

SUMMARY
β-hydroxybutyrate (β-HB) elevation during fasting or caloric restriction is believed to induce antiaging effects and alleviate aging-related neurodegeneration. However, whether β-HB alters the senescence pathway in vascular cells, remains unknown. Here we report that β-HB promotes vascular cell quiescence, which significantly inhibits both stress-induced premature senescence and replicative senescence through p53-independent mechanisms. Further, we identify heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) as a direct binding target of β-HB. β-HB binding to hnRNP A1 markedly enhances hnRNP A1 binding with Octamer-binding transcriptional factor (Oct) 4 mRNA, which stabilizes Oct 4 mRNA and Oct4 expression. Oct4 increases Lamin B1, a key factor against DNA damage-induced senescence. Finally, fasting and intraperitoneal injection of β-HB upregulate Oct4 and Lamin B1 in both vascular smooth muscle and endothelial cells in mice in vivo. We conclude that β-HB exerts anti-aging effects in vascular cells by upregulating an hnRNP A1-induced Oct4-mediated Lamin B1 pathway.
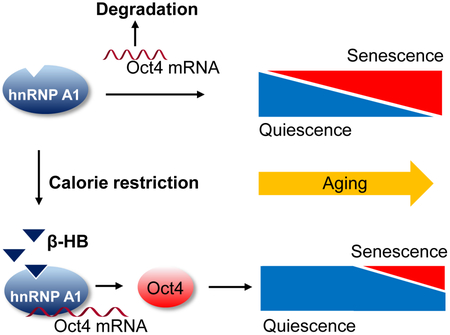
Graphical Abstract
eTOC blurb
β-hydroxybutyrate prevents vascular cell senescence via inducing cell quiescence. β-hydroxybutyrate-induced vascular cell quiescence is dependent on Oct4 expression, which is upregulated by forming hnRNP A1 complex with Oct4 mRNAs. β-hydroxybutyrate-induced Oct4 expression is negatively correlated with hallmarks of vascular senescence in vivo.
INTRODUCTION
Vascular aging is considered as a main risk factor for developing cardiovascular diseases (CVD) (Costantino et al., 2016; North and Sinclair, 2012). Cellular aging, known as senescence, mainly contributes to vascular diseases associated with inflammation and dysfunction of the endothelial and smooth muscle cells (Kovacic et al., 2011; Minamino and Komuro, 2007). Despite years of intense study, therapeutic strategies to mitigate vascular diseases through preventing or removing vascular senescence are still required (Childs et al., 2015).
Cellular senescence is an irreversible cell cycle arrest which restricts the proliferation of abnormal cells. Cellular senescence has multiple physiological functions, including preventing cancer development (Collado and Serrano, 2010) and restricting liver and cardiac fibrosis (Kim et al., 2013). However, the accumulation of senescent cells also contributes to, and can even exacerbate, aging and age-related diseases, such as atherosclerosis (Wang and Bennett, 2012), Alzheimer's disease (Bredesen et al., 2016), and cancer (Finkel et al., 2007). In contrast, cellular quiescence is a reversible cell cycle arrest and usually occurs due to lack of nutrition or growth factors. Maintaining a quiescent state is thought to be a mechanism by which cells can avoid initiating senescence programs (Salmenpera et al., 2016). Thus, identification of endogenous mechanisms that regulate cell senescence and quiescence may provide novel insights into age-related diseases. It is established that fasting and caloric restriction extend both the average and maximal lifespan, as well as prevent age-related diseases through energy sensors dependent on Sirtuin 1 (SIRT1) or Forkhead box O3 (FoxO3) activation (Martin et al., 2006). Additionally, calorie restriction-mediated moderation of cell senescence is reported in recent studies (Anton et al., 2013; Ning et al., 2013). However, it is unknown whether alternative metabolic fuels generated during energy deficit, such as the ketone bodies β-hydroxybutyrate (β-HB) and acetoacetate (AcAc), affect cell senescence and quiescence, nor what the underlying mechanisms may be.
Quiescence not only avoids initiation of the senescent program, but also contributes to the maintenance of stemness, allowing cells to be resistant to genotoxic stress, a major trigger of cellular senescence (Cai et al., 2004; van Deursen, 2014). Octamer-binding transcriptional factor 4 (Oct4, POU5F1), a regulator of pluripotency in embryonic stem cells, was once thought to be permanently silenced in adult somatic cells (Lengner et al., 2008). Interestingly, a recent study suggests that Oct4 is reactivated in adult vascular smooth cells, as demonstrated by its protective role against atherosclerosis (Cherepanova et al., 2016). However, other functions of Oct4 reactivation remain undefined. In addition, the upstream molecules which regulate Oct4 expression at the posttranscriptional level are also poorly characterized.
Recent studies suggest that Oct4 expression can be regulated by heterogeneous nuclear ribonucleoproteins A2/B (hnRNP A2/B), a subfamily of the large A-U hnRNP family (Choi et al., 2013). hnRNP A1 is a member of the hnRNP family that plays a critical role in RNA processing, including alternative mRNA splicing, transcriptional regulation (Jean-Philippe et al., 2013), and stabilization of mRNA under various stress conditions via formation of stress granules (Guil et al., 2006). However, it remains unknown precisely how these proteins interact.
Elevation of ketone bodies, such as β-HB, during fasting or caloric restriction is believed to induce anti-aging effects and alleviate aging-related neurodegeneration (Paoli et al., 2014; Tieu et al., 2003). However, whether ketone body alters pathways in the aging process, specifically the senescence signal in vascular cells, remains unknown. Here we report that β-HB-induced cellular quiescence significantly inhibits both stress-induced premature senescence and replicative senescence through its binding with hnRNP A1, which can bind to and stabilize Oct4 mRNA, thus upregulating Oct4 expression at the posttranscriptional levels.
RESULTS
The preventative effect of β-hydroxybutyrate (β-HB) on stress-induced premature and replicative senescence in vascular cells
Hydrogen peroxide (H2O2) triggers stress-induced premature senescence (SIPS) by increasing oxidative stress (Liu et al., 2014). To test the anti-senescence efficacy of β-HB, primary human umbilical vein endothelial cells (HUVECs) and human aortic smooth muscle cells (hASMCs) (Figure S1A) were exposed to H2O2 with or without β-HB. As expected, H2O2 (150 μM, 3 days) exposure increased numbers of senescence-associated β-galactosidase- (SA β-gal) positive cells (Figure 1A, 1B and S1B) with senescence-related morphological transformations: enlarged, flat, and multinucleated appearance of cells (Figure S1C). The addition of β-HB (4 mM, 3 days) markedly reduced senescence, while acetoacetate (AcAc, 4 mM, 3 days) had no effects on H2O2-induced senescence (Figure 1B). As expected, β-HB also prevented senescence-related morphological transformations (Figure S1C). These results indicate that the anti-senescence effect by ketone bodies is β-HB-specific.
Figure 1. β-hydroxybutyrate (β-HB) prevents cellular senescence.

(A) Representative images of senescence-associated β-galactosidase (SA β-gal) staining. Cultures were exposed to hydrogen peroxide (H2O2, 150 μM, 3 days) with or without β-HB (4 mM) or acetoacetate (AcAc, 4 mM) for 3 days. n = 3. Scale bar, 100 μm.
(B) Quantitative analysis of the SA β-gal-positive cells in Figure 1A. n = 3, NS, non-significant, #p < 0.05, control vs. H2O2, *p < 0.05 H2O2 vs. H2O2 + β-HB.
(C) Quantitative real-time PCR (qRT-PCR) of IL-6 and IL-Ια mRNA after exposing to β-HB (4 mM) with or without H2O2 (150 μM) for 3 days. n = 3, #p < 0.05 control vs. H2O2, *p < 0.05 H2O2 vs. H2O2 + β-HB.
(D) Cell cycle distribution analysis of HUVECs after β-HB (4 mM) treatment for 3 days with or without H2O2 (150 μM). n = 3, *p < 0.05, **p < 0.001 control vs. β-HB, #p< 0.05 H2O2 vs. H2O2 + β-HB.
(E) Time course of the effect of β-HB on SA β-gal activity in HUVECs. n = 3, *p < 0.05 vehicle vs. β-HB, #p<0.05, ##p<0.01 1-week treated vs. 4-week treated.
(F) qRT-PCR analyses of IL-6 and IL-1α mRNA levels in β-HB (4 mM)-treated HUVECs. n = 3, #p < 0.05 1-week control vs. 4-week control, p < 0.05 4-week control vs. 4-week β-HB.
