

Abstract
The functions of sleep remain a mystery. Yet they must be important since sleep is highly conserved, and its chronic disruption is associated with various metabolic, psychiatric, and neurodegenerative disorders. This review will cover our evolving understanding of the mechanisms by which sleep is controlled and the complex relationship between sleep and disease states.
Introduction: Defining and Measuring Sleep
As human beings, we sleep for approximately one-third of our lives, yet nobody knows why we do it, which molecular mechanisms are involved, or why sleep manifests as major changes in the brain’s electrical activity that are associated with unconsciousness. This condition is inherently dangerous, since it prevents animals from protecting themselves and from foraging for food. Therefore, it must serve important functions. This conclusion is underscored by the presence of sleep in all animals in which its essential features have been carefully sought (63) (FIGURE 1). The first of these features is rapidly reversible behavioral quiescence, which distinguishes sleep from seizure, coma, hibernation, and anesthesia. The second feature is reduced arousal. Empirically, this translates to reduced responsiveness to environmental stimuli, which distinguishes sleep from quiet wakefulness. The third feature is homeostatic regulation. This means that a set point exists for daily sleep, with deviations from this set point requiring compensation at a later time. Experimentally, this homeostatic property can be demonstrated by preventing an animal from sleeping and then observing a compensatory increase, or rebound, in sleep shortly afterward (23, 63).
FIGURE 1.
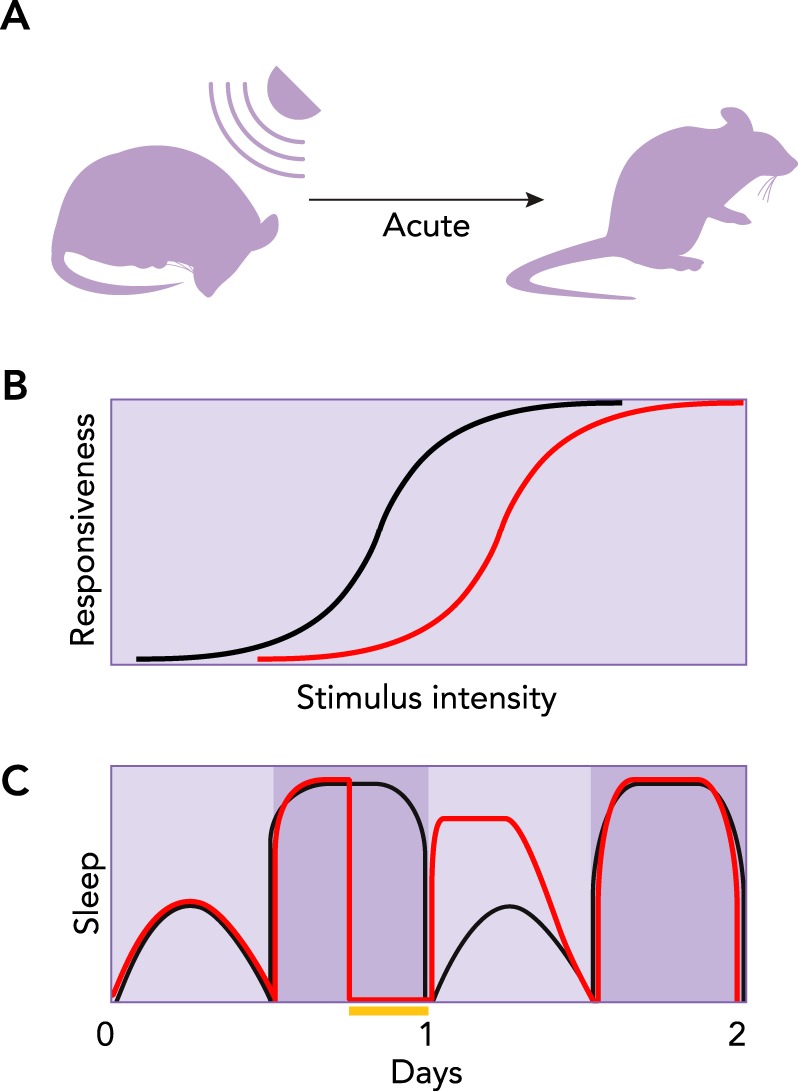
Core behavioral features of sleep
A: rapidly reversible quiescence can be demonstrated with a sufficiently strong stimulus, thus distinguishing sleep from seizure, coma, hibernation, and anesthesia. B: reduced arousal. A greater stimulus intensity is required to evoke a given behavioral response during sleep (red line) than during wakefulness (black line). C: the homeostatic nature of sleep is apparent from animals’ attempts to compensate for lost sleep. Black and red lines illustrate typical sleep cycles for control and sleep-deprived groups, respectively. Yellow bar indicates period of sleep deprivation. The next day, the sleep-deprived group sleeps more (i.e., rebounds) compared with the control.
Researchers have taken an additional approach to characterizing sleep in some animals. This approach involves using electrophysiological techniques, such as the electroencephalogram (EEG) to measure brain activity and the electromyogram (EMG) to measure skeletal muscle tension (22). These techniques have revealed the existence of two major sleep states in mammals, birds, and certain lizards (63). Non-rapid eye movement (NREM) sleep can be distinguished by slowing and synchronization of brain oscillations, whereas rapid eye movement (REM) sleep can be distinguished by brain desynchrony and muscle atonia (22). Importantly, both NREM and REM sleep states are also correlated with the essential behavioral features of sleep described above. Thus these behavioral features alone are considered to be sufficient to define sleep in animals in which electrophysiological recordings are particularly challenging, including fish and many invertebrates (63). Nonetheless, in some of these animals, such as fruit flies, crayfish, and worms, differences in brain activity have been confirmed between low and high arousal states (91, 93, 104), although in these organisms NREM and REM sleep states have not been observed, presumably because invertebrates lack the brain structures required for these phenomena.
Neuroanatomical Basis of Sleep Regulation
Although sleep involves changes in activity throughout the brain, this behavioral state is controlled by specific neural circuits. Mediators of external arousal cues include sensory neurons that project to the thalamus to promote waking (19, 63). In contrast, mediators of internal cues promote waking or sleep, depending on the source of the cue. This concept was elegantly first articulated in 1982, when researchers used EEG recordings of human subjects to tease apart the sleep/wake cycle into temporally distinct components. The results provided evidence for the first time that “two [internally driven] processes play a dominant role in sleep regulation: a sleep-dependent process (process S) and a sleep-independent circadian process (process C)” (17).
Process S represents the sleep-promoting, homeostatic component of sleep regulation. It rises with time spent awake and dissipates with time spent asleep, thus reflecting sleep need (FIGURE 2A) (17, 35). Since the needs fulfilled by sleep are unknown, process S is of great interest to neuroscientists and is being intensely studied using a variety of approaches. For example, process S can be approximated by measuring delta power, which is a mathematical transformation of the low-frequency EEG signature of the deepest stages of NREM sleep (6). Process S can also be approximated by measuring how much sleep the brain attempts to recover following a period of sleep deprivation. This so-called “rebound” manifests as a combination of increased depth and duration of sleep, with the relative changes in the two parameters varying with species. Despite great interest in process S, at the current time, neither its molecular nor its neuroanatomical basis has been identified.
FIGURE 2.

Neuroanatomy of sleep control
A: two process model for control of the sleep/wake cycle by the circadian clock (process C) and the sleep homeostat (process S). The clock drives a cycling pattern of arousal with a period of ~24 h. Process S increases during waking until it exceeds a certain threshold, after which it discharges, leading to suppression of waking and initiation of sleep. B: wake-promoting loci in the mammalian brain, including neuromodulatory centers of the ascending reticular activating system. C: orexin neurons in the lateral hypothalamus excite wake-promoting loci to stabilize the waking state. D: GABAergic inhibition of wake-promoting loci and other unknown targets promotes sleep. Color-coded key: BF, basal forebrain; TC, thalamocortical relay neurons; LH, lateral hypothalamus; VTA, ventral tegmental area; LC, locus coeruleus; DR, dorsal raphe nucleus; VLPO, ventrolateral preoptic nucleus; ZI, zona inserta of the subthalamus; MO, medulla oblongata; LDT/PPT, laterodorsaltegmental and pedunculopontine nuclei. Glut, glutamate; ORX, orexin; HA, histamine; DA, dopamine; NA, noradrenaline; 5HT, serotonin; ACh, acetylcholine.
In contrast, process C represents the wake-promoting contribution of the circadian clock (FIGURE 2A) (17, 35), which is now a mechanistically well-understood phenomenon (see below). Although clocks have been identified in nearly all tissues and cell types, the circadian regulation of the sleep/wake cycle originates in the body’s master clock, located in the suprachiasmatic nucleus (SCN) of the hypothalamus (84, 107). The SCN receives photic input from the retina via the retinohypothalamic tract (RHT). Input from the RHT phase-shifts the endogenous oscillatory activity of the SCN, thus altering the timing of its synaptic output (84, 107). The SCN regulates some brain regions directly (i.e., monosynaptically) and other areas indirectly. Circadian regulation of hormonal release appears to entrain some peripheral endogenous clocks, whereas temperature and feeding act as entrainment cues in other cells (20, 97, 112).
