours, respectively; and 2.2, 1.3, and 1.2-fold at 24 hours respectively. Secretion of the pro-inflammatory cytokine TNF-α was also assessed; however, no differences were seen between Tat treatments or glial strains (data not shown). From this data, it can be inferred that Tat utilizes Beclin1 to induce inflammatory molecule secretion and that 60% reduction in Beclin1 expression level, can decrease this cytokine and chemokine release. To confirm the effects of Tat on cytokine/chemokine secretion were dependent on active protein, Tat was heat inactivated for 30 minutes at 95°C before being treatment to C57BL/6J and Becn1+/− glia and incubated for 24 hours (Fig. S1a–c). Secretion of RANTES was reduced by 7-fold (Fig. S1a), and 3-fold in MCP-1 and IL-6 (Fig. S1b–c) from glia treated with heat inactivated Tat illustrating the specific effects of active Tat on autophagy and autophagy-deficient murine glia. Inflammatory effects of additional viral proteins were also surveyed though secretion was either less than that of Tat or showed no significant difference between the strains (Fig. S2), further supporting the role of Beclin1 as a main facilitator of Tat-mediated inflammation. Numerous studies have reported on the interplay between inflammation and autophagy, with induction of autophagy able to regulate inflammatory molecule secretion while conversely, cytokine secretion also being able to induce autophagy (Deretic et al. 2013; Salminen et al. 2013; Sun et al. 2017). As such, we were interested in the effects of Tat on the expression of key autophagy proteins Beclin1, LC3, and the ubiquitin protein, p62/SQSTM1 since these proteins represent the initiation, autophagosome formation, and maturation stages of the autophagy pathway (Cao et al. 2016b). Correlating to the effects of Tat on cytokine secretion, there were concentration dependent changes in expression of autophagy related proteins Beclin1 and p62/SQSTM1 in C57BL/6J-derived glia (Fig. 2c–e) suggesting induction of the autophagy pathway as well as accumulation of p62/SQSTM1 upon Tat treatment. Becn1+/− glia show reduced expression of Beclin1 as expected; however, these levels were elevated with increasing Tat concentration. Interestingly, p62/SQSTM1 expression was unchanged with increasing concentration of Tat though significantly less than C57BL/6J which suggests autophagic degradation is still ongoing. Taken together, this data indicates that the inflammatory molecule secretion induced by Tat is facilitated somewhat through an association with Beclin1 and possibly the activation of the autophagy pathway.
Fig. 2.
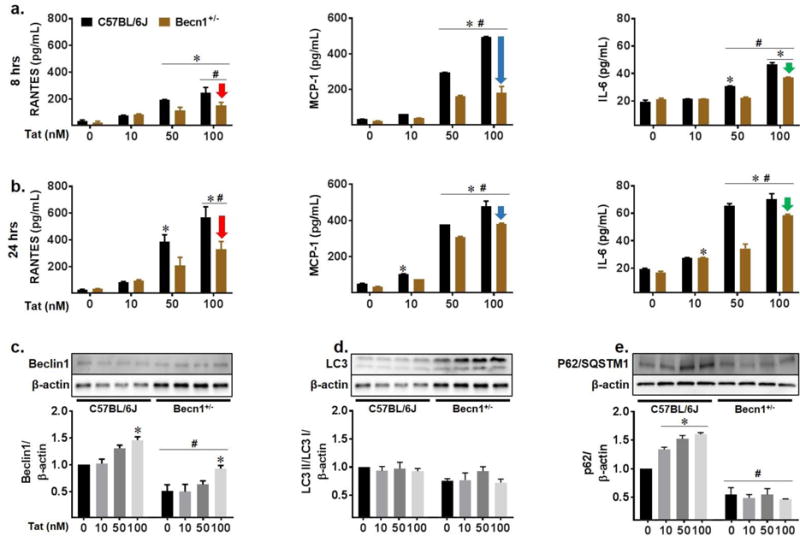
Beclin1 facilitates Tat-induced inflammatory molecule secretion and autophagy dysregulation. Indicated cytokines and chemokines secreted from C57BL/6J and Becn1+/− mixed glia supernatants were measured by ELISA at 8 (a) and 24 hours (b) following increasing concentrations of HIV Tat (10 nM, 50 nM, 100 nM). Arrows indicate decreased Tat-induced secretion of RANTES (red), MCP-1 (blue), and IL-6 (green) from Becn1+/− derived glia. Values were determined from standard curves and are presented as the mean ± SEM of 5 independent experiments. Whole cell lysates from C57BL/6J and Becn1+/− glia after 24-hours of treatment were subjected to immunoblotting with antibodies to Beclin1 (a), LC3 (b), and p62/SQSTM1 (c). Densitometry was performed for quantification, and the ratios of each protein to β-actin are presented graphically. Error bars show the SEM of 3 independent experiments. P < 0.05 * vs. Control; # vs. C57BL/6J
Beclin1 mediates glial inflammation induced by Tat alone and in combination with morphine
Given the co-morbidity of opiate abuse and HIV infection, we were next interested in the role of Beclin1 on the effects of co-exposure to Tat and morphine on cytokine and chemokine secretion. First, we sought to determine the effect of morphine on C57BL/6J and Becn1+/− glial cytokine secretion over a range of morphine concentrations (100 nM, 250 nM, 500 nM, and 1000 nM) for 24 hours of exposure (Fig. 3a–c). While no significant difference to MCP-1 (Fig. 3b) secretion was observed across the various concentrations of morphine, there were concentration dependent fluctuations in RANTES secretion from C57BL/6J glia and IL-6 secretion from both C57BL/6J and Becn1+/− glia (Fig. 3a & c). In addition, Becn1+/− glia showed reduced levels of chemokine secretion as compared to C57BL/6J glia, while strain differences in IL-6 secretion were not significant. Despite these changes, overall, morphine-induced cytokine secretion is quite minimal and not comparable to that of Tat induced secretion making morphine treatment alone experimentally irrelevant (Fig. 2a–c).
Fig. 3.