(G) Effect of β-HB on HUVECs doubling time. n = 3, #p < 0.05 1-week control vs. 4-week control, *p < 0.05 4-week control vs. 4-week β-HB.
(H) Alteration of senescence markers (γH2AX, p21, or p16) and quiescence markers (p27, Lamin B1, or Oct4A) in hASMCs treated with or without β-HB for 24 h. n = 3, *p < 0.05, **p < 0.01, ***p < 0.001 control vs. β-HB treatment.
Data are presented as mean ± standard error of mean (SEM). NS, non-significant; Ctrl, Control.
The proinflammatory cytokines IL-6 and IL-1α are proven markers of the senescence-associated secretory phenotype (SASP) (Tchkonia et al., 2013; Zhu et al., 2014). Quantitative real-time PCR (qRT-PCR) of HUVECs samples indicated that β-HB effectively suppressed the expression of IL-6 and IL-1α triggered by H2O2 (Figure 1C).
Since multi-nuclei formation is frequently observed in senescence (Sadaie et al., 2015), cell cycle analysis was used and demonstrated that β-HB treatment led to accumulation at G0/G1 phase and significant reduction of S phase. Moreover, β-HB strongly prevented H2O2-induced polyploidy formation (Figure 1D and S1D). These results provided initial evidence for the efficacy of β-HB in preventing SIPS.
In order to test if β-HB prevents replicative senescence (RS), SA β-gal activity was measured weekly for 4 weeks. As shown in Figure 1E and Figure S1E, maintaining HUVECs for 4 weeks markedly induced a replicative senescence, but cell culture for 1 week did not induce a senescence. Therefore, β-HB treatment for 4 weeks decreased the doubling time (Figure 1G) and retained higher proliferative potentials (Figure S1F), which may be due to the decreased senescence. On the other hand, cell culture for 1 week did not induce senescence (Figure 1E), while β-HB treatment for 1 week increased the doubling time (Figure 1G) and modestly decreased the cell number, which may be due to the induction of quiescence by β-HB. Additionally, SASP expression of senescent HUVECs was also reduced by 4-week β-HB treatment (Figure 1F). These results suggest that β-HB induces reversible growth arrest, also known as a cellular quiescence, and thus prevents subsequent senescence.
In order to define the molecular mechanisms by which β-HB exerts anti-senescence effects in vascular cells, we monitored DNA damage repair markers and cell cycle regulatory proteins. As shown in Figure S1G, β-HB reduced the levels of γH2AX, a marker of DNA damage (Xu et al., 2015), in both HUVECs and hASMCs. However, β-HB did not affect the phosphorylation of CHK1, BRCA1, or p53 level in either cell line, implying that β-HB-mediated alleviation of senescence is not via enhancement of DNA repair signaling (Williams and Schumacher, 2016; Wu et al., 2010; Yarden et al., 2002). Moreover, β-HB dose-dependently upregulated levels of the cell cycle regulatory proteins p21 (Figure S1G) and p27 (Figure 1H) in normally conditioned cells. Taken together, these results indicate that β-HB-mediated reduction of DNA damage is a consequence of cell cycle retardation caused by quiescence.
To verify the impact of β-HB on cell cycle regulation, we compared levels of multiple proteins involved in senescence or quiescence in non-stressed and oxidant-stressed hASMCs treated with β-HB. β-HB markedly suppressed the oxidant-induced elevation of senescence markers (γH2AX, p21, p16). In the non-stress condition, β-HB significantly upregulated quiescence markers (p27, Lamin B1, Oct4A) (Figure 1H). These findings support the hypothesis that β-HB-induced quiescence may help prevent senescence in vascular cells.
β-HB-induced cellular quiescence is p53-independent
Next, we investigated the effect of β-HB on cellular quiescence. First, we measured the doubling time of HUVECs to determine if growth arrest by β-HB is reversible. Three senescence inducers, H2O2, Aphidicolin (APC), and Nutlin-3a, caused irreversible growth arrest of HUVECs. However, β-HB (4 mM) caused reversible growth arrest, which is the defining feature of quiescent cells (Figure 2A). Cell cycle retardation at the G0/G1 phage was gradually increased by β-HB treatment in a time-dependent manner (Figure 2B). To prove β-HB-induced cell quiescence, flow cytometry analysis was conducted to measure the reduction of the transcriptome as a marker of cellular quiescence (Cheung and Rando, 2013). As expected, β-HB (4 mM) time-dependently decreased the fluorescence intensity of Pyronin Y, used to quantify total RNA (Will et al., 2013) (Figure 2C and S2A, S2B). Quantitation of cell cycle distribution showed that β-HB dramatically induced G0 with simultaneous reductions of G1 and S/G2 phases (Figure 2C). To provide additional evidence of β-HB-mediated quiescence, we tested several molecular markers for quiescence after β-HB treatment, including AMPKα phosphorylation (Thr172) (Saldivia et al., 2016), S6K dephosphorylation (Thr389), and p27 elevation. We also observed that β-HB significantly and time-dependently induced Lamin B1 expression (Figure S2C), which is known to inhibit the G1/S transition (Shimi et al., 2011). These results support β-HB-mediated cell quiescence. Due to ambiguous roles of quiescence and senescence proteins, several specific molecular markers for quiescence were tested to validate cell status. Figure S2D showed that contact inhibition or fetal bovine serum (FBS) deprivation upregulated p27, p21, Lamin B1, and nucleophosmin 1 (NPM1) in quiescent HUVECs, and these increases were negatively associated with proliferation markers, p-p70 (T389) and p-RB (S807, S811). We compared p27, p21, and Lamin B1 levels between FBS deprivation-induced quiescent and H2O2-induced senescent hASMCs in Figure S2E, and SA β-gal activities in quiescence and senescence are shown in Figure S2F. p53 activation and γH2AX upregulation were specifically detected in senescent cells. We found that p27 and Lamin B1, as well as the Oct4, which can activate in vascular smooth muscle cells (Cherepanova et al., 2016), were up- and downregulated in quiescence and senescence, respectively. Based on these results, p27, Lamin B1, and Oct4 were considered useful markers of quiescence.
Figure 2. β-HB induces human umbilical vein endothelial cells (HUVECs) quiescence.

(A) Doubling time of HUVECs treated with β-HB (4 mM, 15 h), H2O2 (150 μM, 15 h), APC (aphidicolin, 2 μM, 15 h), Nutlin-3a (2.5 μM, 15 h), and washout groups for each treatment. n = 3, *p < 0.05 control vs. treated, #p < 0.05 β-HB vs. Washout.
(B) Cell cycle distribution analysis of HUVECs in the G0/G1 phase after β-HB treatment for the indicated time periods.
(C) Cell cycle distribution of HUVECs in the G0, G1, and S/G2 phases after β-HB treatment for the indicated time periods, assessed with flow cytometry after Pyronin Y and Hoechst 33342 staining. n = 3, *p < 0.05 control vs. β-HB-treated (24 h).
(D) Representative images of SA β-gal activity assays of Nutlin-3a (2.5 μM, 15 h) or H2O2 (150 μM, 15 h) -induced senescent HUVECs cultured with nutrient-complete media (2% FBS, EGF, bovine brain extract) or FBS-deprived media (0% FBS, EGF, bovine brain extract). n = 3, Scale bar, 100 μm.
(E) Quantitation of Figure 2D. n = 3, NS, non-significant, #p < 0.05 Control (Ctrl) vs. Nutlin-3a or H2O2 (2% FBS), *p < 0.05, **p < 0.01 H2O2 (2% FBS) vs. H2O2 (0% FBS).
(F) Representative images of SA β-gal staining in Nutlin-3a-treated HUVECs with or without β-HB (4 mM, 15 h). n = 3, Scale bar, 100 μm.
(G) Quantitation of Figure 2F. n = 3, #p < 0.05 Control (Ctrl) vs. Nutlin-3a, *p < 0.05 Nutlin-3a vs. Nutlin-3a + β-HB.
(H) Representative Western blots for Lamin B1 in HUVECs treated with Nutlin-3a (1 or 2.5 μM, 15 h) or H2O2 (75 or 150 μM, 15 h) with or without β-HB (4 mM, 15 h). n = 3.
(I) Effects of β-HB on fluorescein di-β-D-galactopyranoside (FDG) assay. HUVECs were treated with Nutlin-3a (2.5 μM, 15 h) or H2O2 (150 μM, 15 h) in the presence or absence of β-HB (4 mM, 15 h). Excitation/emission = 490/514 (nm), n = 3, #p < 0.05 PBS vs. Nutlin-3a or H2O2, *p < 0.05 H2O2 vs. H2O2 + β-HB.