Part of the lasting appeal of the so-called two-process model for sleep/wake control has been its ability to simply and accurately approximate sleep drive across a 24-h cycle as the difference between sleep-promoting process S and wake-promoting process C. Thus, despite proposed additional elaborations, the two-process model still serves as a major foundation for neuroanatomical and molecular underpinnings of sleep regulation (18).
It is unclear where and how the brain integrates external signals from sensory neurons and internal signals from the homeostat and the clock to determine which arousal state should prevail. However, additional important brain loci are known to help maintain sleep/wake states or the switching between them. The subset of these loci located in the brain stem are known as the ascending reticular activating system (ARAS) and include the dorsal raphe, the ventral tegmental area, the locus coeruleus, and the laterodorsal tegmental and pedunculopontine nuclei, which release serotonin, dopamine, noradrenaline, and acetylcholine, respectively (19, 63, 111) (FIGURE 2B). These neuromodulatory nuclei project through two major pathways to additional arousal-regulating loci in the forebrain. The dorsal pathway modulates the thalamus, which receives much sensory information before passing it on to the cortex for further processing. The ventral pathway extends to the basal forebrain, which also projects to the cortex. The ventral pathway leading through the basal forebrain also includes nodes in the lateral hypothalamus and the tuberomamillary nucleus that release orexin (also known as hypocretin) and histamine, respectively (19, 63, 111). The orexinergic neurons enhance activity in many arousal-promoting nuclei, thus stabilizing and enhancing waking (36, 110) (FIGURE 2C).
The activities of particular brain stem neuromodulatory systems determine which combination of pathways will be activated, and consequently whether waking, REM sleep, or NREM sleep will ultimately result. For example, during waking, cholinergic and monoaminergic brain stem nuclei activate both the dorsal and the ventral pathways. As a result, the cortex is excited, and it can make sense of the external environment because the dorsal pathway maintains thalamic neurons in a firing mode that is conducive to throughput of sensory signaling (63). During REM sleep, cholinergic signaling continues to activate the ventral pathway and thus excites the cortex. However, monoaminergic signaling is simultaneously reduced, resulting in altered firing of thalamic neurons that filters out sensory signaling to the cortex (63). Cholinergic signaling during REM sleep also activates a descending pathway through the subcoeruleus nucleus that terminates with inhibition of motoneurons in the spinal cord, thus causing REM-specific muscle atonia (42). In contrast, during NREM sleep, cholinergic and monoaminergic signaling are both reduced, leading to dampened cortical excitation and heightened filtering of sensory information at the level of the thalamus (63).
The ARAS and the dorsal and ventral pathways to the cortex promote arousal during waking. But other brain loci have been proposed to suppress arousal. These include populations of GABAergic neurons within the cortex (45, 86), the brain stem (7, 8, 129), the ventrolateral preoptic nucleus (4), and other parts of the lateral hypothalamus (50, 60, 73, 124, 126), the basal forebrain (95, 136), and the subthalamus (76) (FIGURE 2D). When these various neuronal clusters are active, REM sleep or NREM sleep results, with switching between sleep states determined by an oscillatory circuit in the brain stem (7, 33, 52, 129, 130). Importantly, many of the sleep-promoting loci appear to form mutually inhibitory connections with arousal-promoting loci. This relationship has been likened to a flip-flop switch that favors rapid transitions and state stability (111).
Molecular Basis of Sleep Regulation
The functions of arousal circuits are determined by specific molecules expressed within those circuits. Therefore, at least some of those molecules must be required to initiate or maintain sleep/wake states. Identifying those molecules has been a goal of researchers interested in understanding sleep physiology and in treating sleep-related disorders. Unfortunately, the kinds of molecules that have been identified so far do not contribute to a coherent cellular function (FIGURE 3). This complexity has made it difficult to predict with any accuracy the identities of additional molecules that contribute to sleep regulation. Nonetheless, such molecules continue to be discovered, largely due to seven main approaches.
FIGURE 3.
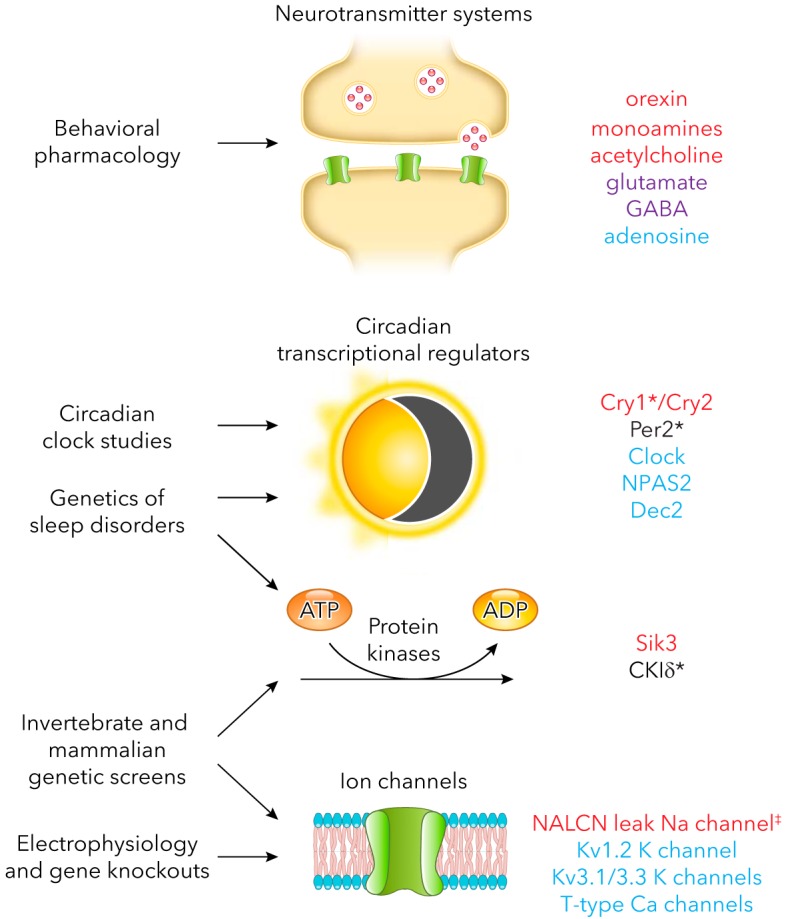
Endogenous molecules that impact sleep and the approaches that led to their discovery
Only mammalian molecules are listed. Red, promotes waking; blue, promotes sleep; purple, promotes waking and sleep; asterisk, regulates sleep timing. Not listed: components of metabolic pathways for neurotransmitters, neurotransmitter receptors, and transporters. ‡A gain-of-function mutation in NALCN decreases REM sleep but has no effect on total sleep duration (44). Loss-of-function mutations in the fly ortholog of NALCN, called narrow abdomen, increase sleep (64). Therefore, NALCN has been provisionally assigned a wake-promoting function.
Behavioral Pharmacology
Various drugs have well-established effects on arousal. The targets of these drugs thus inform our understanding of molecules that impact normal sleep and waking. Care must be taken in confirming such targets, however, since most drugs are selective rather than specific. As a result, a drug’s effects may be mediated by molecules other than an established target. For example, first-generation anti-histamines such as diphenhydramine are thought to promote sleep primarily by antagonizing histamine H1 receptors (67). As discussed above, histamine release by the tuberomamillary nucleus promotes waking. Thus it makes sense that antagonism of histaminergic signaling would promote sleep. However, first-generation anti-histamines also have off-target effects on structurally related muscarinic acetylcholine receptors. Since cholinergic signaling promotes arousal, antagonism of muscarinic receptors by early forms of anti-histamines also probably contributes to the soporific effects of these drugs (67). Other common sleep aids include barbiturates, benzodiazepine, and non-benzodiazepine hypnotics. These three classes of drugs potentiate signaling by GABAA receptors (34), which is consistent with the arousal-suppressing effects of GABA discussed earlier.
Stimulants co-opt endogenous arousal-regulating mechanisms as well. For example, amphetamine and methamphetamine are thought to promote waking by enhancing cortical release of monoamines, especially dopamine (92). In contrast, caffeine is thought to promote waking by preventing the endogenous somnogen, adenosine, from stimulating excitatory adenosine A2 receptors in sleep-active neurons in the basal forebrain (19, 57, 74). Caffeine-insensitive adenosine signaling also appears to promote sleep. For example, adenosine A1 receptors inhibit wake-active neurons in the basal forebrain (10, 51) and brain stem (11, 103). Interestingly, increases in synaptically released adenosine in the basal forebrain correlate with time awake and thus sleep pressure in the cortex (100, 121) (but also see Ref. 16). As a result, adenosine signaling has been proposed to reflect homeostatic sleep drive. Although some studies have suggested that A2 receptors may mediate such signaling, increasing evidence suggests that neuronal A1 receptors are involved (14, 15, 100, 101, 120, 125), perhaps due to increased release of adenosine from astrocytes (14, 48, 113). Also consistent with its proposed function as an endogenous somnogen, adenosine is increased by the sleep-promoting factor prostaglandin D2 (58), whereas genetic manipulations that reduce adenosine also reduce sleep (96).