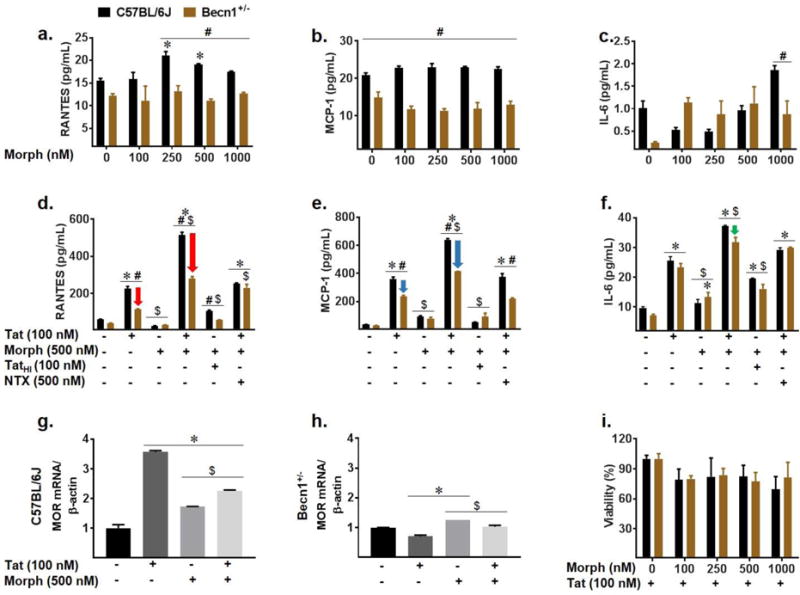
Tat and morphine induced inflammatory effects show contribution of Beclin1. Cell culture supernatants from C57BL/6J and Becn1+/− derived glia after indicated treatment for 24-hours were assessed for cytokine and chemokine secretion by ELISA (a–f). Cytokine secretion was assessed following 24-hour treatment with increasing concentrations of morphine: 100 nM, 250 nM, 500 nM, and 1000 nM (a–c). Tat and morphine co-exposure were assessed after 24-hours (d–f). Arrows indicate reduction in RANTES (d, red), MCP-1 (e, blue), and IL-6 (f, green) secretion from Becn1+/− derived glia relative to C57BL/6J derived glia for given treatment. Values were determined from standard curves and are presented as the mean ± SEM of 4 independent experiments. mRNA levels of murine μ-opioid receptor (MOR) were assessed by qRT-PCR following treatment with Tat and/or morphine (g–h). Values were determined by 2^-ΔΔCT method and normalized to β-actin. Toxicity of combined Tat and increasing concentration (100 nM, 250 nM, 500 nM, 1000 nM) of morphine in C57BL/6J and Becn1+/− derived glia was assessed by MTT assay. Data are presented as percent viability compared to Tat treated cells (i). Error bars show SEM for 3 independent experiments. P < 0.05 * vs. Control; # vs. C57BL/6J; $ vs. Tat
It has been shown that glial co-exposure to Tat and morphine provokes the enhanced secretion of cytokines and chemokines which contribute to the neuroinflammation characteristics of neuroAIDS (El-Hage et al. 2005; El-Hage et al. 2006). Moving forward with 500 nM morphine which has been previously shown to saturate the μ-opioid receptor (El-Hage et al. 2005), we exposed C57BL/6J and Becn1+/− glia to Tat alone and in combination with morphine to confirm whether Beclin1 is involved in the interactive effects of Tat and morphine-induced cytokine/chemokine secretion. MCP-1, RANTES, and IL-6 secretion were induced by Tat treatment to C57BL/6J and significantly less in Becn1+/− glia (Fig. 3d–f; Fig. 2b). Co-exposure with morphine significantly enhanced Tat-induced cytokine secretion from both strains of glia. Interestingly, co-exposure with Tat and morphine in Becn1+/− glia showed a significant reduction of about 1.8-fold in RANTES and a significant reduction of about 1.5 fold in MCP-1 secretion when compared to similarly treated C57BL/6J glia (Fig. 3d–e; red and blue arrows) suggesting that 60% reduction in Beclin1 protein expression is sufficient to significantly reduce Tat and morphine-induced RANTES and MCP-1 secretion. Despite this reduction in cytokine secretion from Becn1+/− glia, these cells are still expressing approximately 40% of the Beclin1 protein which may explain the enhancement in inflammatory molecules detected with combination Tat and morphine treatment. Co-treating the murine glia with heat inactivated Tat and morphine showed significant reduction of RANTES and MCP-1 secretion and minimal induction of IL-6 secretion, further supporting the interactive effects of morphine and active Tat. Using the μ-opioid receptor antagonist naltrexone prior to morphine administration was able to significantly decrease RANTES, MCP-1 and IL-6 secretion to the levels of Tat alone indicating the interaction is acting through the μ-opioid receptor (MOR). Of note, Tat and morphine enhanced MCP-1 release in Becn1+/− glia was significantly reduced to the level of Tat alone with pre-treatment of naltrexone, implying Beclin1 and MOR dependent mechanisms (Fig. 3e).
Looking further at the actions of Tat and morphine occurring at the MOR which is widely present throughout the brain, in particular the striatum, mRNA levels were measured by qRT-PCR (Fig. 3g–h). Analysis showed that with Tat treatment, mRNA levels of MOR were increased by 3.5-fold in C57BL/6J glia whereas in Becn1+/− glia, there was only a slight decrease in MOR as compared to the untreated control. The co-exposure of Tat and morphine to C57BL/6J glia also caused an increase in MOR mRNA, albeit, not as robust as Tat alone treatment (Fig. 3g). Interestingly in Becn1+/− glia, Tat and morphine treatment led to higher MOR mRNA levels than that of Tat alone; however, this was not significantly different from untreated control (Fig. 3h). To ensure the combined treatment was not drastically or differentially reducing the viability of the glia and therefore reducing cytokine/chemokine production or mRNA, an MTT assay was performed to determine cell viability across a range of morphine concentrations (Fig. 3i). Approximately 20% cell death was observed with each of the treatments, with no significant difference between concentrations of morphine. Overall, from the data it can be taken that the autophagy pathway intercedes in Tat and morphine-induced cytokine production. Of note is that, 60% reduction of Beclin1 protein expression (Fig. 1) is sufficient to reduce Tat-induced inflammation but not sufficient to prevent enhanced inflammation detected in Becn1+/− glia when co-exposed with morphine and Tat as compared to Tat-treated glia, suggesting that additional mechanism(s) may be at work in regulating the release of inflammatory molecules in conjunction with Beclin1 and the autophagy pathway, or that complete or >60% protein reduction is required for better inhibition of inflammation.