Data are presented as mean ± standard error of mean (SEM). NS, non-significant.
To test if β-HB inhibition of senescence is dependent on the p53 pathway, we used the MDM2 (E3 ubiquitin ligase for p53) inhibitor Nutlin-3a to induce p53-dependent senescence. Serum deprivation-induced quiescent cells were resistant to senescence induced by H2O2. However, quiescence did not prevent the senescence trigged by Nutlin-3a (2.5 μM, 15 h), which induces robust upregulation of p53 and p21 causing p53-dependent senescence (Figure 2D and 2E). Additionally, serum deprivation promoted Nutlin 3a-induced cell death, but prevented H2O2-induced cell death (Figure S2G and S2H). These results indicate that anti-senescence triggered by quiescence accumulation is p53-independent.
We further evaluated the anti-senescence efficacy of β-HB in case of p53-medicated senescent HUVECs. Interestingly, β-HB accelerated the senescence stimulated by Nutlin-3a treatment (Figure 2F and 2G) and did not affect Nutlin-3a-induced growth arrest (Figure S2I). Taken together, β-HB-mediated alleviation of senescence is p53-independent.
Serum deprivation- and contact inhibition-induced quiescent cells exhibited p27 and Lamin B1 upregulation. The levels of p27 and Lamin B1 were distinctly upregulated in quiescent cells induced by either serum deprivation or contact inhibition, compared to proliferative cells (Figure S2D and S2E). Additionally, expression of NPM1, which is involved in chromatin organization (Martin et al., 2009), was also associated with Lamin B1 expression (Figure S2D). Lamin B1 was down-regulated in senescent HUVECs triggered by Nutlin-3a or H2O2 treatment. Hence, H2O2-mediated Lamin B1 loss was partially recovered by co-treatment with β-HB, but not in the case of Nutlin-3a-induced senescence (Figure 2H). Additionally, Lamin B1 level was inversely proportional to cell senescence (Figure 2H and 2I). Taken together, these results suggest that β-HB induces cell quiescence via Lamin B1 upregulation, which prevents oxidative stress-induced senescence in a p53-independent manner.
hnRNP A1 is a direct target of β-HB in regulating cell quiescence
There are several derivatives of β-HB including S-β-HB, β-HB ester, β-HB salt, and butyrate (Figure 3A). Interestingly, only S-β-HB which is an enantiomer of β-HB, and β-HB salt which is a pH-neutralized form of β-HB, both inhibited the H2O2-induced SASP (IL-6 and IL-1α) transcriptional expression in a similar manner to β-HB treatment (Figure S3A). These results suggest that both the beta hydroxyl and carboxylic acid groups are required for the activity of β-HB. However, lower pH caused by β-HB treatment, as well as changes in the R-S configuration to a gamma carbon of β-HB, did not affect the activity of β-HB. Therefore, the gamma carbon of β-HB was modified to elongate the linker for affinity chromatography.
Figure 3. Proteomics approach to identify the target protein of β-HB action.

(A) Structures of β-HB and its analogues: S-β-HB, Butyrate, β-HB ester, and β-HB salt.
(B) Structures of negative control beads (SP-Me), β-HB, and butyrate-conjugated Sepharose beads (SP-β-HB and SP-BA).
(C) Representative image of affinity chromatography using SP-β-HB, SP-BA, and SP-Me. Binding proteins were eluted with β-HB gradient buffer (0.1 Μ Tris-HCl, β-HB 0.5–8 mM), then stained with Coomassie brilliant blue dye. Four proteins (SFPQ, Heat shock protein 70, FUS, and hnRNP A1) were identified by MALDI TOF MS.
(D) Representative Western blots obtained from three individual experiments. Pull-down assays using SP-Me (negative control) and SP-β-HB (positive control). Cell lysates were pretreated with competitors (β-HB, S-β-HB, β-HB ester, and Butyrate) to validate specific binding with hnRNP A1. n = 3.
(E) Quantitation of Figure 3D. **p < 0.01, ***p < 0.001 vehicle vs. treated.
(F) Representative Western blots for hnRNP A1 showing the knockdown efficiency of siRNA targeting hnRNP A1.
(G) Representative images of SA β-gal staining and quantitation of SA β-gal-positive cells in hnRNP A1-silenced HUVECs treated with or without H2O2 (150 μM). Scale bar, 1 mm. #p < 0.05 Control vs. H2O2, *p < 0.05 H2O2 + si-Ctrl vs. H2O2 + si-A1.
Data are presented as mean ± standard error of mean (SEM). NS, non-significant; si-Ctrl, control siRNA; si-A1, hnRNP A1 siRNA.
Two negative control beads, SP-Me and SP-BA, were compared with β-HB-conjugated beads (SP-β-HB) (Figure 3B). To identify β-HB-binding protein(s), affinity chromatography was performed using β-HB-conjugated Sepharose beads compared to a negative control (Figure 3C). We identified hnRNP A1 as a β-HB-binding protein using MALDI-TOF mass analysis. To confirm binding affinity, a pull-down assay was performed using SP-β-HB beads, and hnRNP A1 was validated with a specific antibody. The binding between hnRNP A1 and β-HB-conjugated beads was disturbed when the interaction was challenged with excess β-HB, S-β-HB, β-HB ester, or butyrate acting as binding competitors (Figure 3D and 3E). Furthermore, silencing hnRNP A1 triggered senescence of HUVECs and accelerated the response to H2O2 (Figure 3F and3G).
As depicted in Figure 3C, β-HB also weakly bound two additional proteins, identified by mass spectrometry as Splicing factor proline- and glutamine-rich (SFPQ) and RNA-binding protein FUS (FUS). However, silencing SFPQ or FUS did not significantly induce senescence (Figure S3B and S3C). 2-D proteomics analysis (Figure S3D) showed that β-HB treatment barely altered proteomes with the exception of NPM1. Because NPM1 regulates quiescence (Ito et al., 2010) and cooperatively acts with Lamin B1 (Martin et al., 2009), NPM1 upregulation might be involved in quiescence associated with β-HB-mediated Lamin B1 upregulation.
Oct4A upregulation by hnRNP A1
To determine the association between hnRNP A1 and Oct4 mRNAs, RNA-immunoprecipitation assays were performed in the presence or absence of β-HB. The binding of hnRNP A1 with Oct4 mRNA was enriched by treatment of β-HB or S-β-HB. In addition, β-HB ester and butyrate, which has no effect on SASP inhibition, did not affect the interactions between hnRNP A1 and Oct4A mRNA (Figure 4A).
Figure 4. hnRNP A1 mediates Oct4 mRNA stabilization in the presence of β-HB.

(A) RNA-immunoprecipitation (RIP) using cytoplasmic hnRNP A1 to assess binding of Oct4, Lamin B1, and p27 mRNAs with hnRNP A1 in the presence of β-HB (4 mM, 15 h) or β-HB analogues (S-β-HB, β-HB ester, and butyrate, 4 mM, 15 h). n = 3, p < 0.001 vehicle vs. treated groups.
(B) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis of Oct4A mRNA expression in hASMCs and human umbilical vein endothelial cells (HUVECs) treated with β-HB at the indicated concentrations for 15 h. n = 3, p < 0.05, p < 0.01 control vs. β-HB treated groups.
(C) Representative Western blots for Oct4A and Lamin B1 in HUVECs and hASMCs after β-HB treatment for the indicated time points. n = 3.
(D) qRT-PCR analysis of Oct4A and Oct4B mRNA in HUVECs transfected with si-Ctrl or hnRNP A1 siRNA. n = 3, #p < 0.05 si-Ctrl vs. si-A1.
(E) qRT-PCR analysis of Oct4A mRNA in control siRNA-transfected or hnRNP A1-silenced HUVECs treated with or without β-HB (4 mM, 15 h). n = 3, #p < 0.05, ##p < 0.01 si-Ctrl vs. si-A1, *p < 0.05 control vs. β-HB-treated group.
(F) Regulation of Oct4 mRNA stability by β-HB treatment (4 mM, 15 h) in HUVECs treated with actinomycin D. By blocking transcription with actinomycin D (6.5 μg/ml), the half-life of Oct4A mRNA was demonstrated at various times after adding actinomycin D. n = 3, *p < 0.05 control vs. β-HB.
(G) Oct4 mRNA stability analysis in hnRNP A1-silenced HUVECs by actinomycin D application in a time-dependent manner. n = 3, #p < 0.05 si-Ctrl vs. si-A1.