Circadian Clock Studies
Based on research from both flies and mice, we also know the molecular composition of the core circadian oscillator, a major regulator of the timing of arousal. In mammals, this oscillator utilizes the transcription factor CLOCK or its homolog NPAS2 and another transcription factor called BMAL1, which heterodimerize to activate transcription of clock-controlled genes (37, 84). Many of these genes control bodily functions at certain times of day. However, some of these genes function together with CLOCK/NPAS2::BMAL1 to allow the molecular clock to cycle. For example, the Period (Per) and Cryptochrome (Cry) translation products heterodimerize as PER::CRY to repress CLOCK/NPAS2::BMAL1. The net effects of this negative feedback loop are waxing and waning of CLOCK/NPAS2::BMAL1 activity and therefore cycling transcription of clock-controlled genes, including Per and Cry (37, 84, 122). In a second loop, CLOCK/NPAS2::BMAL1 upregulate transcription of Ror and Rev-erb α/β, which feed back to maintain appropriate transcription of Clock and Bmal1 (54). Other essential components of the molecular clock regulate the stabilities, interaction partners, and subcellular localizations of components of these loops (54, 122). The coordinate function of all of these core clock molecules leads to transcriptional [or, in some cases, indirect posttrancriptional (46)] cycling of downstream effector genes over a period of ~24 h. Since the clock regulates the timing of the sleep/wake cycle, it is not surprising that some core clock genes are also required for normal sleep timing and duration. For example, knockout of Clock or Npas2 causes a reduction in sleep (43, 89), and double knockouts of the two Cry homologs Cry1 and Cry2 cause an increase in sleep (132). The downstream transcriptional targets of these genes that are ultimately responsible for effects on arousal have yet to be identified.
Genetics of Sleep Disorders
A third approach to understanding the molecular basis of sleep regulation has been to identify families suffering from heritable sleep disorders, map the responsible genes, and confirm their identities by phenocopying disorders in mutant animal models. This approach has historically been very time-consuming, but it has also benefitted from circadian clock research, which has provided candidate genes to inspect for aberrations in patient populations. For example, patients with familial delayed sleep phase disorder (DSPD) typically wake up late in the morning and fall asleep late at night (65). This behavior has been mapped to a gain-of-function mutation in the gene encoding the core circadian clock gene Cry1 that causes its translation product to constitutively repress the circadian transcriptional activators CLOCK and BMAL1 (98). In contrast, patients with familial advanced sleep phase disorder (ASPD) typically wake up and fall asleep very early. Behavior in a subset of these patients has been mapped to loss-of-function mutations in another core circadian repressor, Per2, and in a kinase that controls PER2’s stability called CKIδ (49). Last, some naturally short sleepers rise early in the morning, like patients with ASPD, but exhibit normal timing of sleep onset in the evening. Some of these individuals have been shown to carry a mutation in Dec2, a clock-controlled repressor of various other cycling genes (53). Mice engineered with the same mutation have elevated levels of the wake-promoting neuropeptide orexin, and their excess sleep can be attenuated with an orexin receptor antagonist, suggesting that the DEC2 protein normally limits orexin levels to facilitate sleep (55). Rapid advances in whole-genome sequencing promise to accelerate progress in human sleep genetics in the near future.
Forward Genetic Screens in Mammals
Identifying additional molecules involved in sleep regulation has been difficult for many reasons. In particular, such molecules have often been unpredictable, so some researchers have attempted to discover them by using unbiased screens for mutants that exhibit altered sleep phenotypes. In mammals, such screens are very time-consuming, labor-intensive, and expensive. For these reasons, only one forward genetic screen for sleep mutants has been successfully conducted in mice. So far, only two mutants have emerged from this screen. The first, called Sleepy, contains a splicing mutation in a kinase-encoding gene called Sik3 that causes increased NREM sleep by mechanisms that are not yet clear. The second sleep mutant, called Dreamless, appears to possess a gain-of-function mutation in a gene encoding the leak sodium channel NALCN. Consistent with the selective reduction in REM sleep that it causes, this mutant exhibits increased electrical activity in part of the brain stem oscillator responsible for shutting off REM sleep (44).
Studies in Genetically Tractable Invertebrates
The discovery that NALCN is required for mammalian sleep regulation was actually preceded by the discovery that an NALCN homolog called narrow abdomen stabilizes sleep and wake states in flies, with loss-of-function mutations leading to an increase in total sleep (64). These findings illustrate the power of invertebrate behavioral genetics when it comes to predicting the identities of novel mammalian sleep-regulating genes. In fact, although many genes have now been implicated in sleep regulation in flies (2), only a subset of these has been tested for homologous functions in mammals. One of these is the Shaker potassium channel. Loss-of-function mutations in Shaker lead to a near abolition of sleep in flies (30), and knockout of the mammalian Shaker homolog Kv1.2 also reduces sleep in mice (40). Shaker was identified in a forward genetic screen for sleep mutants (30). Molecules identified in such screens tend to have the most profound phenotypes. Other examples include sleep-promoting positive regulators of Shaker, such as Hyperkinetic (21) and Sleepless/Quiver (72, 135) [which also downregulates nicotinic acetylcholine receptors (134)]; the sleep-promoting E3 ubiquitin ligase insomniac (119); the sleep-promoting cyclin A (106), its regulator taranis (1) and regulator of cyclin A1 (106), and its likely effector cyclin-dependent kinase 1 (1); and the wake-promoting RNA-editing enzyme ADAR (105). Except for Shaker and its positive regulators, which reduce excitability, and to some extent for ADAR, which restricts prolonged presynaptic release of glutamate, the mechanisms by which these sleep-regulating molecules modify neuronal function are unknown.
Electrophysiological Studies Combined With Gene Knockouts
The genes described above illustrate another point: molecules that regulate excitability and synaptic physiology are highly represented among genes that have large effects on sleep. To some extent, this finding is expected, since excitability and synaptic transmission are essential to establishing and maintaining the properties of neural circuits. This idea has led several groups to identify ion channels known to contribute to the firing properties of arousal-controlling brain loci, genetically ablate the corresponding genes, and measure the resulting sleep phenotypes of mutant animals. As a result, it is now known that T-type calcium channels and Kv3-type potassium channels are required for burst firing of thalamic neurons that contribute to brain oscillations during NREM sleep (9, 12, 27, 41, 99). It seems likely that additional ion channels will emerge as essential to sleep/wake control as other arousal-regulating nuclei are characterized electrophysiologically and their underlying currents are manipulated in knockout studies.
Microarrays
Forward genetic screens offer the advantage of starting out with an established phenotype. However, mapping the responsible gene, then figuring out where and how it functions can be slow, expensive, and labor-intensive. An alternative screening approach that largely avoids these problems has been to use microarrays to identify genes that are differentially expressed during sleep and wake states. Such studies have demonstrated that, relative to extended waking, sleep causes upregulation of transcripts implicated in macromolecular biosynthesis and synaptic depression, and sleep causes downregulation of brain transcripts implicated in cellular stress responses, energy homeostasis, and synaptic potentiation (29, 31, 79, 81, 123).
Importantly, these patterns of altered gene expression are conserved among flies, birds, and mammals, suggesting that they reflect core rather than species-specific mechanisms (28). However, adrenalectomized mice show fewer changes in gene expression across the sleep/wake cycle (85). This result illustrates a major disadvantage of microarray studies over other approaches to identify molecules involved in sleep regulation; i.e., it is difficult to distinguish between transcriptional changes to stressful responses, which include extended waking and sleep disruption, and transcriptional changes to more specific mechanisms involved in sleep/wake control and function. Thus microarrays should be viewed as a starting point for formulating a hypothesis about general mechanisms. But demonstrating a causal relation between a gene and its role in sleep regulation requires validation, often by recapitulating a relevant phenotype using pharmacological or genetic ablation of the molecule in question. So far at least, data obtained by microarrays has not reached this stage.
Connections Between Sleep and Disease
Disturbances in circadian functions and sleep have long been associated with metabolic, psychiatric, and neurodegenerative disorders. However, increasing evidence suggests that such disturbances may not always be secondary symptoms but may in fact contribute to the etiology and severity of disease states (FIGURE 4). In some cases, the causal relation can be traced to dysregulation of one or more neurotransmitter systems that control arousal, or to a disruption in circadian timing of sleep (see above). In other cases, however, the relation is unclear, in part because the mechanisms underlying sleep regulation and sleep-related disorders are poorly understood. Below is a list of common types of these disorders and a brief description of how they impact or are impacted by the circadian and sleep systems.