Tat and morphine use Beclin1 to alter the autophagy pathway and JNK signaling
After determining the effects of Tat and morphine on cytokine release and the concentration dependent effects on autophagy protein expression we observed with Tat treatment (Fig. 2c–e), we were interested to see how the autophagy pathway might be altered upon co-exposure with morphine. Beclin1 expression was elevated with Tat treatment in both strains, 2-fold in C56BL/6J and 1.4-fold in Becn1+/− (Fig. 4a–b). Co-exposure to morphine increased C57BL/6J Beclin1 expression as compared to control; however, there was no enhancing interactive effect as compared to Tat. Morphine was seen to cause a slight elevation in Beclin1 expression in both C57BL/6J and Becn1+/− derived glia. Interestingly, Tat and morphine co-exposure to Becn1+/− glia caused no change in Beclin1 expression compared to control. The expression ratio of LC3 II/LC3 I showed small fluctuations following Tat and morphine treatments alone or in combination from which conclusions on autophagosome formation may not be drawn (Fig. 4c–d). Surprisingly, morphine treatment to Becn1+/− glia reduced the LC3 II/LC3 I ratio which may be suggestive of either reduced lipidation and reduced autophagosome formation or enhanced autophagic flux. Upon analysis of p62/SQSTM1, we observed Tat caused increases in expression within C57BL/6J glia that were not seen in Becn1+/− glia (Fig. 4e–f) taken as accumulation of p62/SQSTM1 and halt of autophagic degradation. Similar to what was seen with Beclin1 expression, no additive effects on p62/SQSTM1 expression were seen with Tat and morphine co-exposure for either C57BL/6J or Becn1+/− glia; however, where C57BL/6J was slightly elevated with co-exposure, the Becn1+/− was on level with control. Coupling C57BL/6J LC3 and p62/SQSTM1 expression, the data suggests that where Tat alone can prevent p62/SQSTM1 and thus autophagosome degradation, when in combination with morphine, the increase in LC3II/I and p62/SQSTM1 may indicate increased number of autophagosomes which are also not being degraded. By comparison, the reduced LC3 II/I and unchanged p62/SQSTM1 expression in Becn1+/− glia treated with Tat alone or in combination with morphine, imply a degree of ongoing autophagic flux despite the reduction of Beclin1. The data provide evidence that Tat alone or in combination with morphine can initiate the autophagy pathway but prevent autophagosome degradation by the lysosome, which in the context of HIV, would allow for enhanced viral replication. However, by reducing the levels of Beclin1, autophagosome degradation may persist, albeit at lowered levels.
Fig. 4.
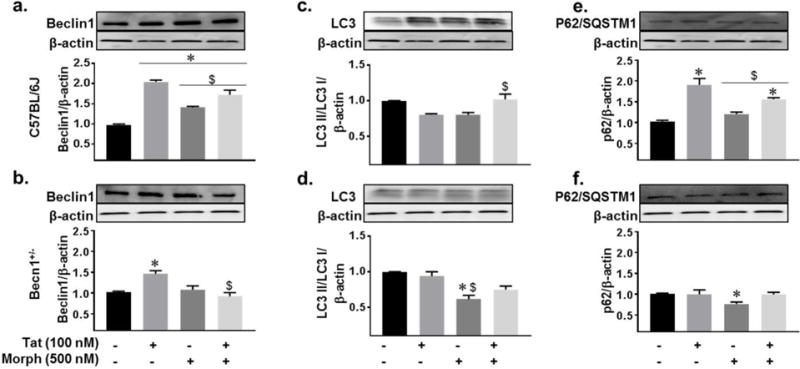
Beclin1-mediated autophagy dysregulation by Tat and morphine. Whole cell lysates from C57BL/6J and Becn1+/− derived glia following 24 hours of the indicated treatments were subjected to immunoblotting with antibodies to Beclin1 (a–b), LC3 (c–d), and p62/SQSTM1 (e–f). Densitometry was performed for quantification on separate blots, and the ratios of each protein to β-actin are presented graphically. Error bars show the SEM of 3 independent experiments. P < 0.05 * vs. Control; # vs. C57BL/6J; $ vs. Tat
Given the observation that reducing Beclin1 protein expression by approximately 60% was not sufficient to completely attenuate Tat and morphine-induced inflammatory molecule secretion (Fig. 3d–f), we were interested to examine whether other signaling pathways were potentially working with Beclin1 and the autophagy pathway to influence Tat and morphine-induced cytokine release. Mitogen-activated protein kinase signaling serves many roles in cell survival with c-Jun N-terminal kinase (JNK) and p38 MAPK being shown to be activated by pro-inflammatory cytokines and environmental stress (Davis 2000). In addition, HIV or viral protein exposure can activate JNK and/or p38 MAPKs with inhibition of these pathways reducing toxic effects (Barber et al. 2004; D’Aversa et al. 2004). In this study, expression of JNK in C57BL/6J was increased 2-fold when treated with Tat whereas in Becn1+/− glia, differences were slight (Fig. S3a–b). Co-treatment with morphine yielded vastly differing expression of JNK between C57BL/6J and Becn1+/− glia. Tat and morphine treatment to C57BL/6J raised JNK expression 1.2-fold but in Becn1+/−, significantly reduced JNK expression 2-fold. In both C57BL/6J and Becn1+/− glia, morphine, to our surprise, reduced JNK expression. Finally, we looked at the expression of the MAPK p38. Tat exposure to C57BL/6J glia was the only treatment to show an effect by raising p38 expression 1.5-fold (Fig. S3c–d). No significant differences were seen in Becn1+/−glia with any of the treatments. From this data it can be gathered that Tat and morphine also target kinase pathways and may work with Beclin1 to regulate their inflammatory effects.
Tat and morphine induced effects on intracellular calcium are mediated by Beclin1
Calcium excitotoxicity is a feature of HIV-induced neuronal death pathology which can be stimulated by inflammatory and oxyradical stressors potentially causing altered glutamate and NMDAR signaling and excessive release of calcium stores from the endoplasmic reticulum (Barger et al. 2007; Haughey et al. 2001; Kruman et al. 1999; Norman et al. 2008). Previous studies, as well as our own recently published data, have shown that increases in intracellular calcium caused by HIV in human astrocytes can lead to neuronal injury (Haughey et al. 1999; Rodriguez et al. 2017b). In these studies, we use Tat, which we have previously shown to induce calcium release in brain cells (El-Hage et al. 2008; El-Hage et al. 2005). To assess the role of autophagy in glial calcium homeostasis we use the fluorescent indicator Fura-2. As expected, we observed a significant increase in [Ca2+]i release in C57BL/6J glia treated with Tat, which was significantly increased in the presence of morphine (Fig. 5a). In contrast, Tat-induced [Ca2+]i release was significantly reduced in Becn1+/− glia and further exacerbation with morphine was abrogated when compared to similarly treated C57BL/6J glia (Fig. 5b). To ensure that the increased Fura-2 ratios corresponded to intracellular calcium release, we pre-treated murine glia with the intracellular calcium chelator, BAPTA/AM. Pre-treatment with BAPTA abrogated calcium levels in C57BL/6J and Becn1+/− derived glia confirming that the increased levels in Fura-2 ratios were due to the release of intracellular calcium. This data suggests that the reduction of Beclin1 expression allows primary murine glia to prevent Tat and morphine-induced excessive [Ca2+]i release.