(H) Representative Western blots for subcellular hnRNP A1 localization after β-HB treatment (4 mM, 2-8 h). α-Tubulin and Lamin A/C were used to normalize results for the cytoplasm and nucleus, respectively.
(I) Quantification of Figure 4H. n = 3, *p < 0.05, ***p < 0.001 control vs. β-HB treatment. ###p < 0.01 β-HB-treated (8 h) vs. washout.
(J) Representative immunofluorescence labeling for hnRNP A1 (green) and eIF4E (red) in hASMCs after β-HB treatment and washout. Linear trajectories crossing the cell with the two signals were shown by yellow line. White triangles indicate colocalization of eIF4E and hnRNP A1. Quantification of colocalization is presented at the right side of image. n = 3, Scale bar, 20 μm. p < 0.01, p < 0.01 control vs. β-HB treatment. ###p < 0.001 15 h vs. washout.
Data are presented as mean ± standard error of mean (SEM).
Although β-HB has biphasic induction of Oct4 mRNA in HUVECs, β-HB (2-4 mM) markedly increased Oct4 mRNA levels in both HUVECs and hASMCs (Figure 4B). A dual luciferase assay indicated that transcriptional activity of Oct4A was enhanced by β-HB and S-β-HB, whereas butyrate and niacin did not induce significant changes (Figure S4A). Additionally, Oct4A and Lamin B1 levels in β-HB-treated HUVECs or hASMCs peaked at 15 h (Figure 4C).
Then the alternative splicing regulation of Oct4 gene by hnRNP A1 was assessed. As shown in Figure S4B, β-HB (4 mM) did not regulate alternative splicing of the Oct4 gene by showing upregulation of both Oct4A and Oct4B. Likewise, both Oct4A and Oct4B mRNA were reduced by hnRNP A1 silencing in HUVECs (Figure 4D). Moreover, the total absence of β-HB-enhanced Oct4 upregulation in hnRNP A1-silenced cells suggested that hnRNP A1 is critical for β-HB-induced Oct4 activation (Figure 4E).
Next, we conducted mRNA stability analysis. The half-life of Oct4 mRNA was effectively extended from 90 min to 5 hours by β-HB treatment (Figure 4F) in transcriptional inhibitor actinomycin D-pretreated HUVECs. Moreover, silencing of hnRNP A1 shorten the half-life of Oct4 mRNA even under normal condition (Figure 4G). These data indicate that Oct4 mRNA is stabilized by hnRNP A1 in the presence of β-HB.
We next determined if Oct4 mRNA stabilization is associated with stress granule formation. Subcellular fractionation results showed that β-HB induced translocation of hnRNP A1 from the nucleus to the cytoplasm (Figure 4H, 4I and S4C). Furthermore, immunofluorescent staining of hnRNP A1 with stress granule markers, eIF4E and TIA-1, also supported β-HB-mediated transport of hnRNP A1 from the nucleus to the cytoplasm to form stress granules (Figure 4J, S4D and S4E). Plot of line scan showed a significant correlation between the spatial localization of eIF4E and hnRNP A1 (Figure 4J), or TIA-1 and hnRNP A1 (Figure S4E) in a time-dependent manner of β-HB treatment. These results define the molecular mechanism of β-HB/hnRNP A1-mediated elevation of Oct4.
Functional role of Oct4A in cellular senescence
To validate the new functional role of Oct4 in cellular senescence, we employed the CRISPR/Cas9 knocking down system and a pCMV ectopic expression system. Oct4 positively regulated the expression of p21, p27, and Lamin B1, while negatively regulating the levels of γH2AX. The negative correlation of γH2AX with Oct4 expression indicates that loss of Oct4 triggers cellular senescence by inducing DNA damage. On the contrary, ectopic expression of Oct4 prevents senescence by reducing DNA damage (Figure 5A).
Figure 5. Oct4 knockdown and ectopic expression in human umbilical vein endothelial cells (HUVECs).

(A) Representative Western blots for quiescence- and senescence-associated proteins (Lamin B1, p27, p21, and γH2AX) after Oct4 gene knockdown or overexpression. n = 5.
(B) HUVECs growth rates after Oct4 knockdown or ectopic expression. Following transfection with GFP, Oct4-myc, or CRISPR/cas9-Oct4 plasmid, equal numbers of HUVECs were seeded on plates, and cells were counted daily. n = 3, #p < 0.05 1 day vs. days after seeding.
(C) Cell cycle distribution of quiescent (G0 phase) HUVECs transfected with/without CRISPR/cas9-Oct4. n = 3, #p < 0.05 Mock vs. CRISPR/cas9-Oct4, *p < 0.05 vehicle vs. β-HB. NS, non-significant.
(D) SA β-gal assays of mock or Oct4-knockdown HUVECs after H2O2 (150 μM, 24 h) stimulation with or without β-HB treatment (4 mM, 24 h). n = 3, *p < 0.05 H2O2 (Stone et al.) vs. β-HB + H2O2 (Stone et al.), #p<0.05 Mock vs. CRISPR/cas9-Oct4.
(E) Representative immunofluorescent labeling for γH2AX (green) in mock or Oct4 knockdown HUVECs (CRISPR/cas9-Oct4). n = 3, Scale bar, 10 μm.
(F) Apoptosis analysis of mock or Oct4-knockdown HUVECs (CRISPR/cas9-Oct4) stimulated by H2O2 (200 μM) for 3 days. n = 3.
(G) Representative Western blots for the quiescence markers Lamin B1, p-eIF2α, and p27 in si-Ctrl- or Oct4 siRNA-transfected HUVECs with or without β-HB (4 mM, 15 h) treatment. n = 4.
(H) Senescence-associated FDG assay. HUVECs were transfected with myc or Oct4-myc plasmids, and treated with Nutlin-3a (2.5 μM, 15 h), H2O2 (150 μM, 15 h), or ApC (2 μM, 15 h) 2 days after transfection. n = 3, #p < 0.05 myc vs. Oct4-myc, *p < 0.05 control vs. treated, NS, non-significant.
(I) qRT-PCR analyses for Oct4, SOX2, KLF4, c-Myc, and Nanog in HUVECs after β-HB (4 mM) treatment for the indicated times. n = 3, p < 0.01, p < 0.05 control vs. treated.
Data are presented as mean ± standard error of mean (SEM).
Consistent with the observation that Oct4 increased p27 and Lamin B1 levels (Figure 5A), ectopic Oct4 overexpression retarded cell growth (Figure 5B). Conversely, selective Oct4 knockdown promoted cell growth (Figure 5B). These data imply that Oct4-mediated cell cycle regulation is strongly related with p27 and Lamin B1 controlled cell quiescence. Oct4 knockdown disrupted β-HB-induced cell cycle arrest in G0/G1 (Figure S5A), suggesting that Oct4 induces cellular quiescence. Additionally, silencing Oct4 expression could activate p-p70 S6K and p-RB, which are exhibited as proteins synthesis and G1/S transition signaling (Figure S5B). Staining of Pyronin Y and Hoechst33342 further demonstrated that Oct4 knockdown promoted G1/S transition by reducing the quiescent state (Figure 5C and S5C) while inducing senescence (Figure S5D). These data suggest that β-HB-induced quiescence is Oct4 dependent.
To further study the Oct4-dependent preventative effect of β-HB, cell senescence was assessed using Oct4-knockdown HUVECs. SA β-gal assays further demonstrated that the anti-senescence effect of β-HB is mediated by Oct4A (Figure 5D and S5E). Likewise, loss of Oct4A significantly increased SASP expression and countered the preventative effect of β-HB (Figure S5F). To further validate that reduction of quiescence or induction of rapid cell growth is through Oct4 loss and therefore is vulnerable to genotoxic stress, DNA damage and apoptosis were tested using Oct4-silenced HUVECs. As expected, Oct4A knockdown significantly enhanced H2O2-mediated DNA damages (Figure 5E) and promoted apoptosis of HUVECs (Figure 5F and S5G).
As depicted in Figure 5G, Oct4A-knockdown in HUVECs markedly alleviated β-HB-induced upregulation of the quiescence regulatory proteins Lamin B1, p27, and p-eIF2α. Consistently, Oct4A silencing abolished Lamin B1 transcription (Figure S5H), suggesting that Oct4A is a crucial transcriptional factor for this protein. Conversely, ectopic Oct4A overexpression reduced H2O2- and APC-induced senescence. Importantly, Oct4A overexpression did not alter Nutlin-3a-induced senescence, implying that Oct4-mediated senescence alleviation does not require p53 (Figure 5H).