FIGURE 4.
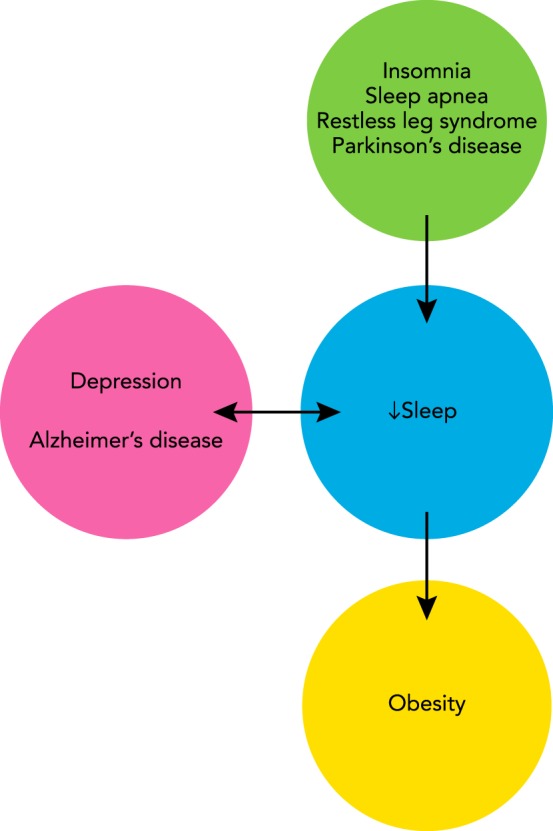
Interaction between sleep and disorders of the nervous system
Many disorders disrupt sleep (arrow from green to blue circle). In other cases, low sleep exacerbates existing health problems (arrow from blue to yellow circle). Positive feedback may exist between poor-quality sleep and still other disorders (bidirectional arrow between blue and pink circles).
Insomnia
People suffering from insomnia have trouble falling asleep, staying asleep, or feeling refreshed following sleep (5). Up to 10% of adults are afflicted with insomnia severely enough for it to be referred to as a chronic disorder, and at least another 40% of the population is estimated to suffer from its symptoms intermittently (87). Its causes are multivariate, but ultimately they lead to hyperactivity of subcortical arousal-promoting neural circuits during sleep (94). The American College of Physicians recommends cognitive behavioral therapy, which involves a combination of mental exercises and healthy sleeping habits as a primary approach to treating insomnia (102). Non-benzodiazepines are a common alternative that function by elevating inhibitory GABAergic signaling, thus suppressing arousal. More recently, orexin receptor antagonists have also been approved for treatment of insomnia (56).
Although insomnia is moderately heritable, specific genetic risk factors for it are largely unknown (75). One exception is fatal familial insomnia (FFI), a rare autosomal dominant disease caused by a specific mutation in the prion protein gene PRNP. Symptoms of FFI initially include insomnia and autonomic dysfunctions (e.g., tachycardia), followed by progressive cognitive and motor impairment, and finally death. These symptoms result from neuronal and astrocytic cell death in brain regions through which PRNP rapidly spreads, including the thalamus, a known locus for control of sleep (77).
Narcolepsy
Patients with narcolepsy exhibit excessive daytime sleepiness associated with frequent intrusions of REM sleep into the waking state. Narcolepsy is also often accompanied by cataplexy, which is a temporary state of paralytic muscle weakness whose onset is triggered by positive emotions such as excitement or laughter. During these periods of paralysis, patients are conscious of the world around them. Thus the cataplectic state of narcolepsy has been described as a hybrid between REM paralysis and the waking state (24). Patients with narcolepsy have trouble maintaining complete wakefulness for more than a few hours at a time, and they have disrupted nighttime sleep as well, thus leading to the characterization of narcolepsy as a wake and sleep maintenance disorder. Narcolepsy is caused by reduced signaling involving the neuropeptide orexin, possibly due to autoimmune destruction of the neurons that produce it (138). Treatments include stimulants such as amphetamine to facilitate monoamine release and thus increase wakefulness during the day, hypnotics to promote sleep at night and thus reduce daytime sleepiness, and anti-depressants that reduce REM sleep drive and cataplexy (13).
Metabolic Disorders and Obesity
Thirty percent of the U.S. workforce reports sleeping 6 h or less per night (78), which is significantly less than the 7–8 h recommended by the American Academy of Sleep Medicine (128). These statistics are particularly concerning considering that reduced sleep quality and quantity increase the risk of weight gain and Type 2 diabetes (25, 26, 59). Although the reasons for this relation are unproven, several studies provide plausible explanations. For example, restricted sleep has been reported to increase food consumption and plasma levels of leptin, a hunger-promoting hormone, while lowering levels of ghrelin, a satiety-promoting hormone (90, 115, 116, 118). The underlying mechanisms by which sleep restriction might lead to such molecular changes are unknown.
Sleep Apnea
Another contributing factor to metabolic disorders is the prevalence of obstructive sleep apnea (OSA), a type of sleep-disordered breathing that afflicts ~15% of adults (137). OSA is caused by repeated collapse of the upper airway, which results in bouts of intermittent hypoxia. To compensate, patients undergo repeated brief arousals, resulting in poor-quality sleep and subsequent daytime sleepiness. These repeated cycles also upregulate the sympathetic nervous system and production of reactive oxygen species (ROS). Although the underlying molecular mechanisms are not well-understood, elevated sympathetic tone, ROS, and sleep fragmentation are known to increase the risk of inflammation and metabolic dysfunction, including obesity (71, 83).
Restless Leg Syndrome
Another common sleep disorder is restless leg syndrome (RLS), which is characterized by a strong urge to move one’s legs at night that is exacerbated by lack of movement. Although RLS can be secondary to other disorders, primary (or idiopathic) RLS has a strong genetic component that allows it to exist on its own. Based on multiple genome-wide association studies, six genes have been implicated in primary RLS. However, their molecular functions appear to be diverse and unrelated, and alleles for high risk of RLS are excluded from their coding regions (61, 109). Drugs that elevate levels of dopamine in the brain are first-line treatment options for RLS, suggesting that dysfunction arises from disrupted dopamine signaling in patients. However, attempts to correlate risk for RLS with variants of genes involved in dopamine function have so far proven unsuccessful (38, 69). Thus the molecular mechanisms that contribute to RLS continue to remain elusive.
Depression
Disrupted sleep is a common symptom of depression and a major risk factor for suicide (3, 70). Paradoxically, however, sleep deprivation is sometimes used as a temporary way to treat depression (133). Debate continues as to the possible causal relation between these two health problems. However, it seems likely that both involve altered monoaminergic signaling. For example, many antidepressants that elevate synaptic levels of serotonin and/or noradrenaline also cause or exacerbate symptoms of insomnia (131). Depending on other symptoms, some physicians may thus opt to supplement treatment of patients suffering from both depression and insomnia with sedatives, at least in the short-term, or with antidepressants that have sedative properties of their own (62, 131). In patients for whom insomnia is not a problem, compounds in the latter category may be avoided since they can lead to oversedation. These examples illustrate that the desired effect on sleep may be an important consideration when selecting the optimal type of pharmacotherapy for treatment of depression.
Neurodegenerative Diseases
Poor sleep quality and excessive daytime sleepiness unrelated to medications and comorbidities are common symptoms among patients with Parkinson’s disease (PD) and Alzheimer’s disease (AD). These symptoms are often reported very early in disease progression, thus suggesting a mechanistic relation to the earliest stages of the diseases (82, 114). In PD, it has been hypothesized that excessive daytime sleepiness is secondary to loss of wake-promoting dopaminergic neurons during disease progression (138). However, in AD, sleep/wake dysfunction may have a more causal relation to the development of the disease state. In AD, amyloid beta (Aβ) and phosphorylated tau protein are believed to contribute to disease pathology. Notably, extracellular accumulation of Aβ peaks during the waking period and reaches a nadir during sleep, with sleep deprivation exaggerating and sleep-promoting drugs ameliorating these effects (68). Still other studies have shown that sleep deprivation exacerbates tau pathology and loss of synapses (39, 108). Although the specific mechanisms by which disrupted sleep might contribute to AD pathology are unknown, one possibility is that extended waking promotes Aβ plaque formation. In support of this hypothesis, Aβ release is upregulated by synaptic activity (32, 66), which is generally higher during waking than during sleep (127). In humans, short sleep duration and poor sleep quality are also correlated with elevated Aβ load (117). Thus disrupted sleep may be a proxy for disease progression. Since neurodegeneration during AD also contributes to disrupted sleep, it has been suggested that the bidirectional relationship between poor-quality sleep and AD leads to a vicious cycle that exacerbates both conditions (47, 80, 88).
Concluding Remarks
The pace of discovery of new sleep-regulating molecules and brain loci continues at a rapid pace. Because of the growing recognition that sleep is not just affected by but also contributes to disease states, these discoveries present new opportunities to intervene in many common, and in some cases tragically debilitating, health problems. These discoveries also illustrate the growing synergy between basic research and the rapidly evolving field of sleep medicine.