Fig. 5.
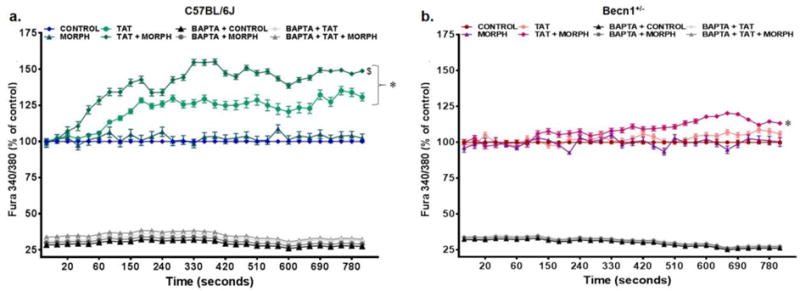
Tat and morphine enhanced intracellular calcium release is prevented by reduction of Beclin1 expression. Intracellular calcium release from C57BL/6J (a) and Becn1+/− (b) glia was assessed by Fura-2 over 800 seconds following treatment with Tat ± morphine. The calcium channel blocker BAPTA was used as a control. Results represent the percentage of control values and are the mean ± SEM from 3 independent experiments. P < 0.05 * vs. Control; # vs. C57BL/6J; $ vs. Tat
Limited Tat and morphine-interactive effects on oxidative stress are not mediated by Beclin1
In conjunction with neuroinflammation, glial oxidative stress is an additional pathological effect common to HAND and is often characteristic of Tat-induced glial dysfunction (D’Aversa et al. 2005; Shi et al. 1998). The production of reactive oxygen and nitrogen species, has not only been associated with the activation of intracellular inflammatory signaling, but also has been shown to cause lipid peroxidation, DNA damage, and facilitate excitotoxicity through the release of glutamate, ultimately leading to neuronal damage or death (Barger et al. 2007; Droge 2002; Kaul and Forman 1996; Pocernich et al. 2005; Steiner et al. 2006; Streit et al. 1999). To evaluate the role of autophagy in mediating the effects of Tat and morphine on oxidative stress in mixed glia, intracellular ROS formation was assessed by DCF reactivity and reactive nitrogen species (RNS) determined nitrite accumulation. Peak levels of DCF fluorescence were recorded after 16 hours of treatment for both C57BL/6J and Becn1+/− glia (Fig. 6a–b). At 16 hours, Tat treatment was seen to illicit the highest level DCF fluorescence for both C57BL/6J and Becn1+/− glia with no apparent additive effect with morphine. Notably, though the increases in ROS caused by Tat were not substantial, within the strains, Tat evoked a 35% increase in ROS in C57BL/6J glia compared to untreated control (Fig. 6a) whereas there was only a 20% increase in Becn1+/− glia (Fig. 6b). In addition, although no enhancing effect with Tat and morphine was observed in either strain, when compared to untreated control, Tat and morphine raised ROS by 20% in C57BL/6J derived glia compared to a 13% increase in Becn1+/− derived glia. RNS assessed by NO production was measured by Griess assay, which is based upon the accumulation of nitrite as the product of NO metabolism. C57BL/6J glia showed no significant differences in NO accumulation upon treatment with Tat alone or in combination with morphine (Fig. 6c) compared to untreated control. In Becn1+/− glia, we observed a temporary spike in NO accumulation early after Tat treatment which stabilizes at later time points (Fig. 6d). In addition, Tat and morphine causes a gradual accumulation of NO in Becn1+/− glia, which is only significantly different after 24 hours of treatment. These studies suggest there is an effect on ROS and NO production upon exposure to Tat but detect no interactive effect when combined with morphine with minimal intervention by Beclin1.
Fig. 6.
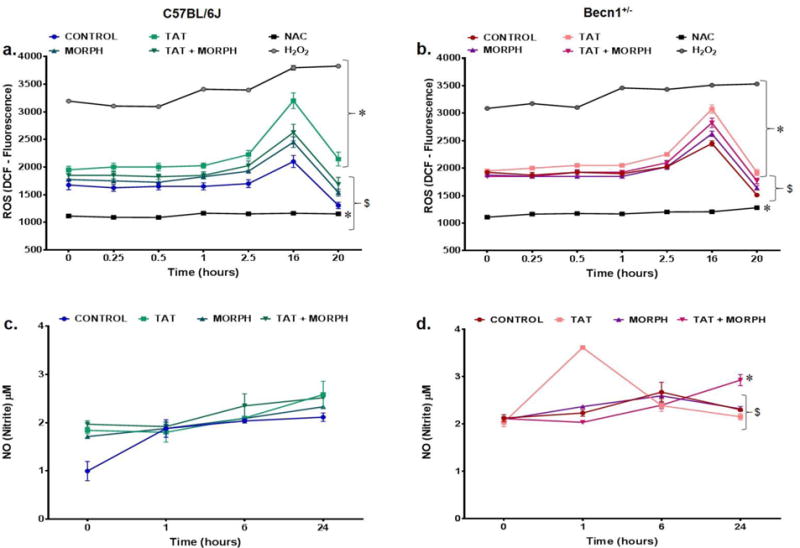
Beclin1 is independent of oxidative stress induced by Tat with limited contribution of morphine ROS production was assessed by dichlorofluorescein diacetate fluorescence (DCF) over a period of 24 hours following the indicated treatments to C57BL/6J (a) and Becn1+/− (b) glia. Mean DCF relative fluorescence was used as an estimate of ROS and was compared for each treatment. Results are the mean ± SEM from 6 independent experiments. NO production by C57BL/6J (c) and Becn1+/− (d) glia were examined at 0, 1, 6 and 24 hours following the indicated treatments. Nitrite levels were assessed using the Griess reaction with conversion of known concentrations of nitrate to nitrite as standards. Results represent the mean ± SEM of 3 independent experiments. P < 0.05 * vs. Control; # vs. C57BL/6J; $ vs. Tat
Discussion
In this study, we use glial cells from mouse possessing a monoallelic deletion of the Becn1 gene to examine to consequences of autophagy deficiency in regulating HIV and morphine-induced inflammation, specifically through Tat. Given that homozygous deletion of the Becn1 allele results in embryonic lethality, using a murine model with heterozygous deletion of the Becn1 allele provides a reduced autophagic environment while maintaining sufficient expression for growth and development. This model not only showed reduced expression of the autophagy protein Beclin1 (Fig. 1d), but also showed less LC3\ upon induction of autophagy by the mTOR inhibitor rapamycin (Fig 1f–h). Neuroinflammation is a well-documented characteristic of HIV associated dementia-like symptoms and plays a key role in HIV-1-induced neuropathogenesis (Kaul et al. 2001). In addition, studies have shown in vitro exacerbation of cytokine and chemokine secretion from astrocytes and microglia treated with HIV or Tat with co-exposure of morphine (Bokhari et al. 2009; El-Hage et al. 2008; El-Hage et al. 2005; El-Hage et al. 2006). Kyei