We next assessed whether β-HB treatment alters other Yamanaka factors including SOX2, KLF4, and c-Myc (Takahashi and Yamanaka, 2006). Significant elevation of Oct4A mRNA was observed 4 h after β-HB treatment (Figure 5I). In contrast, the mRNA levels of SOX2, KLF4, c-Myc, and Nanog were not elevated. Our findings strongly indicate that β-HB-alleviated senescence is Oct4A dependent and does not involve other Yamanaka factors.
Oct4A upregulation in vascular tissue by acute elevation of β-HB
To further determine β-HB’s effect on Oct4 upregulation in vivo, C57BL/6J mice were intraperitoneally injected with β-HB salt or S-β-HB salt (1.5 g/kg body weight) (Figure 6A). As shown in Figure 6B, both β-HB and S-β-HB were able to upregulate Oct4A, Lamin B1, p27, and phospho-eIF2α in thoracic aorta, indicating cell quiescence. Interestingly, β-HB-induced Oct4 protein elevation was only observed in the aorta, brain, and heart (Figure S6A). Oct4A mRNA was also increased by β-HB and S-β-HB injection, especially in the aorta, brain cortex, and heart (Figure 6C). Additionally, S-β-HB, which was not utilized in the ketolysis pathway (Figure S6B and S6C), triggered more significant Oct4A induction than β-HB did (Figure 6C), indicating that Oct4 upregulation is independent of energy homeostasis. Moreover, β-HB injection enhanced Oct4A expression, as well as the number of Oct4 positive cells in aorta (Figure 6D).
Figure 6. Oct4A upregulation in vascular tissues by β-HB injection or fasting.

(A) Blood β-HB levels in C57BL/6J mice after intraperitoneal (I.P.) injection of β-HB (1.5 g/kg body weight) or S-β-HB (1.5 g/kg). n = 6/group, *p < 0.05, **p < 0.01, ***p < 0.001 vehicle vs. β-HB injection.
(B) Western blotting analysis for Oct4A and other quiescence markers (Lamin B1, p27, and p-eIF2α) in C57BL/6J mice aorta after I.P. injection with PBS, β-HB salt (1.5 g/kg body weight), or S-β-HB salt (1.5 g/kg body weight) dissolved in PBS. n = 6 mice/group.
(C) qRT-PCR analysis of Oct4A expression in various organs after I.P. injection with PBS, β-HB salt, or S-β-HB salt (1.5 g/kg body weight). n = 6 mice/group, *p < 0.05, **p < 0.01 PBS vs. β-HB or S-β-HB.
(D) Representative immunofluorescent labeling with a FITC-conjugated Oct4A antibody in C57BL/6J mouse thoracic aorta 4 h after β-HB injection (1.5 g/kg). Quantitation of Oct4A-positive cells, normalized by DAPI, are shown at right. n = 6 mice/group, *p < 0.05 PBS vs. β-HB.
(E) Blood β-HB levels in C57BL/6J mice during fasting for the indicated time periods. n = 6 mice, *p < 0.05, **p < 0.01 feeding vs. fasting.
(F) Western blotting analyses of Oct4A and Lamin B1 in thoracic aorta from fed or fasted mice. n = 8 mice/group.
(G) Representative immunofluorescent labeling for Oct4A and CD31 in fed or fasted (72 h) mice aortas. Oct4A-expressing endothelial cells and smooth muscle cells are indicated with yellow and white arrows, respectively. Nuclei were stained with DAPI. Scale bar, 100 μm. n = 4 mice/group.
(H) Representative immunofluorescent labeling for Oct4A and SM α-Actin in fed or fasted (72 h) mice aortas. Scale bar, 100 μm. n = 4 mice/group. p < 0.01 feeding vs. fasting.
(I) Representative immunohistochemical staining for Oct4A in fed or fasted (72 h) mice aorta. Red arrow indicates endothelial cells and black arrow indicates smooth muscle cells. Scale bar, 50 μm. n = 5 mice/group.
Data are presented as mean ± standard error of mean (SEM).
Blood levels of β-HB in C57BL/6J mice was upregulated from 0.7 mM to 2.6 mM by fasting in a time-dependent manner (Figure 6E). Oct4A and Lamin B1 protein levels in thoracic aorta tissue were dramatically elevated by fasting for 72 h (Figure 6F and S6D). Immunofluorescent staining using anti-CD31, anti-smooth muscle (SM) α-Actin, or anti-Oct4 antibodies demonstrated that three-day fasting elevated Oct4 expression in endothelial and vascular smooth muscle cells (Figure 6G and 6H). Immunohistochemistry assay also provided evidences for Oct4 upregulation in endothelial and smooth muscle cells during fasting (Figure 6I). These results indicate that acute induction of circulating β-HB upregulates Oct4 and Lamin B1 expression in vivo.
Chronic effect of β-HB on preventing senescence in intermediate aged mice
Experiments were performed to determine if maintaining appropriate β-HB blood levels alleviates vascular aging. As shown in Figure S7A, IL-1α, an upstream regulator of IL-6, was elevated in middle-aged mice, whereas IL-6, a major SASP marker, was elevated in aged mice (96 weeks old) (Figure S7B). The concentration of serum β-HB varied in the middle-aged and aged-mice from 0.3 to 1.2 mM with a negative correlation with body weight (Figure S7C).
We evaluated if blood β-HB is linked to SASP expression in aged mice (n = 38) with a wide range of blood β-HB (0.3–1.2 mM). As depicted in Figure S7D, low IL-1α levels were found in mice with β-HB concentrations between 0.8 and 1.2 mM, whereas high levels were observed in mice with β-HB concentrations between 0.3 and 0.5 mM. Similarly, the level of IL-6 was negatively associated with β-HB. The expression of both Oct4A and Lamin B1 was positively associated with higher blood β-HB (Figure S7E). Furthermore, staining of whole mouse aorta supported the conjecture that SA β-gal positivity was negatively associated with blood β-HB level (Figure 7A and 7B).
Figure 7. Circulating β-HB chronically affects Oct4 signaling in aged mice.

(A) SA β-gal activity assays of whole aorta from aged mice (80 weeks old, n = 10) stratified by blood β-HB level.
(B) Quantification of SA β-gal positivity in Figure 7A. SA β-gal activity in mice aorta negatively correlated with blood β-HB level (R2 = 0.8201).
(C) Fluorogenic β-gal activity assays of multiple sections of aged mice aorta (Arch and Thoracic). Mice were grouped according to blood β-HB level (0.4 and 1.1 mM, n = 6 mice/group), then aorta tissues were reacted with SPiDer β-gal. *p < 0.05, **p < 0.01 0.4 mM vs. 1.1 mM.
(D) Representative immunofluorescence staining for Lamin B1 and Oct4A in 64-week-old mice stratified by blood β-HB concentration (0.6 and 1.1 mM, n = 4 mice/group). Linear trajectories (yellow line) crossing the cell with two signals were presented right side of image. White arrows and black triangles indicate colocalization of Lamin B1 and Oct4A.
(E) Western blotting analyses of Oct4A and Lamin B1 in aged mice (80 weeks old) stratified by blood β-HB concentration (0.3-0.4 mM and 1.0-1.1 mM, n = 6 mice/group).
(F) Flow cytometry analysis of mice primary smooth muscle cells. Aortic smooth muscle cells were isolated from mice stratified by blood β-HB concentration (1.1 mM and 0.4 mM). Isolated aortic smooth muscle cells were reacted with SPiDer β-gal assay, then fixed and stained with Oct4 antibody. n = 6 mice/group.
(G) Quantitative analysis of Figure 7H. SA β-gal positivity according to the level of Oct4. n = 6 mice/group. p < 0.01 Oct4 + vs. Oct4 −.
(H) SA β-gal assay at the aortic root of Apoe −/− mice with western diet for 8 weeks (upper panel). SPiDer β-gal assay with Oct4 immunostaining (bottom panels) at the aortic roots. n = 5 mice/group.
Data are presented as mean ± standard error of mean (SEM).
To further assess whether β-HB distinctly affects different parts of the aorta, whole mouse aortas were separated into the aortic arch (Arch) and thoracic aorta (TA), multiple sections of Arch and TA were examined with SPiDer β-gal assay. As shown in Figure 7C and S7F, the mice with high level of blood β-HB (1.1 mM) exhibited significant reduction of SA β-gal activity in the Arch and moderate reduction in the TA, compared to those with low level of β-HB (0.4 mM).