Acknowledgments
I thank Kendall Satterfield and Meilin Wu for helpful discussion and comments on this manuscript.
This work was supported by National Institute of General Medical Sciences Grant GM-125080.
No conflicts of interest, financial or otherwise, are declared by the author.
W.J. prepared figures; W.J. drafted manuscript; W.J. edited and revised manuscript; W.J. approved final version of manuscript.
References
- 1.Afonso DJ, Liu D, Machado DR, Pan H, Jepson JE, Rogulja D, Koh K. TARANIS functions with cyclin A and Cdk1 in a novel arousal center to control sleep in Drosophila. Curr Biol 25: 1717–1726, 2015. doi: 10.1016/j.cub.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afonso DJ, Machado DR, Koh K. Control of sleep by a network of cell cycle genes. Fly (Austin) 9: 165–172, 2015. doi: 10.1080/19336934.2016.1153776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ağargün MY, Kara H, Solmaz M. Sleep disturbances and suicidal behavior in patients with major depression. J Clin Psychiatry 58: 249–251, 1997. doi: 10.4088/JCP.v58n0602. [DOI] [PubMed] [Google Scholar]
- 4.Alam MA, Kumar S, McGinty D, Alam MN, Szymusiak R. Neuronal activity in the preoptic hypothalamus during sleep deprivation and recovery sleep. J Neurophysiol 111: 287–299, 2014. doi: 10.1152/jn.00504.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.American Psychiatric Association., and American Psychiatric Association. DSM-5 Task Force Diagnostic and Statistical Manual of Mental Disorders: DSM-5. Washington, D.C.: American Psychiatric Association, 2013, p. xliv. [Google Scholar]
- 6.Amzica F, Steriade M. Electrophysiological correlates of sleep delta waves. Electroencephalogr Clin Neurophysiol 107: 69–83, 1998. doi: 10.1016/S0013-4694(98)00051-0. [DOI] [PubMed] [Google Scholar]
- 7.Anaclet C, Ferrari L, Arrigoni E, Bass CE, Saper CB, Lu J, Fuller PM. The GABAergic parafacial zone is a medullary slow wave sleep-promoting center. Nat Neurosci 17: 1217–1224, 2014. doi: 10.1038/nn.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anaclet C, Lin JS, Vetrivelan R, Krenzer M, Vong L, Fuller PM, Lu J. Identification and characterization of a sleep-active cell group in the rostral medullary brainstem. J Neurosci 32: 17970–17976, 2012. doi: 10.1523/JNEUROSCI.0620-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, Scammell TE, Tonegawa S. Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci USA 102: 1743–1748, 2005. doi: 10.1073/pnas.0409644102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arrigoni E, Chamberlin NL, Saper CB, McCarley RW. Adenosine inhibits basal forebrain cholinergic and noncholinergic neurons in vitro. Neuroscience 140: 403–413, 2006. doi: 10.1016/j.neuroscience.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 11.Arrigoni E, Rainnie DG, McCarley RW, Greene RW. Adenosine-mediated presynaptic modulation of glutamatergic transmission in the laterodorsal tegmentum. J Neurosci 21: 1076–1085, 2001. doi: 10.1523/JNEUROSCI.21-03-01076.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Astori S, Wimmer RD, Prosser HM, Corti C, Corsi M, Liaudet N, Volterra A, Franken P, Adelman JP, Lüthi A. The Ca(V)3.3 calcium channel is the major sleep spindle pacemaker in thalamus. Proc Natl Acad Sci USA 108: 13823–13828, 2011. doi: 10.1073/pnas.1105115108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattarai J, Sumerall S. Current and future treatment options for narcolepsy: a review. Sleep Sci 10: 19–27, 2017. doi: 10.5935/1984-0063.20170004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjorness TE, Dale N, Mettlach G, Sonneborn A, Sahin B, Fienberg AA, Yanagisawa M, Bibb JA, Greene RW. An adenosine-mediated glial-neuronal circuit for homeostatic sleep. J Neurosci 36: 3709–3721, 2016. doi: 10.1523/JNEUROSCI.3906-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bjorness TE, Kelly CL, Gao T, Poffenberger V, Greene RW. Control and function of the homeostatic sleep response by adenosine A1 receptors. J Neurosci 29: 1267–1276, 2009. doi: 10.1523/JNEUROSCI.2942-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blanco-Centurion C, Xu M, Murillo-Rodriguez E, Gerashchenko D, Shiromani AM, Salin-Pascual RJ, Hof PR, Shiromani PJ. Adenosine and sleep homeostasis in the Basal forebrain. J Neurosci 26: 8092–8100, 2006. doi: 10.1523/JNEUROSCI.2181-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borbély AA. A two process model of sleep regulation. Hum Neurobiol 1: 195–204, 1982. [PubMed] [Google Scholar]
- 18.Borbély AA, Daan S, Wirz-Justice A, Deboer T. The two-process model of sleep regulation: a reappraisal. J Sleep Res 25: 131–143, 2016. doi: 10.1111/jsr.12371. [DOI] [PubMed] [Google Scholar]
- 19.Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev 92: 1087–1187, 2012. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buhr ED, Yoo SH, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Science 330: 379–385, 2010. doi: 10.1126/science.1195262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bushey D, Huber R, Tononi G, Cirelli C. Drosophila hyperkinetic mutants have reduced sleep and impaired memory. J Neurosci 27: 5384–5393, 2007. doi: 10.1523/JNEUROSCI.0108-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campbell IG. EEG recording and analysis for sleep research. Curr Protoc Neurosci 10: 2, 2009. doi: 10.1002/0471142301.ns1002s49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell SS, Tobler I. Animal sleep: a review of sleep duration across phylogeny. Neurosci Biobehav Rev 8: 269–300, 1984. doi: 10.1016/0149-7634(84)90054-X. [DOI] [PubMed] [Google Scholar]
- 24.Cao M, Guilleminault C. Hypocretin and its emerging role as a target for treatment of sleep disorders. Curr Neurol Neurosci Rep 11: 227–234, 2011. doi: 10.1007/s11910-010-0172-9. [DOI] [PubMed] [Google Scholar]
- 25.Cappuccio FP, D’Elia L, Strazzullo P, Miller MA. Quantity and quality of sleep and incidence of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care 33: 414–420, 2010. doi: 10.2337/dc09-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cappuccio FP, Taggart FM, Kandala NB, Currie A, Peile E, Stranges S, Miller MA. Meta-analysis of short sleep duration and obesity in children and adults. Sleep 31: 619–626, 2008. doi: 10.1093/sleep/31.5.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi S, Yu E, Lee S, Llinás RR. Altered thalamocortical rhythmicity and connectivity in mice lacking CaV3.1 T-type Ca2+ channels in unconsciousness. Proc Natl Acad Sci USA 112: 7839–7844, 2015. doi: 10.1073/pnas.1420983112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cirelli C. Cellular consequences of sleep deprivation in the brain. Sleep Med Rev 10: 307–321, 2006. doi: 10.1016/j.smrv.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Cirelli C. A molecular window on sleep: changes in gene expression between sleep and wakefulness. Neuroscientist 11: 63–74, 2005. doi: 10.1177/1073858404270900. [DOI] [PubMed] [Google Scholar]
- 30.Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, Tononi G. Reduced sleep in Drosophila Shaker mutants. Nature 434: 1087–1092, 2005. doi: 10.1038/nature03486. [DOI] [PubMed] [Google Scholar]
- 31.Cirelli C, Tononi G. Gene expression in the brain across the sleep-waking cycle. Brain Res 885: 303–321, 2000. doi: 10.1016/S0006-8993(00)03008-0. [DOI] [PubMed] [Google Scholar]
- 32.Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48: 913–922, 2005. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 33.Cox J, Pinto L, Dan Y. Calcium imaging of sleep-wake related neuronal activity in the dorsal pons. Nat Commun 7: 10763, 2016. doi: 10.1038/ncomms10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.D’Hulst C, Atack JR, Kooy RF. The complexity of the GABAA receptor shapes unique pharmacological profiles. Drug Discov Today 14: 866–875, 2009. doi: 10.1016/j.drudis.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 35.Daan S, Beersma DG, Borbély AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol 246: R161–R183, 1984. [DOI] [PubMed] [Google Scholar]