et al has previously shown that autophagy is used to augment viral biogenesis in macrophages by HIV (Kyei et al. 2009) which provides a basis for studying the role of autophagy in mediating neurodegeneration both by HIV and in the context of drugs of abuse. Further, studies on viral proteins have illustrated not only their role in inducing neuroinflammation through the secretion of inflammatory cytokines, but also intervening in various stages of the autophagy pathway (Campbell et al. 2015; Kyei et al. 2009; Liu et al. 2017; Mabrouk et al. 1991; Ronaldson and Bendayan 2006; Saribas et al. 2015; Silverstein et al. 2012; Trillo-Pazos et al. 2000). The viral protein Tat has long been demonstrated to induce damage to the brain in murine studies through cytokine driven inflammation and gliosis (Jones et al. 1998). The penetrative capabilities of the recombinant protein across cell membranes is well documented. Extracellular Tat is able to be internalized through its basic domain within exon 1 and its RGD domain which is able to bind integrin receptors (Vogel et al. 1993). Given that Tat can enter the cell and localize to the nucleus, there are a number of pathways such as NFκB which may be activated to induce these inflammatory effects (Conant et al. 1996). In our present study, Tat was shown to have a concentration dependent effect on neuroinflammation through the secretion of cytokines and chemokines, with lack of Beclin1 able to significantly attenuate these effects (Fig. 2a–b). This is consistent with previous studies by our lab showing that silencing of Beclin1 in microglia and astrocytes reduces HIV replication as well as HIV and morphine-induced inflammation (El-Hage et al. 2015; Rodriguez et al. 2017a; Rodriguez et al. 2017b). In addition, here we provide evidence that Tat, an established promoter of neuroinflammation, can also be modulated by autophagy within brain glia using Beclin1. We observe Tat concentration dependent changes to autophagy-associated proteins Beclin1 and p62/SQSTM1 in C57BL/6J glia which are limited in the Becn1+/− glia (Fig. 2c–e). Studies have shown that the effects of Tat on autophagy are largely dependent on conditions and cell type, either activating or suppressing autophagy function in glial cells or neurons (Bruno et al. 2014; Fields et al. 2015). While activation of autophagy by Tat should direct the viral protein for degradation as shown in CD4+ T lymphocytes (Sagnier et al. 2015), dysregulation in the degradation of the autophagolysosome may allow for persistence of Tat and autophagosome accumulation (Hui et al. 2012). The study by Sagnier et al also found it is possible for Tat to exhibit ubiquitin-independent, direct interaction with p62/SQSTM1. Though there is a possibility the changes to p62/SQSTM1 within our studies may be independent of ubiquitination and LC3, this would require further study. Notably, the study by Sagnier was performed in CD4+ T cells and it is likely that these interactions of Tat may be cell-type specific.
The interactive effects of Tat and opiates on glial function have been previously reported by our lab and others (El-Hage et al. 2008; Hauser et al. 2007), yet the mechanistic role of Beclin1-mediated autophagy has not been fully confirmed. Examining the interactive effects of Tat and morphine on cytokine and chemokine production, we observed that morphine was able to exacerbate the levels of RANTES, MCP-1, and IL-6 secretion induced by Tat, which were mediated by the μ-opioid receptor (Fig. 3d–f). Although diminished Beclin1 expression was able to significantly reduce Tat and morphine-induced cytokine secretion as compared to C57BL/6J derived glia, this reduction alone was not sufficient to abrogate RANTES and MCP-1 release. This agrees with our previous studies which showed significant but not complete abrogation of cytokine and chemokine release in HIV and morphine-exposed microglia and astrocytes transfected with siRNA against Beclin1 (El-Hage et al. 2015; Rodriguez et al. 2017b). Of note, previous studies by Turchan-Cholewo et al on the effects of Tat and morphine on μ-opioid receptor surface expression and mRNA levels imply cell-specificity with differing effects between primary murine astrocytes and microglia and cell lines (Turchan-Cholewo et al. 2008). That group suggested the cell type specificity of Tat and morphine action at the μ-opioid receptor may determine the role of opioid receptors in brain inflammatory signaling responses, which is supported by our qRT-PCR results (Fig. 3g–h). In addition, differential induction of chemokines and their receptors by Tat and morphine has been suggested to have cell type-specific bidirectional, cross-sensitization with opioid receptors which may also modulate their expression (Rogers and Peterson 2003). Therefore, it is possible that the Beclin1-dependent responses to Tat and morphine are cell-type specific with the relative presence of microglia and astrocytes potentially masking the ameliorating effects of autophagy reduction in our mixed-glial model.