The middle-aged mice (64 weeks-old) that maintained high level of β-HB (1.1 mM) displayed significantly increased expression of Oct4 and Lamin B1 over mice maintained with a 0.6 mM concentration of β-HB. Additionally, the majority of Oct4A expression was mainly co-localized with Lamin B1 (Figure 7D). Plot of line scan demonstrated an increase of expression, and a colocalization of Lamin B1 with Oct4A in TA, influenced by high level of β-HB (1.1 mM). Consistently, Aged mice (80 weeks old) with high circulating β-HB (1.0-1.1 mM) had higher Oct4A and Lamin B1 levels than mice with 0.3-0.4 mM circulating β-HB. In addition, thoracic aortas from older animals displayed higher Oct4A expression than aortic arches with senescent phenotypes (Figure 7E).
To further validate a negative relationship of senescence and Oct4A, and to confirm a β-HB-mediated Oct4A elevation, flow cytometry analysis was performed using isolated mouse primary smooth muscle cells (Figure 7F). Here, we demonstrated that high level β-HB (1.1 mM) upregulated Oct4 expression in primary smooth muscle cells compared to cells with 0.4 mM β-HB (Figure S7G). Oct4 upregulated cells (Oct4 +) exhibited significant reduction of SA β-gal activity compared to Oct4A downregulated cells (Oct4 −) (Figure 7G).
Since advanced atherosclerotic lesions exhibit senescent cells (Childs et al., 2016), we conducted co-staining of SPiDer β-gal and Oct4 in mice aortic roots, which develop atherosclerosis. Most of senescence positive areas in lesion, were not colocalized with Oct4 expressing cells (Figure 7H). All the data suggest that chronic upregulation of β-HB consequently reduces or delays cellular senescence via upregulating Oct4A expression.
DISCUSSION
In this study, we have demonstrated that β-HB prevents both replicative- and stress-induced senescence in vascular cells and reduces serum levels of the pro-senescence marker IL-1α in vivo. This prevention of senescence by β-HB is Oct4A-dependent and p53-independent. β-HB mediates Oct4A elevation by direct binding with hnRNP A1, induces vascular cell quiescence, thus prevents cellular senescence.
Our research provides a novel insight into the mechanism of β-HB-mediated anti-senescence in vascular tissues. Although many studies have focused on the beneficial effects of caloric restriction, which can extend lifespan and delay aging in various species (Colman et al., 2014; Minor et al., 2010), the effect and mechanism of caloric restriction on vascular cell quiescence and senescence remain poorly defined (Minamino and Komuro, 2007; Tian and Li, 2014; Uryga and Bennett, 2016). The principal finding of our study is the identification of the molecular target of β-HB using a ligand fishing approach. Ligand fishing approaches can still be highly effective in identifying the target molecules of small molecules such as pharmaceutic drugs or bio-active metabolites (Spring, 2005). With this approach, we identified that hnRNP A1 is a direct binding target of β-HB, and regulates the affinity for its target mRNAs.
Stress granule formation is an important cellular process to stabilize mRNAs under various stress conditions. Many RNA binding proteins, such as TDP-43 and hnRNPs, are aggregated with RNAs to prevent a stress-induced breakage and function as a storage for RNAs, which can cause re-initiation of gene expression (Guil et al., 2006; Hamilton et al., 1997; Henics et al., 1994). Based on recent studies demonstrating the regulation of Oct4 expression by hnRNP A2 in stem cells (Choi et al., 2013), we have tested that whether β-HB can modulate the binding affinity of hnRNP A1 with Oct4 mRNA in vascular cells. Our results show that the interaction of β-HB with hnRNP A1 enables hnRNP A1 to form a complex with Oct4 mRNAs, resulting in stabilization of Oct4 mRNA in vitro.
Our second important finding is the functional role of Oct4 in adult somatic cells, specifically vascular tissues. Oct4 activation induced by β-HB strongly triggers quiescent vascular tissue via upregulating p21. As a result of Oct4 activation by β-HB under caloric restriction, increased cell quiescence markers such as mTOR inhibition, AMPK activation, Lamin B1 elevation, and low transcriptional activity were observed, while decreased senescence indictors such as induction of γH2AX, SASP expression, and SA β-gal activity were noted. Although previous studies have concluded that Oct4 is permanently silenced in adult somatic cells (Buganim et al., 2013; Cherepanova et al., 2016), the role of Oct4 should be reconsidered because it can be reactivated in certain adult somatic cells. Our results along with recent reports support Oct4 reactivation, contrary to previous dogma. Recent evidence shows that DNA demethylation can be a mechanism of Oct4 activation (Cherepanova et al., 2016; Kellner and Kikyo, 2010). Our results suggest another possibility for Oct4 activation via hnRNP A1-mediated stabilization of Oct4 mRNA in normal and oxidative stress condition.
We have demonstrated that the vascular cell quiescence triggered by β-HB and hnRNP A1-mediated Oct4 activation exhibits strong oxidative stress resistance and protects against cellular senescence. Cellular quiescence is a crucial process for maintaining the stemness of embryonic stem cells (ESCs) through inducing autophagy. We did not show sufficient results to prove β-HB induces autophagy and autophagy-dependent cellular quiescence (An et al., 2014; Garcia-Prat et al., 2016). However, we predict that β-HB can induce autophagy in vascular cells, as well as ESCs, based on our observations of AMPK activation and mTOR inhibition by Oct4 upregulation after treating with β-HB in vascular cells. Future study will explore this hypothesis.
Interestingly, the anti-senescence effect of β-HB is exhibited more drastically in young cells. These observations can be explained when considering passage dependent reduction of Oct4 mRNA. Knowing that the mechanism of β-HB-induced Oct4 elevation is via enhanced stability of Oct4 mRNA, the function of β-HB is limited in the total absence of any mRNA. Therefore, we assume that the anti-senescence effect of β-HB will not occur when aging is progressed or in case of Oct4 genetic deletion. Additionally, as shown in Figure 2, quiescent cells induced by β-HB or serum deprivation promoted cell senescence in p53 hyper-activated cells stimulated by Nutlin-3a. Further studies are required to validate the role of β-HB in p53-dependent senescence or aging in vivo and in vitro.
Considering that aging is the most important non-modifiable risk factors for CVD and delaying or preventing vascular aging can protect again cardiovascular dysfunction, our study is significant because ketone body, produced during calorie restriction, delays vascular aging by preventing both replicative- and stress-induced senescence in vascular cells. This prevention of senescence by β-HB is via its direct binding of hnRNP A1 resulting in the stabilization of Oct4A mRNA and Oct4A expression. Targeting Oct4A by β-HB or β-HB-like compounds might be effective in preventing the occurrences or delaying the progression of CVD.
In summary, our studies reveal an important role for β-HB in cellular anti-senescence mediated Oct4 upregulation in the vasculature system. It provides novel strategy to prevent senescence-associated vascular aging via accumulating or maintaining quiescent vascular cells. Thus our research identifies Oct4 as a potent therapeutic target and β-HB and S-β-HB as a potent treatment for anti-aging or age-related vascular diseases.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to the Lead Contact, Ming-Hui Zou (mzou@gsu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Maintenance
Human umbilical vein endothelial cells (HUVECs, catalog no. LSC0035C) and human aortic smooth muscle cells (hASMCs, catalog no. C0075C), isolated from an individual donor were originally obtained from the Thermo Fisher Scientific (Waltham, MA, USA). HUVECs were cultured in EBM Basal Medium (Lonza, catalog no. CC-3121) supplemented with EGM SingleQuats (Lonza, catalog no. CC-4133) and hASMCs were cultured in Medium 231 (Gibco, catalog no. M231500) supplemented with SMGS (Gibco, catalog no. S00725). All culture media were supplemented with 5% heat-inactivated fetal bovine serum (FBS, Gibco), 100 U mL−1 penicillin, and 0.1 mg mL−1 streptomycin (Sigma-Aldrich) in a 37°C incubator with humidity and 5% CO2. HUVECs and hASMCs were obtained from single donor. And mice primary cells were isolated from 6 mice/group at the same day. The cells were maintained and regularly tested for mycoplasma contamination by PCR and confirmed to be free of any contamination.
Mice strains
C57BL/6J mice were purchased from the Jackson Laboratory. All mice were housed in temperature-controlled rooms with a 12 h light-dark cycle and given free access to water and food. After starvation for 5 h, 8 weeks-old male mice were injected intraperitoneally with PBS, β-HB, or S-β-HB (1.5 g/kg body weight in PBS). In parallel, 8 weeks-old male mice were randomized and fed a regular chow diet or fasted for 72 h. Then, the mice were sacrificed. Tissues were collected for gene expression evaluation in various organs including the aorta, liver, heart, brain, adipose, and kidney (n = 6). All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at Georgia State University.