- 36.de Lecea L, Huerta R. Hypocretin (orexin) regulation of sleep-to-wake transitions. Front Pharmacol 5: 16, 2014. doi: 10.3389/fphar.2014.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeBruyne JP, Weaver DR, Reppert SM. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat Neurosci 10: 543–545, 2007. doi: 10.1038/nn1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Desautels A, Turecki G, Montplaisir J, Ftouhi-Paquin N, Michaud M, Chouinard VA, Rouleau GA. Dopaminergic neurotransmission and restless legs syndrome: a genetic association analysis. Neurology 57: 1304–1306, 2001. doi: 10.1212/WNL.57.7.1304. [DOI] [PubMed] [Google Scholar]
- 39.Di Meco A, Joshi YB, Praticò D. Sleep deprivation impairs memory, tau metabolism, and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol Aging 35: 1813–1820, 2014. doi: 10.1016/j.neurobiolaging.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 40.Douglas CL, Vyazovskiy V, Southard T, Chiu SY, Messing A, Tononi G, Cirelli C. Sleep in Kcna2 knockout mice. BMC Biol 5: 42, 2007. doi: 10.1186/1741-7007-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Espinosa F, Torres-Vega MA, Marks GA, Joho RH. Ablation of Kv3.1 and Kv3.3 potassium channels disrupts thalamocortical oscillations in vitro and in vivo. J Neurosci 28: 5570–5581, 2008. doi: 10.1523/JNEUROSCI.0747-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fraigne JJ, Torontali ZA, Snow MB, Peever JH. REM sleep at its core—circuits, neurotransmitters, and pathophysiology. Front Neurol 6: 123, 2015. doi: 10.3389/fneur.2015.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Franken P, Dudley CA, Estill SJ, Barakat M, Thomason R, O’Hara BF, McKnight SL. NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: genotype and sex interactions. Proc Natl Acad Sci USA 103: 7118–7123, 2006. doi: 10.1073/pnas.0602006103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Funato H, Miyoshi C, Fujiyama T, Kanda T, Sato M, Wang Z, Ma J, Nakane S, Tomita J, Ikkyu A, Kakizaki M, Hotta-Hirashima N, Kanno S, Komiya H, Asano F, Honda T, Kim SJ, Harano K, Muramoto H, Yonezawa T, Mizuno S, Miyazaki S, Connor L, Kumar V, Miura I, Suzuki T, Watanabe A, Abe M, Sugiyama F, Takahashi S, Sakimura K, Hayashi Y, Liu Q, Kume K, Wakana S, Takahashi JS, Yanagisawa M. Forward-genetics analysis of sleep in randomly mutagenized mice. Nature 539: 378–383, 2016. doi: 10.1038/nature20142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerashchenko D, Wisor JP, Burns D, Reh RK, Shiromani PJ, Sakurai T, de la Iglesia HO, Kilduff TS. Identification of a population of sleep-active cerebral cortex neurons. Proc Natl Acad Sci USA 105: 10227–10232, 2008. doi: 10.1073/pnas.0803125105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Green CB. Circadian posttranscriptional regulatory mechanisms in mammals. Cold Spring Harb Perspect Biol 10: a030692, 2018. doi: 10.1101/cshperspect.a030692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guarnieri B, Sorbi S. Sleep and cognitive decline: a strong bidirectional relationship. it is time for specific recommendations on routine assessment and the management of sleep disorders in patients with mild cognitive impairment and dementia. Eur Neurol 74: 43–48, 2015. doi: 10.1159/000434629. [DOI] [PubMed] [Google Scholar]
- 48.Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 61: 213–219, 2009. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hallows WC, Ptáček LJ, Fu YH. Solving the mystery of human sleep schedules one mutation at a time. Crit Rev Biochem Mol Biol 48: 465–475, 2013. doi: 10.3109/10409238.2013.831395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hassani OK, Henny P, Lee MG, Jones BE. GABAergic neurons intermingled with orexin and MCH neurons in the lateral hypothalamus discharge maximally during sleep. Eur J Neurosci 32: 448–457, 2010. doi: 10.1111/j.1460-9568.2010.07295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hawryluk JM, Ferrari LL, Keating SA, Arrigoni E. Adenosine inhibits glutamatergic input to basal forebrain cholinergic neurons. J Neurophysiol 107: 2769–2781, 2012. doi: 10.1152/jn.00528.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayashi Y, Kashiwagi M, Yasuda K, Ando R, Kanuka M, Sakai K, Itohara S. Cells of a common developmental origin regulate REM/non-REM sleep and wakefulness in mice. Science 350: 957–961, 2015. doi: 10.1126/science.aad1023. [DOI] [PubMed] [Google Scholar]
- 53.He Y, Jones CR, Fujiki N, Xu Y, Guo B, Holder JL Jr, Rossner MJ, Nishino S, Fu YH. The transcriptional repressor DEC2 regulates sleep length in mammals. Science 325: 866–870, 2009. doi: 10.1126/science.1174443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirano A, Fu YH, Ptáček LJ. The intricate dance of post-translational modifications in the rhythm of life. Nat Struct Mol Biol 23: 1053–1060, 2016. doi: 10.1038/nsmb.3326. [DOI] [PubMed] [Google Scholar]
- 55.Hirano A, Hsu PK, Zhang L, Xing L, McMahon T, Yamazaki M, Ptáček LJ, Fu YH. DEC2 modulates orexin expression and regulates sleep. Proc Natl Acad Sci USA 115: 3434–3439, 2018. doi: 10.1073/pnas.1801693115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holst SC, Valomon A, Landolt HP. Sleep pharmacogenetics: personalized sleep-wake therapy. Annu Rev Pharmacol Toxicol 56: 577–603, 2016. doi: 10.1146/annurev-pharmtox-010715-103801. [DOI] [PubMed] [Google Scholar]
- 57.Huang ZL, Qu WM, Eguchi N, Chen JF, Schwarzschild MA, Fredholm BB, Urade Y, Hayaishi O. Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nat Neurosci 8: 858–859, 2005. doi: 10.1038/nn1491. [DOI] [PubMed] [Google Scholar]
- 58.Huang ZL, Urade Y, Hayaishi O. The role of adenosine in the regulation of sleep. Curr Top Med Chem 11: 1047–1057, 2011. doi: 10.2174/156802611795347654. [DOI] [PubMed] [Google Scholar]
- 59.Jackson CL, Redline S, Kawachi I, Hu FB. Association between sleep duration and diabetes in black and white adults. Diabetes Care 36: 3557–3565, 2013. doi: 10.2337/dc13-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jego S, Glasgow SD, Herrera CG, Ekstrand M, Reed SJ, Boyce R, Friedman J, Burdakov D, Adamantidis AR. Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat Neurosci 16: 1637–1643, 2013. doi: 10.1038/nn.3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiménez-Jiménez FJ, Alonso-Navarro H, García-Martín E, Agúndez JAG. Genetics of restless legs syndrome: an update. Sleep Med Rev 39: 108–121, 2018. doi: 10.1016/j.smrv.2017.08.002. [DOI] [PubMed] [Google Scholar]
- 62.Jindal RD, Thase ME. Treatment of insomnia associated with clinical depression. Sleep Med Rev 8: 19–30, 2004. doi: 10.1016/S1087-0792(03)00025-X. [DOI] [PubMed] [Google Scholar]
- 63.Joiner WJ. Unraveling the evolutionary determinants of sleep. Curr Biol 26: R1073–R1087, 2016. doi: 10.1016/j.cub.2016.08.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Joiner WJ, Friedman EB, Hung HT, Koh K, Sowcik M, Sehgal A, Kelz MB. Genetic and anatomical basis of the barrier separating wakefulness and anesthetic-induced unresponsiveness. PLoS Genet 9: e1003605, 2013. doi: 10.1371/journal.pgen.1003605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones CR, Huang AL, Ptáček LJ, Fu YH. Genetic basis of human circadian rhythm disorders. Exp Neurol 243: 28–33, 2013. doi: 10.1016/j.expneurol.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron 37: 925–937, 2003. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 67.Kaneko Y, Shimada K, Saitou K, Sugimoto Y, Kamei C. The mechanism responsible for the drowsiness caused by first generation H1 antagonists on the EEG pattern. Methods Find Exp Clin Pharmacol 22: 163–168, 2000. [PubMed] [Google Scholar]
- 68.Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326: 1005–1007, 2009. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang SG, Lee HJ, Choi JE, Park YM, Park JH, Han C, Kim YK, Kim SH, Lee MS, Joe SH, Jung IK, Kim L. Association study between antipsychotics-induced restless legs syndrome and polymorphisms of dopamine D1, D2, D3, and D4 receptor genes in schizophrenia. Neuropsychobiology 57: 49–54, 2008. doi: 10.1159/000129667. [DOI] [PubMed] [Google Scholar]