The autophagy pathway can be modulated by HIV or Tat exposure, with other HIV proteins also able to modulate autophagy. Other studies have reported that upon HIV treatment, human astrocytes show increased protein expression of Beclin1 indicative of autophagy initiation in conjunction with elevated LC3 (Mehla and Chauhan 2015). In addition, it has been reported that Tat is able to stimulate autophagy by increasing the levels of Bcl-2 associated athanogene 3 (BAG3) in human glial cells (Bruno et al. 2014). Our studies are in agreement as we detected elevated expression of Beclin1 in C57BL/6J derived glia treated with Tat; however, though also increased relative to untreated control, no interactive effect was detected when co-administered with morphine (Fig. 4a). It is of note that Tat exposure to Becn1+/− derived glia moderately increased Beclin1 protein expression, albeit not as significantly, when compared with C57BL/6J derived glia and even less so with Tat and morphine treated Becn1+/− derived glia (Fig. 4b). It is important to note that LC3 expression levels alone are not sufficient to determine autophagic flux due to the fact that LC3 II is in equilibrium in terms of its formation and degradation. As such, data must be analyzed in conjunction with other markers such as the adaptor protein p62/SQSTM1 or in the presence of autophagosomes (Mizushima and Yoshimori 2007). Protein expression of LC3 and p62/SQSTM1 in Tat treated C57BL/6J derived glia was suggestive of reduced LC3 leading to the accumulation of p62/SQSTM1. When combined with morphine, both protein levels were elevated compared to untreated control, which may indicate a reduction in autophagosomes and autophagic clearance (Fig. 4c & e). Notably, these effects were significantly minimized in glia derived from the Becn1+/− mouse (Fig. 4d & f). Studies in human astrocytes demonstrated that late stage autophagy could be interrupted by the HIV protein Nef as characterized by LC3 and p62/SQSTM1 accumulation (Saribas et al. 2015). Similarly, although in neurons, Hui et al has reported that Tat can also disrupt endolysosome degradation (Hui et al. 2012). As such, it is possible that Tat is able to induce Beclin1 expression to initiate autophagy in glia while concurrently preventing its own degradation through targeting of late stage autophagy. This type of hijacking of the autophagy pathway allows for the protection of viruses within the autophagosome and allows for the enhanced viral replication observed in HIV infected brain cells (Meulendyke et al. 2014). The exact mechanism by which Tat induces autophagy in glial cells is the current focus of work presently being done in our lab. Our findings show that by reduction of Beclin1-mediated autophagy, Tat and Tat and morphine-induced alterations to autophagy protein expression can be prevented and ultimately limit their effects; however, additional studies on the events at the lysosomal stage are still needed to first determine whether the lysosomal fusion is occurring, and second determine any effects on autophagolysosome acidification. Other signaling pathways such as p38 MAPK and JNK have also been implicated in HIV and Tat pathogenesis and have been shown to not only activate pro-apoptotic pathways but also inflammatory cascades (Hauser et al. 2006). Our studies (Fig S3 a–d) on the expression of these proteins suggest that Tat and morphine-induced activation of MAPK pathways like JNK, and possibly to a lesser extent p38, are able to induce Beclin1-mediated initiation of the autophagy pathway while also supporting activation of inflammatory immune responses as well as other detrimental processes, which are inhibited by reduction of Beclin1-mediated autophagy (Fig. 7). Provided that JNK has the capability of phosphorylating Bcl-2, separating it from Beclin1, it is possible that such pathways may explain the Tat-mediated activation of the autophagy pathway (Wei et al. 2008).
Fig. 7.
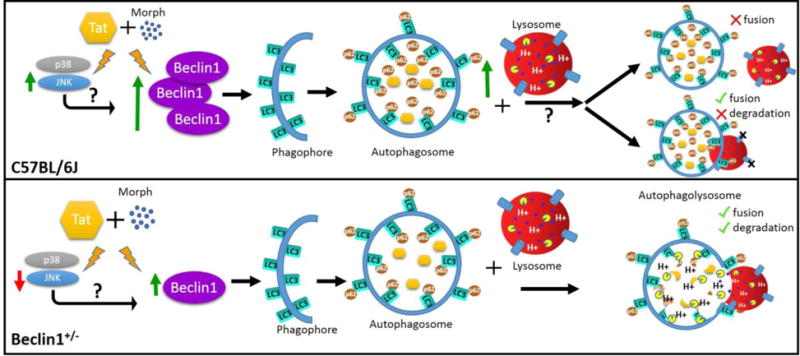
Schematic diagram summarizing the differential effects of Tat and morphine on the autophagy pathway in C57BL/6J and Beclin1-deficient mouse glia. Green arrows indicate an induction/accumulation, red arrows indicate a reduction. Reducing the expression of Beclin1 in mouse glia prevented excessive Tat and morphine induction of the autophagy pathway while also limiting the accumulation of the adaptor protein p62/SQSTM1. The kinase proteins JNK and p38 are also induced by Tat and morphine exposure which may contribute to the activation of Beclin1 and the autophagy pathway. Future studies on the fate of the autophagosome are currently pending to determine the mechanism by which the autophagy pathway is initiated by Tat and morphine but the process halted before degradation may occur.
Tat-induced neurotoxicity is largely facilitated through the activation of the brain glia which leads to oxidative stress through excitotoxic mechanisms often involving calcium (Pocernich et al. 2005). Becn1+/− deficient glia provided further evidence supporting the role of Beclin1 in mediating calcium release in astrocytes. Similar to what was shown in HIV-infected human astrocytes, Tat was able to induce calcium release from C57BL/6J derived glia, which was significantly enhanced by co-exposure to morphine (Fig. 5a–b). Moreover, Tat or Tat and morphine-induced calcium release was significantly decreased in Becn1+/− derived glia which paralleled the siBeclin1 treatment decreases shown in HIV-infected human astrocytes. Provided that intracellular Ca2+ is a key second messenger to numerous signaling pathways and is essential to the maintenance of glial homeostasis, it was of key importance to this study to validate the role of Beclin1 as a potential alleviating factor for Tat and morphine induced excitotoxicity. We also recently showed using gene silencing against Beclin1 and the autophagy inducer, rapamycin, a role of autophagy in buffering the release of calcium, ROS, and NO in HIV-infected human astrocytes exposed to morphine (Rodriguez et al. 2017b). Analysis of ROS production in the present study did not detect an enhancing effect upon Tat and morphine treatment of either C57BL/6J or Becn1+/− derived glia (Fig. 6a–b) contradictory to our previous study with HIV-infected astrocytes or studies in microglia (Turchan-Cholewo et al. 2009). Interestingly, we were able detect a slight, albeit significant, interactive effect of Tat and morphine on RNS production in Becn1+/− derived glia (Fig. 6c–d) similarly to what was seen previously seen in HIV-infected human astrocytes (Rodriguez et al. 2017b). It has been previously shown that interruption of the autophagy pathway may also aggravate toxicities due to the virus (Cao et al. 2016a). Using glia derived from an autophagy-deficient mouse we further confirmed that Tat and morphine-induced calcium, but not ROS and NO release, are mediated via a Beclin1-mediated pathway. In conclusion, using glial cells derived from Beclin1-deficient animal, we observe an association between the autophagy protein Beclin1 and the HIV protein Tat, which may underlie a mechanism for glial neuropathology in the context of opiate abuse and provide insight into targeting of future therapeutics.
Supplementary Material
Acknowledgments
We gratefully acknowledge the support of the National Institutes of Health (NIH)-National Institute on Drug Abuse (NIDA) grants R01 DA036154 awarded to NEH for funding the study. We also acknowledge the financial support of NIH/NIDA R01 DA036154-S1 Diversity Supplement awarded to NEH to support JL and Presidential Fellowship provided to CRO by the Florida International University Graduate School.
Footnotes
Compliance with Ethical Standards
Conflict of Interest
Author Jessica Lapierre declares that she has no conflict of interest. Author Myosotys Rodriguez declares that she has no conflict of interest. Author Chet Raj Ojha declares that he has no conflict of interest. Author Nazira El-Hage declares that she has no conflict of interest.