Mouse model of atherosclerosis
8 weeks old male Apoe−/− mice (n = 10) were fed a western diet (21% milk fat, 0.15% cholesterol) for 8 weeks to establish aortic lesions. Mice aortic roots were fixed overnight in 4% paraformaldehyde, then embedded in optimum cutting temperature compound (OCT).
METHOD DETAILS
SA β-galactosidase assay
SA β-gal staining kit (Cell Signaling Technology, catalog no. 9860) was used to stain senescent cells. According to the manufacturer’s instructions (Cell Signaling Technology), HUVECs and hASMCs were fixed for 10 min, stained for 1 day and analyzed. Senescent cells, identified as blue-stained cells, were captured with light microscopy (Olympus IX73), then images were processed using imaging software (CellSens Dimension). Mice aortas were fixed for 10 min, stained for 1week and captured with light microscopy.
Fluorescein Di-β-D-Galactopyranoside assay (FDG)
Fluorescein Di-β-D-Galactopyranoside (FDG) was used to quantify the SA β-gal activity in HUVECs. FDG was dissolved in DMSO solution in which the sock solution (FDG, 200 mM) was stored at −20°C. Cells were cultured in 96-well plates, washed with PBS, then fixed with solution (2% formaldehyde, 0.2% glutaraldehyde). Staining solution (37 mM citric acid, 126 mM Na2HPO4, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 150 mM NaCl, 2 mM MgCl2) was added to each well, then 10 μL of 2 mM FDG was added to each well. After incubation in the dark at 37°C for 24 h without CO2 supply, the supernatant was transferred to a 96-well plate and fluorescein fluorescence was measured using the fluorometer (Tecan M1000) with a 485 nm excitation and a 535 nm emission wavelength. Genomic DNA of attached cells was measured with Qubit dsDNA HS assay kit in order to normalize β-galactosidase activity.
SPiDer β-galactosidase assay
Cells were washed with PBS, and SPiDer β-Gal working solution was added to the culture dish for 30 min at 37°C. After reaction, cells were washed with PBS, then fixed with 4% PFA/PBS solution. SA β-galactosidase positive cells were captured under fluorescence microscope with a 488 nm excitation and 533 nm emission wavelength. The mice aortas were separated into aortic arch and thoracic aorta to performed SPiDer β-gal assay. Aortic arch and thoracic aorta were embedded in optimum cutting temperature compound (OCT) compound. Then, 2.5 mm of aortic arch was sectioned every 0.5 mm from innominate artery to left subclavian, and 5 mm of thoracic aorta was sectioned every 1 mm from the middle portion of the descending thoracic aorta to diaphragm. Tissue slides were washed with PBS, and reacted by SPiDer β-gal solution for one hour at 37°C. After washing slides with PBS, SA β-galactosidase positive areas were captured under fluorescence microscope with a 488 nm excitation and 533 nm emission wavelength.
Synthesis of β-HB, Butyrate-conjugated beads, and S-β-HB salt
(S)-(+)-4-Amino-3-hydroxybutyric acid was dissolved in coupling buffer (0.2 M NaHCO3, 0.5 M NaCl, pH 8.3), and prewashed NHS-activated sepharose beads (10 mL) were added to the solution. The mixture was incubated in a 4°C shaker for 24 h. Then, the remaining NHS-activated groups were blocked with blocking buffer (0.5 M ethanolamine, 0.5 M NaCl, pH 8.3). β-HB and Butyrate-conjugated beads were washed with washing buffer (0.1 M Tris-HCl, pH 8.9). In order to prepare S-β-HB salt, (S)-hydroxybutyric acid was dissolved in acetone, then 0.1 mM of NaOH solution was added dropwise. Crystalized white power was washed by acetone and dried. 1H NMR (D2O): δ 4.07 (m, 1H), 2.33 (t, 1H), 2.21 (t, 1H), 1.12 (d, 3H).
Affinity chromatography
Cultured HUVECs were washed with PBS and then homogenized with a 26-gauge syringe in binding buffer (10 mM Tris-HCl, pH 7.4, 50 mM KCl, 5 mM MgCl2, 1 mM EDTA, and 0.1 mM N3VO4). The cell lysate was centrifuged at 13,000 rpm for 20 min at 4°C, and the supernatant was collected. In total, 4 mg of protein was obtained. The cell lysate was precleared by incubation with non-conjugated Sepharose beads and then loaded into a 15 mL volume of β-HB or Butyrate-conjugated Sepharose beads. After washing with 3 volumes of washing buffer (0.1 M Tris-HCl, pH 8.9), affinity chromatography was performed with β-HB gradient elution buffer (0.1 M Tris-HCl, β-HB 0.5 mM–8 mM). Samples were concentrated and separated by SDS-PAGE and visualized by Coomassie Brilliant Blue staining. The samples were then digested with trypsin, and peptide tandem mass spectrometry analysis of the digested peptides was performed using a Matrix-assisted laser desorption/ionization (MALDI) time-of-flight mass spectrometer.
Real-time RT-PCR
Total RNA was isolated using the RNeasy Mini Kit (QIAGEN, Cat no. 74106) and purified according to the manufacturer’s instruction. Synthesis of cDNA was conducted using iScript cDNA Synthesis Kit (Bio-Rad, Cat no. 1708890) according to the manufacturer’s instruction. Gene expression was quantified with the CFX96 real-time system (Bio-Rad) using the iQ SYBR Green supermix (Bio-Rad). The relative amounts of each mRNA were calculated by the comparative ΔCt method using GAPDH as house-keeping genes. Relative expression was calculated using the cycling threshold (Ct) method as 2−ΔCt.
Immunofluorescence staining
After β-HB treatment to the HUVECs, hASMCs or prepared thoracic aorta tissue of mice, samples were fixed with 4% paraformaldehyde (v/v) in PBS for 12 min at room temperature (RT), permeabilized with 0.2% Triton X-100 (v/v) in PBS for 10 min at RT, and blocked with 5% normal goat serum for 30 min at RT. Cells were incubated with primary antibodies (1: 500) at 37°C for 30 min, then followed by Alexa Fluor® 488 or 647 conjugated secondary antibodies (1: 500) at 37°C for 60 min. Images were captured using confocal microscope or fluorescence microscope.
Western blotting
Cell lysates were prepared in RIPA lysis buffer (50 mM Tris, pH 7.0, 150 mM NaCl, 5 mM EDTA, 1% deoxycholic acid, 0.1% SDS, 30 mM Na2HPO4, 50 mM NaF, and 1 mM NaVO4) containing protease inhibitor cocktail (Roche Applied Science). Proteins (40 μg) were resolved by 8 or 12% SDS-PAGE and transferred to PVDF membranes (Millipore). The membrane was blocked with 5% nonfat dry milk in TBS-T (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, and 0.1% Tween-20) and probed with primary antibodies overnight at 4°C. The secondary antibodies used were horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Cell Signaling Technology). The membranes were washed 3 times with TBS-T and then visualized with chemiluminescent peroxidase reagents (Roche, Germany).
Subcellular fractionation
Cells were harvested and homogenized by pass through a 27 gauge needle 10-times with fractionation buffer (150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM, protease inhibitors). Centrifuge the cell lysate at 800×G for 10 min. The pellet contains nuclei proteins and the supernatant contains cytoplasmic, mitochondrial and plasma-membrane proteins. Centrifuge supernatant at 10,000×G for 20min. The pellet contains mitochondrial proteins and the supernatant contains cytoplasmic and plasma-membrane proteins. Finally, Centrifuge supernatant at 100,000×G for 1 h. The supernatant contains pure cytoplasmic proteins and the pellet contains plasma-membrane proteins.
RNA-immunoprecipitation (RIP)
Cytoplasmic proteins were obtained through subcellular fractionation method described in Method part. Cell lysate was resuspended in freshly prepared RIP buffer (150 mM KCl, 25 mM Tris pH 7.4, 5 mM EDTA, 0.5 mM DTT, 0.5% NP40, 100 U/mL RNAase inhibitor, and protease inhibitor). Antibodies to hnRNP A1 were added to supernatant and incubated for overnight at 4°C with gentle rotation. Protein A/G beads were added, then incubated for 2 h at 4°C with gentle rotation. Precipitated beads were washed three times and resuspended with RIP buffer. Bound RNAs were extracted with TRIzol according to manufacturer’s instruction. Synthesis of cDNA was conducted using iScript cDNA Synthesis Kit according to the manufacturer’s instruction. Gene expression was quantified on the CFX96 real-time system (Bio-Rad) using the iQ SYBR Green supermix (Bio-Rad).