- 70.Kay DB, Dombrovski AY, Buysse DJ, Reynolds CF, Begley A, Szanto K. Insomnia is associated with suicide attempt in middle-aged and older adults with depression. Int Psychogeriatr 28: 613–619, 2016. doi: 10.1017/S104161021500174X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Khalyfa A, Kheirandish-Gozal L, Gozal D. Circulating exosomes in obstructive sleep apnea as phenotypic biomarkers and mechanistic messengers of end-organ morbidity. Respir Physiol Neurobiol. doi: 10.1016/j.resp.2017.06.004. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koh K, Joiner WJ, Wu MN, Yue Z, Smith CJ, Sehgal A. Identification of SLEEPLESS, a sleep-promoting factor. Science 321: 372–376, 2008. doi: 10.1126/science.1155942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Konadhode RR, Pelluru D, Blanco-Centurion C, Zayachkivsky A, Liu M, Uhde T, Glen WB Jr, van den Pol AN, Mulholland PJ, Shiromani PJ. Optogenetic stimulation of MCH neurons increases sleep. J Neurosci 33: 10257–10263, 2013. doi: 10.1523/JNEUROSCI.1225-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lazarus M, Shen HY, Cherasse Y, Qu WM, Huang ZL, Bass CE, Winsky-Sommerer R, Semba K, Fredholm BB, Boison D, Hayaishi O, Urade Y, Chen JF. Arousal effect of caffeine depends on adenosine A2A receptors in the shell of the nucleus accumbens. J Neurosci 31: 10067–10075, 2011. doi: 10.1523/JNEUROSCI.6730-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lind MJ, Gehrman PR. Genetic pathways to insomnia. Brain Sci 6: e64, 2016. doi: 10.3390/brainsci6040064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu K, Kim J, Kim DW, Zhang YS, Bao H, Denaxa M, Lim SA, Kim E, Liu C, Wickersham IR, Pachnis V, Hattar S, Song J, Brown SP, Blackshaw S. Lhx6-positive GABA-releasing neurons of the zona incerta promote sleep. Nature 548: 582–587, 2017. doi: 10.1038/nature23663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Llorens F, Zarranz JJ, Fischer A, Zerr I, Ferrer I. Fatal familial insomnia: clinical aspects and molecular alterations. Curr Neurol Neurosci Rep 17: 30, 2017. doi: 10.1007/s11910-017-0743-0. [DOI] [PubMed] [Google Scholar]
- 78.Luckhaupt SE, Tak S, Calvert GM. The prevalence of short sleep duration by industry and occupation in the National Health Interview Survey. Sleep 33: 149–159, 2010. doi: 10.1093/sleep/33.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mackiewicz M, Shockley KR, Romer MA, Galante RJ, Zimmerman JE, Naidoo N, Baldwin DA, Jensen ST, Churchill GA, Pack AI. Macromolecule biosynthesis: a key function of sleep. Physiol Genomics 31: 441–457, 2007. doi: 10.1152/physiolgenomics.00275.2006. [DOI] [PubMed] [Google Scholar]
- 80.Mander BA, Winer JR, Jagust WJ, Walker MP. Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer’s disease? Trends Neurosci 39: 552–566, 2016. doi: 10.1016/j.tins.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maret S, Dorsaz S, Gurcel L, Pradervand S, Petit B, Pfister C, Hagenbuchle O, O’Hara BF, Franken P, Tafti M. Homer1a is a core brain molecular correlate of sleep loss. Proc Natl Acad Sci USA 104: 20090–20095, 2007. doi: 10.1073/pnas.0710131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McCurry SM, Ancoli-Israel S. Sleep dysfunction in alzheimer’s disease and other dementias. Curr Treat Options Neurol 5: 261–272, 2003. doi: 10.1007/s11940-003-0017-9. [DOI] [PubMed] [Google Scholar]
- 83.Mesarwi OA, Sharma EV, Jun JC, Polotsky VY. Metabolic dysfunction in obstructive sleep apnea: a critical examination of underlying mechanisms. Sleep Biol Rhythms 13: 2–17, 2015. doi: 10.1111/sbr.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mohawk JA, Takahashi JS. Cell autonomy and synchrony of suprachiasmatic nucleus circadian oscillators. Trends Neurosci 34: 349–358, 2011. doi: 10.1016/j.tins.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mongrain V, Hernandez SA, Pradervand S, Dorsaz S, Curie T, Hagiwara G, Gip P, Heller HC, Franken P. Separating the contribution of glucocorticoids and wakefulness to the molecular and electrophysiological correlates of sleep homeostasis. Sleep 33: 1147–1157, 2010. doi: 10.1093/sleep/33.9.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morairty SR, Dittrich L, Pasumarthi RK, Valladao D, Heiss JE, Gerashchenko D, Kilduff TS. A role for cortical nNOS/NK1 neurons in coupling homeostatic sleep drive to EEG slow wave activity. Proc Natl Acad Sci USA 110: 20272–20277, 2013. doi: 10.1073/pnas.1314762110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morin CM, Jarrin DC. Insomnia and healthcare-seeking behaviors: impact of case definitions, comorbidity, sociodemographic, and cultural factors. Sleep Med 14: 808–809, 2013. doi: 10.1016/j.sleep.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 88.Musiek ES, Holtzman DM. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354: 1004–1008, 2016. doi: 10.1126/science.aah4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Naylor E, Bergmann BM, Krauski K, Zee PC, Takahashi JS, Vitaterna MH, Turek FW. The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci 20: 8138–8143, 2000. doi: 10.1523/JNEUROSCI.20-21-08138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nedeltcheva AV, Kilkus JM, Imperial J, Kasza K, Schoeller DA, Penev PD. Sleep curtailment is accompanied by increased intake of calories from snacks. Am J Clin Nutr 89: 126–133, 2009. doi: 10.3945/ajcn.2008.26574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nichols ALA, Eichler T, Latham R, Zimmer M. A global brain state underlies C. elegans sleep behavior. Science 356: eaam6851, 2017. doi: 10.1126/science.aam6851. [DOI] [PubMed] [Google Scholar]
- 92.Nishino S, Mao J, Sampathkumaran R, Shelton J. Increased dopaminergic transmission mediates the wake-promoting effects of CNS stimulants. Sleep Res Online 1: 49–61, 1998. [PubMed] [Google Scholar]
- 93.Nitz DA, van Swinderen B, Tononi G, Greenspan RJ. Electrophysiological correlates of rest and activity in Drosophila melanogaster. Curr Biol 12: 1934–1940, 2002. doi: 10.1016/S0960-9822(02)01300-3. [DOI] [PubMed] [Google Scholar]
- 94.Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, Kupfer DJ. Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry 161: 2126–2128, 2004. doi: 10.1176/appi.ajp.161.11.2126. [DOI] [PubMed] [Google Scholar]
- 95.Oishi Y, Xu Q, Wang L, Zhang BJ, Takahashi K, Takata Y, Luo YJ, Cherasse Y, Schiffmann SN, de Kerchove d’Exaerde A, Urade Y, Qu WM, Huang ZL, Lazarus M. Slow-wave sleep is controlled by a subset of nucleus accumbens core neurons in mice. Nat Commun 8: 734, 2017. doi: 10.1038/s41467-017-00781-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Palchykova S, Winsky-Sommerer R, Shen HY, Boison D, Gerling A, Tobler I. Manipulation of adenosine kinase affects sleep regulation in mice. J Neurosci 30: 13157–13165, 2010. doi: 10.1523/JNEUROSCI.1359-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Panda S. Circadian physiology of metabolism. Science 354: 1008–1015, 2016. doi: 10.1126/science.aah4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Patke A, Murphy PJ, Onat OE, Krieger AC, Özçelik T, Campbell SS, Young MW. Mutation of the human circadian clock gene CRY1 in familial delayed sleep phase disorder. Cell 169: 203–215.e13, 2017. doi: 10.1016/j.cell.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pellegrini C, Lecci S, Lüthi A, Astori S. Suppression of sleep spindle rhythmogenesis in mice with deletion of CaV3.2 and CaV3.3 T-type Ca(2+) channels. Sleep 39: 875–885, 2016. doi: 10.5665/sleep.5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Porkka-Heiskanen T, Strecker RE, McCarley RW. Brain site-specificity of extracellular adenosine concentration changes during sleep deprivation and spontaneous sleep: an in vivo microdialysis study. Neuroscience 99: 507–517, 2000. doi: 10.1016/S0306-4522(00)00220-7. [DOI] [PubMed] [Google Scholar]
- 101.Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science 276: 1265–1268, 1997. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qaseem A, Kansagara D, Forciea MA, Cooke M, Denberg TD; Clinical Guidelines Committee of the American College of Physicians . Management of chronic insomnia disorder in adults: a clinical practice guideline from the American College of Physicians. Ann Intern Med 165: 125–133, 2016. doi: 10.7326/M15-2175. [DOI] [PubMed] [Google Scholar]