Author contributions
JL performed and analyzed the experiments shown in Fig. 1-7, Fig. S1-3, and wrote the manuscript. MR performed and analyzed the experiment shown in Fig. 1e and Fig. 5a–b. CRO assisted in the editing of the manuscript. NEH designed, analyzed, and coordinated the study and edited the manuscript. All authors reviewed the results and approved the final version of the manuscript.
References
- Barber SA, Uhrlaub JL, DeWitt JB, Tarwater PM, Zink MC. Dysregulation of mitogen-activated protein kinase signaling pathways in simian immunodeficiency virus encephalitis. Am J Pathol. 2004;164:355–362. doi: 10.1016/S0002-9440(10)63125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhari SM, et al. Morphine enhances Tat-induced activation in murine microglia. J Neurovirol. 2009;15:219–228. doi: 10.1080/13550280902913628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno AP, et al. HIV-1 Tat protein induces glial cell autophagy through enhancement of BAG3 protein levels. Cell Cycle. 2014;13:3640–3644. doi: 10.4161/15384101.2014.952959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell GR, Rawat P, Bruckman RS, Spector SA. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015;11:e1005018. doi: 10.1371/journal.ppat.1005018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Fu M, Kumar S, Kumar A. Methamphetamine potentiates HIV-1 gp120-mediated autophagy via Beclin-1 and Atg5/7 as a pro-survival response in astrocytes. Cell Death Dis. 2016a;7:e2425. doi: 10.1038/cddis.2016.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Glazyrin A, Kumar S, Kumar A. Role of Autophagy in HIV Pathogenesis and Drug Abuse Mol Neurobiol. 2016b doi: 10.1007/s12035-016-0118-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, et al. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant K, Ma M, Nath A, Major EO. Extracellular human immunodeficiency virus type 1 Tat protein is associated with an increase in both NF-kappa B binding and protein kinase C activity in primary human astrocytes. J Virol. 1996;70:1384–1389. doi: 10.1128/jvi.70.3.1384-1389.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aversa TG, Eugenin EA, Berman JW. NeuroAIDS: contributions of the human immunodeficiency virus-1 proteins Tat and gp120 as well as CD40 to microglial activation. J Neurosci Res. 2005;81:436–446. doi: 10.1002/jnr.20486. [DOI] [PubMed] [Google Scholar]
- D’Aversa TG, Yu KO, Berman JW. Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein Tat. J Neurovirol. 2004;10:86–97. doi: 10.1080/13550280490279807. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, Hauser KF. Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca(2+)](i), NF-kappaB trafficking and transcription. PLoS One. 2008;3:e4093. doi: 10.1371/journal.pone.0004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1. Tat Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Rodriguez M, Dever SM, Masvekar RR, Gewirtz DA, Shacka JJ. HIV-1 and Morphine Regulation of Autophagy in Microglia: Limited Interactions in the Context of HIV-1 Infection and Opioid Abuse. Journal of virology. 2015;89:1024–1035. doi: 10.1128/JVI.02022-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, et al. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensoli B, et al. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espert L, et al. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS One. 2009;4:e5787. doi: 10.1371/journal.pone.0005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields J, et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: implications for HIV-associated neurocognitive disorders. J Neurosci. 2015;35:1921–1938. doi: 10.1523/JNEUROSCI.3207-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman H, Pross S, Klein TW. Addictive drugs and their relationship with infectious diseases FEMS. Immunol Med Microbiol. 2006;47:330–342. doi: 10.1111/j.1574-695X.2006.00097.x. [DOI] [PubMed] [Google Scholar]
- Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience. 2001;102:555–563. [Google Scholar]
- Haughey NJ, Holden CP, Nath A, Geiger JD. Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J Neurochem. 1999;73:1363–1374. doi: 10.1046/j.1471-4159.1999.0731363.x. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Hauser KF, El-Hage N, Buch S, Nath A, Tyor WR, Bruce-Keller AJ, Knapp PE. Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol. 2006;1:98–105. doi: 10.1007/s11481-005-9000-4. [DOI] [PubMed] [Google Scholar]
- Hauser KF, et al. HIV-1 neuropathogenesis: glial mechanisms revealed through substance abuse. J Neurochem. 2007;100:567–586. doi: 10.1111/j.1471-4159.2006.04227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser KF, Fitting S, Dever SM, Podhaizer EM, Knapp PE. Opiate drug use and the pathophysiology of neuroAIDS. Curr HIV Res. 2012;10:435–452. doi: 10.2174/157016212802138779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui L, Chen X, Haughey NJ, Geiger JD. Role of endolysosomes in HIV-1 Tat-induced neurotoxicity. ASN Neuro. 2012;4:243–252. doi: 10.1042/AN20120017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M, Olafson K, Del Bigio MR, Peeling J, Nath A. Intraventricular injection of human immunodeficiency virus type 1 (HIV-1) tat protein causes inflammation, gliosis, apoptosis, and ventricular enlargement. J Neuropathol Exp Neurol. 1998;57:563–570. doi: 10.1097/00005072-199806000-00004. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kaul N, Forman HJ. Activation of NF kappa B by the respiratory burst of macrophages Free. Radic Biol Med. 1996;21:401–405. doi: 10.1016/0891-5849(96)00178-5. [DOI] [PubMed] [Google Scholar]
- Kiriyama Y, Nochi H. The Function of Autophagy in Neurodegenerative. Diseases Int J Mol Sci. 2015;16:26797–26812. doi: 10.3390/ijms161125990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. (2nd) 2011;7:1273–1294. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer-Hammerle S, Rothenaigner I, Wolff H, Bell JE, Brack-Werner R. Cells of the central nervous system as targets and reservoirs of the human immunodeficiency virus. Virus Res. 2005;111:194–213. doi: 10.1016/j.virusres.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Kruman II, et al. Evidence that Par-4 participates in the pathogenesis of HIV encephalitis. Am J Pathol. 1999;155:39–46. doi: 10.1016/S0002-9440(10)65096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Kyei GB, et al. Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol. 2009;186:255–268. doi: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, et al. ASPP2 Plays a Dual Role in gp120-Induced Autophagy and Apoptosis of Neuroblastoma Cells. Front Neurosci. 2017;11:150. doi: 10.3389/fnins.2017.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabrouk K, Van Rietschoten J, Vives E, Darbon H, Rochat H, Sabatier JM. Lethal neurotoxicity in mice of the basic domains of HIV and SIV Rev proteins. Study of these regions by circular dichroism FEBS. Lett. 1991;289:13–17. doi: 10.1016/0014-5793(91)80898-d. [DOI] [PubMed] [Google Scholar]