- 103.Rainnie DG, Grunze HC, McCarley RW, Greene RW. Adenosine inhibition of mesopontine cholinergic neurons: implications for EEG arousal. Science 263: 689–692, 1994. doi: 10.1126/science.8303279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ramón F, Hernández-Falcón J, Nguyen B, Bullock TH. Slow wave sleep in crayfish. Proc Natl Acad Sci USA 101: 11857–11861, 2004. doi: 10.1073/pnas.0402015101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Robinson JE, Paluch J, Dickman DK, Joiner WJ. ADAR-mediated RNA editing suppresses sleep by acting as a brake on glutamatergic synaptic plasticity. Nat Commun 7: 10512, 2016. doi: 10.1038/ncomms10512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rogulja D, Young MW. Control of sleep by cyclin A and its regulator. Science 335: 1617–1621, 2012. doi: 10.1126/science.1212476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rosenwasser AM, Turek FW. Neurobiology of circadian rhythm regulation. Sleep Med Clin 10: 403–412, 2015. doi: 10.1016/j.jsmc.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 108.Rothman SM, Herdener N, Frankola KA, Mughal MR, Mattson MP. Chronic mild sleep restriction accentuates contextual memory impairments, and accumulations of cortical Aβ and pTau in a mouse model of Alzheimer’s disease. Brain Res 1529: 200–208, 2013. doi: 10.1016/j.brainres.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rye DB. The molecular genetics of restless legs syndrome. Sleep Med Clin 10: 227–233, 2015. doi: 10.1016/j.jsmc.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 110.Saper CB, Fuller PM, Pedersen NP, Lu J, Scammell TE. Sleep state switching. Neuron 68: 1023–1042, 2010. doi: 10.1016/j.neuron.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature 437: 1257–1263, 2005. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- 112.Schibler U, Gotic I, Saini C, Gos P, Curie T, Emmenegger Y, Sinturel F, Gosselin P, Gerber A, Fleury-Olela F, Rando G, Demarque M, Franken P. Clock-talk: interactions between central and peripheral circadian oscillators in mammals. Cold Spring Harb Symp Quant Biol 80: 223–232, 2015. doi: 10.1101/sqb.2015.80.027490. [DOI] [PubMed] [Google Scholar]
- 113.Schmitt LI, Sims RE, Dale N, Haydon PG. Wakefulness affects synaptic and network activity by increasing extracellular astrocyte-derived adenosine. J Neurosci 32: 4417–4425, 2012. doi: 10.1523/JNEUROSCI.5689-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schrempf W, Brandt MD, Storch A, Reichmann H. Sleep disorders in Parkinson’s disease. J Parkinsons Dis 4: 211–221, 2014. [DOI] [PubMed] [Google Scholar]
- 115.Spaeth AM, Dinges DF, Goel N. Sex and race differences in caloric intake during sleep restriction in healthy adults. Am J Clin Nutr 100: 559–566, 2014. doi: 10.3945/ajcn.114.086579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Spiegel K, Tasali E, Penev P, Van Cauter E. Brief communication: Sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med 141: 846–850, 2004. doi: 10.7326/0003-4819-141-11-200412070-00008. [DOI] [PubMed] [Google Scholar]
- 117.Spira AP, Gamaldo AA, An Y, Wu MN, Simonsick EM, Bilgel M, Zhou Y, Wong DF, Ferrucci L, Resnick SM. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol 70: 1537–1543, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.St-Onge MP, Roberts AL, Chen J, Kelleman M, O’Keeffe M, RoyChoudhury A, Jones PJ. Short sleep duration increases energy intakes but does not change energy expenditure in normal-weight individuals. Am J Clin Nutr 94: 410–416, 2011. doi: 10.3945/ajcn.111.013904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Stavropoulos N, Young MW. insomniac and Cullin-3 regulate sleep and wakefulness in Drosophila. Neuron 72: 964–976, 2011. doi: 10.1016/j.neuron.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stenberg D, Litonius E, Halldner L, Johansson B, Fredholm BB, Porkka-Heiskanen T. Sleep and its homeostatic regulation in mice lacking the adenosine A1 receptor. J Sleep Res 12: 283–290, 2003. doi: 10.1046/j.0962-1105.2003.00367.x. [DOI] [PubMed] [Google Scholar]
- 121.Strecker RE, Morairty S, Thakkar MM, Porkka-Heiskanen T, Basheer R, Dauphin LJ, Rainnie DG, Portas CM, Greene RW, McCarley RW. Adenosinergic modulation of basal forebrain and preoptic/anterior hypothalamic neuronal activity in the control of behavioral state. Behav Brain Res 115: 183–204, 2000. doi: 10.1016/S0166-4328(00)00258-8. [DOI] [PubMed] [Google Scholar]
- 122.Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet 18: 164–179, 2017. doi: 10.1038/nrg.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Terao A, Wisor JP, Peyron C, Apte-Deshpande A, Wurts SW, Edgar DM, Kilduff TS. Gene expression in the rat brain during sleep deprivation and recovery sleep: an Affymetrix GeneChip study. Neuroscience 137: 593–605, 2006. doi: 10.1016/j.neuroscience.2005.08.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tsunematsu T, Ueno T, Tabuchi S, Inutsuka A, Tanaka KF, Hasuwa H, Kilduff TS, Terao A, Yamanaka A. Optogenetic manipulation of activity and temporally controlled cell-specific ablation reveal a role for MCH neurons in sleep/wake regulation. J Neurosci 34: 6896–6909, 2014. doi: 10.1523/JNEUROSCI.5344-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Urade Y, Eguchi N, Qu WM, Sakata M, Huang ZL, Chen JF, Schwarzschild MA, Fink JS, Hayaishi O. Sleep regulation in adenosine A2A receptor-deficient mice. Neurology 61, Suppl 6: S94–S96, 2003. doi: 10.1212/01.WNL.0000095222.41066.5E. [DOI] [PubMed] [Google Scholar]
- 126.Vetrivelan R, Kong D, Ferrari LL, Arrigoni E, Madara JC, Bandaru SS, Lowell BB, Lu J, Saper CB. Melanin-concentrating hormone neurons specifically promote rapid eye movement sleep in mice. Neuroscience 336: 102–113, 2016. doi: 10.1016/j.neuroscience.2016.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vyazovskiy VV, Cirelli C, Pfister-Genskow M, Faraguna U, Tononi G. Molecular and electrophysiological evidence for net synaptic potentiation in wake and depression in sleep. Nat Neurosci 11: 200–208, 2008. doi: 10.1038/nn2035. [DOI] [PubMed] [Google Scholar]
- 128.Watson NF, Badr MS, Belenky G, Bliwise DL, Buxton OM, Buysse D, Dinges DF, Gangwisch J, Grandner MA, Kushida C, Malhotra RK, Martin JL, Patel SR, Quan SF, Tasali E. Recommended amount of sleep for a healthy adult: a joint consensus statement of the american academy of sleep medicine and sleep research society. Sleep 38: 843–844, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Weber F, Chung S, Beier KT, Xu M, Luo L, Dan Y. Control of REM sleep by ventral medulla GABAergic neurons. Nature 526: 435–438, 2015. doi: 10.1038/nature14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Weber F, Hoang Do JP, Chung S, Beier KT, Bikov M, Saffari Doost M, Dan Y. Regulation of REM and non-REM sleep by periaqueductal GABAergic neurons. Nat Commun 9: 354, 2018. doi: 10.1038/s41467-017-02765-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wichniak A, Wierzbicka A, Walęcka M, Jernajczyk W. Effects of Antidepressants on Sleep. Curr Psychiatry Rep 19: 63, 2017. doi: 10.1007/s11920-017-0816-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wisor JP, O’Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, Edgar DM, Franken P. A role for cryptochromes in sleep regulation. BMC Neurosci 3: 20, 2002. doi: 10.1186/1471-2202-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wolf E, Kuhn M, Normann C, Mainberger F, Maier JG, Maywald S, Bredl A, Klöppel S, Biber K, van Calker D, Riemann D, Sterr A, Nissen C. Synaptic plasticity model of therapeutic sleep deprivation in major depression. Sleep Med Rev 30: 53–62, 2016. doi: 10.1016/j.smrv.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 134.Wu M, Robinson JE, Joiner WJ. SLEEPLESS is a bifunctional regulator of excitability and cholinergic synaptic transmission. Curr Biol 24: 621–629, 2014. doi: 10.1016/j.cub.2014.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wu MN, Joiner WJ, Dean T, Yue Z, Smith CJ, Chen D, Hoshi T, Sehgal A, Koh K. SLEEPLESS, a Ly-6/neurotoxin family member, regulates the levels, localization and activity of Shaker. Nat Neurosci 13: 69–75, 2010. doi: 10.1038/nn.2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xu M, Chung S, Zhang S, Zhong P, Ma C, Chang WC, Weissbourd B, Sakai N, Luo L, Nishino S, Dan Y. Basal forebrain circuit for sleep-wake control. Nat Neurosci 18: 1641–1647, 2015. doi: 10.1038/nn.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Young T, Palta M, Dempsey J, Peppard PE, Nieto FJ, Hla KM. Burden of sleep apnea: rationale, design, and major findings of the Wisconsin Sleep Cohort study. WMJ 108: 246–249, 2009. [PMC free article] [PubMed] [Google Scholar]
- 138.Zeitzer JM. Control of sleep and wakefulness in health and disease. Prog Mol Biol Transl Sci 119: 137–154, 2013. doi: 10.1016/B978-0-12-396971-2.00006-3. [DOI] [PubMed] [Google Scholar]