- Mayne M, Bratanich AC, Chen P, Rana F, Nath A, Power C. HIV-1 tat molecular diversity and induction of TNF-alpha: implications for HIV-induced neurological disease. Neuroimmunomodulation. 1998;5:184–192. doi: 10.1159/000026336. [DOI] [PubMed] [Google Scholar]
- McKnight NC, et al. Beclin 1 is required for neuron viability and regulates endosome pathways via the UVRAG-VPS34 complex. PLoS Genet. 2014;10:e1004626. doi: 10.1371/journal.pgen.1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehla R, Chauhan A. HIV-1 differentially modulates autophagy in neurons and astrocytes. J Neuroimmunol. 2015;285:106–118. doi: 10.1016/j.jneuroim.2015.06.001. [DOI] [PubMed] [Google Scholar]
- Meulendyke KA, Croteau JD, Zink MC. HIV life cycle, innate immunity and autophagy in the central nervous system. Curr Opin HIV AIDS. 2014;9:565–571. doi: 10.1097/COH.0000000000000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002;186(Suppl 2):S193–198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- New DR, Ma M, Epstein LG, Nath A, Gelbard HA. Human immunodeficiency virus type 1 Tat protein induces death by apoptosis in primary human neuron cultures. J Neurovirol. 1997;3:168–173. doi: 10.3109/13550289709015806. [DOI] [PubMed] [Google Scholar]
- Norman JP, et al. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS One. 2008;3:e3731. doi: 10.1371/journal.pone.0003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippon V, et al. The basic domain of the lentiviral Tat protein is responsible for damages in mouse brain: involvement of cytokines. Virology. 1994;205:519–529. doi: 10.1006/viro.1994.1673. [DOI] [PubMed] [Google Scholar]
- Pocernich CB, Sultana R, Mohmmad-Abdul H, Nath A, Butterfield DA. HIV-dementia, Tat-induced oxidative stress, and antioxidant therapeutic considerations. Brain Res Brain Res Rev. 2005;50:14–26. doi: 10.1016/j.brainresrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Pulliam L, Herndier BG, Tang NM, McGrath MS. Human immunodeficiency virus-infected macrophages produce soluble factors that cause histological and neurochemical alterations in cultured human brains. J Clin Invest. 1991;87:503–512. doi: 10.1172/JCI115024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, Kaushik A, Lapierre J, Dever SM, El-Hage N, Nair M. Electro-Magnetic Nano-Particle Bound Beclin1 siRNA Crosses the Blood-Brain Barrier to Attenuate the Inflammatory Effects of HIV-1 Infection in Vitro. J Neuroimmune Pharmacol. 2017a;12:120–132. doi: 10.1007/s11481-016-9688-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, et al. Importance of Autophagy in Mediating Human Immunodeficiency Virus (HIV) and Morphine-Induced Metabolic Dysfunction and Inflammation in Human Astrocytes Viruses. 2017b;9:201. doi: 10.3390/v9080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M, et al. Intranasal drug delivery of small interfering RNA targeting Beclin1 encapsulated with polyethylenimine (PEI) in mouse brain to achieve HIV attenuation. Sci Rep. 2017c;7:1862. doi: 10.1038/s41598-017-01819-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers TJ, Peterson PK. Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol. 2003;24:116–121. doi: 10.1016/s1471-4906(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Ronaldson PT, Bendayan R. HIV-1 viral envelope glycoprotein gp120 triggers an inflammatory response in cultured rat astrocytes and regulates the functional expression of P-glycoprotein. Mol Pharmacol. 2006;70:1087–1098. doi: 10.1124/mol.106.025973. [DOI] [PubMed] [Google Scholar]
- Sagnier S, et al. Autophagy restricts HIV-1 infection by selectively degrading Tat in CD4+ T lymphocytes. J Virol. 2015;89:615–625. doi: 10.1128/JVI.02174-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K, Kauppinen A. Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: impact on the aging process. Ageing Res Rev. 2013;12:520–534. doi: 10.1016/j.arr.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Saribas AS, Khalili K, Sariyer IK. Dysregulation of autophagy by HIV-1 Nef in human astrocytes. Cell Cycle. 2015;14:2899–2904. doi: 10.1080/15384101.2015.1069927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi B, Raina J, Lorenzo A, Busciglio J, Gabuzda D. Neuronal apoptosis induced by HIV-1 Tat protein and TNF-alpha: potentiation of neurotoxicity mediated by oxidative stress and implications for HIV-1 dementia. J Neurovirol. 1998;4:281–290. doi: 10.3109/13550289809114529. [DOI] [PubMed] [Google Scholar]
- Silverstein PS, Shah A, Weemhoff J, Kumar S, Singh DP, Kumar A. HIV-1 gp120 and drugs of abuse: interactions in the central nervous system. Curr HIV Res. 2012;10:369–383. doi: 10.2174/157016212802138724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Henderson EE, Rogers TJ. Mu-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology. 2003;309:99–107. doi: 10.1016/s0042-6822(03)00015-1. [DOI] [PubMed] [Google Scholar]
- Steiner J, et al. Oxidative stress and therapeutic approaches in HIV dementia. Antioxid Redox Signal. 2006;8:2089–2100. doi: 10.1089/ars.2006.8.2089. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and autophagy regulation - a two-way street. Mol Med. 2017;23 doi: 10.2119/molmed.2017.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trillo-Pazos G, McFarlane-Abdulla E, Campbell IC, Pilkington GJ, Everall IP. Recombinant nef HIV-IIIB protein is toxic to human neurons in culture. Brain Res. 2000;864:315–326. doi: 10.1016/s0006-8993(00)02213-7. [DOI] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga FO, Ding Q, Keller JN, Hauser KF, Knapp PE, Bruce-Keller AJ. Cell-specific actions of HIV-Tat and morphine on opioid receptor expression in glia. J Neurosci Res. 2008;86:2100–2110. doi: 10.1002/jnr.21653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga FO, Gupta S, Keller JN, Knapp PE, Hauser KF, Bruce-Keller AJ. Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem. 2009;108:202–215. doi: 10.1111/j.1471-4159.2008.05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel BE, Lee SJ, Hildebrand A, Craig W, Pierschbacher MD, Wong-Staal F, Ruoslahti E. A novel integrin specificity exemplified by binding of the alpha v beta 5 integrin to the basic domain of the HIV Tat protein and vitronectin. J Cell Biol. 1993;121:461–468. doi: 10.1083/jcb.121.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Sinha S, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4:949–951. doi: 10.4161/auto.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Spector SA. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS (London, England) 2008;22:695–699. doi: 10.1097/QAD.0b013e3282f4a836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, Knapp PE. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at mu-opioid receptor-expressing glia. Brain. 2011;134:3616–3631. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.