Cell cycle analysis
HUVECs and hASMCs were trypsinized from the culture dish after β-HB treatment and gathered by centrifugation. Precipitated cells were washed twice by repeating suspension and precipitation in PBS buffer. Precipitated cells were suspended with 500 μL PBS and fixed with 5 mL of ice-cold 70% ethanol overnight. Fixed cells were harvested and resuspended in PBS and treated with 100 μg/mL of RNase A at 37°C for 20 min. Propidium iodide (PI) was then added at a final concentration of 50 μg/mL for DNA staining, and 10,000 fixed cells were analyzed on a FACS Fortessa (BD Biosciences). For G0 phase analysis, Pyronin Y and Hoechst 33342 were used to stain mRNA and DNA separately. Cells were harvested in the same manner as described above, then re-suspend in culture media containing 10 μg/mL Hoechst 33342, incubated at 37°C for 15 min, then 5 μL of 100 ug/mL Pyronin Y was added. After a 15 min incubation, cells were transferred to ice and 10,000 cells were analyzed on a FACS Fortessa (BD Biosciences).
Oct4 knockdown by CRISPR-Cas9
Oct4A knockout HUVECs were prepared by CRISPR-Cas9-mediated gene editing. We used CRISPR design tool (http://tools.genome-engineering.org) to select Oct4 target sequences. Potential target oligomers for sgRNA constructs was designed with high efficiency score. Oct4 sgRNA sequence was 5’- CACCGGCACTAGCCCCACTCCAACC-3 ‘ and 5’-AAACGGTTGGAGTGGGGCTAGTGCC-3 ‘. Oct4 targeting gRNA was inserted into PX459 plasmid, then HUVECs were transfected with this plasmid. After transfection, the cells were selected with medium containing 2.0 μg/mL Puromycin.
Primary aortic smooth muscle cell isolation
Mice aortas were prepared from 6 mice to obtain sufficient cells for flow cytometry analysis. Dissected aortas are rinsed with PBS and placed into a dish with enzyme solution (1 mg/ml Collagenase Type II, 0.744 units/ml Elastase, PBS) containing 1% of penicillin-streptomycin. After incubation at 37°C for 10 minutes, aortas are placed into dish with DMEM/F12 media. Adventitia is removed by forceps, then aortas are chopped and incubated at 37°C in 5% CO2 incubator for 1 hour with enzyme solution. Chopped aortas are washed with media by centrifugation and placed into collagen coated plate. After 2 days, floating debris are removed by changing media, and cluster of primary cells were maintained up to two passages. 80% confluence of smooth muscle cells were fixed and analyzed by flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed at least three times and multiple samples represent biological replicates. All animal experiments were performed using randomly assigned mice. Statistical significance was tested using two-tailed Student’s t-test when the two group were compared. Statistical significance was tested using Two-way ANOVA with Bonferroni post hoc tests when multiple groups were compared. Values are the mean ± S.E. of three independent experiment. Values of p < 0.05, p < 0.01 and p < 0.001 are denoted by *, ** and *** or #, ##, ### respectively.
DATA AND SOFTWARE AVAILABILITY
The accession number for the original images reported in this paper is http://dx.doi.org/10.17632/9yzxwnf8ws.1
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-p16 | Abcam | Cat#ab189034 |
| Rabbit polyclonal anti-p21 | Cell Signaling | Cat#2947S; RRID: AB_823586 |
| Rabbit polyclonal anti-p27 | Cell Signaling | Cat#2552S; RRID: AB_10693314 |
| Rabbit polyclonal anti-p53 | Cell Signaling | Cat#9282S; RRID: AB_10693944 |
| Rabbit monoclonal anti-Oct4A | Thermo Fisher | Cat#A13998; RRID: AB_2534182 |
| Rabbit monoclonal anti-Oct4-FITC | NOVUS | Cat#NB100-2379F |
| Rabbit polyclonal anti-Lamin B1 | Cell Signaling | Cat#12586S; RRID: AB_2534182 |
| Rabbit polyclonal anti-Lamin A/C | Cell Signaling | Cat#2032S; RRID: AB_2136278 |
| Rabbit polyclonal anti-hnRNP A1 | Cell Signaling | Cat#8443S |
| Rabbit monoclonal anti-NPM1 | Thermo Fisher | Cat#MA5-12508; RRID: AB_10981922 |
| Rabbit polyclonal anti-IL-6 | Cell Signaling | Cat#12153S; RRID: AB_2687897 |
| Rabbit polyclonal anti-γH2AX | Cell Signaling | Cat#2577S; RRID: AB_2118010 |
| Rabbit polyclonal anti-p-CHK1 (S345) | Cell Signaling | Cat#2348S; RRID: AB_331212 |
| Rabbit polyclonal anti-p-BRCA1 (S1524) | Cell Signaling | Cat#9009S; RRID: AB_491003 |
| Rabbit polyclonal anti-p-RB (S780) | Cell Signaling | Cat#9307S; RRID: AB_330015 |
| Rabbit polyclonal anti-p-RB (S807, S811) | Cell Signaling | Cat#9308S; RRID: AB_331472 |
| Rabbit polyclonal anti-p-P70 (T389) | Cell Signaling | Cat#9205S; RRID: AB_330944 |
| Rabbit polyclonal anti-eIF4E | Cell Signaling | Cat#9742S; RRID: AB_823488 |
| Rabbit polyclonal anti-TIA-1 | Santa Cruz Bio | Cat#SC-166247; RRID: AB_2201545 |
| Rat monoclonal anti-CD31 | Abcam | Cat#ab56299; RRID: AB_940884 |
| Mouse monoclonal anti-a-smooth muscle Actin | Abcam | Cat#ab7817; RRID: AB_262054 |
| Mouse monoclonal anti-β-Actin | Santa Cruz Bio | Cat#SC-47778; RRID: AB_2714189 |
| Mouse monoclonal anti-α-Tubulin | Cell Signaling | Cat#3873S; RRID: AB_1904178 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| (R)-3-Hydroxybutyric acid | Sigma-Aldrich | 54920; CAS: 625-72-9 |
| (S)-3-Hydroxybutyric acid | Sigma-Aldrich | 54925; CAS: 6168-83-8 |
| Litium acetoacetate | Sigma-Aldrich | A8509; CAS: 3483-11-2 |
| Sodium butyrate | Sigma-Aldrich | 303410; CAS: 156-54-7 |
| Hydrogene Peroxide | Fisher Scientific | H325-500; CAS: 7722-84-1 |
| Propidium iodide | Sigma-Aldrich | P4170; CAS: 25535-16-4 |
| Aphidicolin | Sigma-Aldrich | A0781; CAS: 38966-21-1 |
| Nutlin-3a | Sigma-Aldrich | SML0580; CAS: 675576-98-4 |
| Actinomycin D | Sigma-Aldrich | A1410; CAS: 50-76-0 |
| NHS-Activated Sepharose 4 Fast Flow | GE Healthcare | Cat#17090601 |
| (S)-(+)-4-Amino-3-hydroxybutyric acid | Sigma-Aldrich | 542946; CAS: 7013-05-0 |
| γ-Aminobutyric acid | Sigma-Aldrich | A2129; CAS: 56-12-2 |
| Ethyl (R)-(−)-3-hydroxybutyrate | Sigma-Aldrich | 347329; CAS: 24915-95-5 |
| Protein A/G Magnetic Beads | Thermo Fisher | Cat#88802 |
| Fluorescein Di-β-D-Galactopyranoside (FDG) | Thermo Fisher | F1179; CAS: 17817-20-8 |
| SPiDer-β-Gal | Dojindo | SG02-10; CAS: 1824699-57-1 |
| Sucrose | Sigma-Aldrich | S1888; CAS: 57-50-1 |
| Albumin, Bovine Fraction V (BSA) | RPI | A30075-100; CAS: 9048-46-8 |
| Fetal bovine sereum (FBS) | Sigma-Aldrich | Cat#12303C |
| Penicillin-Streptomycin | Thermo Fisher | 15140122; CAS: 69-57-8 |
| EBM basal Medium | Lonza | Cat#CC-3121 |
| Media 231 | Gibco | Cat#M-231-500 |
| DMEM/F12 medium | Gibco | Cat#11320 |
| Collagenase Type II | Worthington Biochemical |
LS004174; CAS: 9001-12-1 |
| Elastase | Worthington Biochemical |
LS002279; CAS: 39445-21-1 |
| EGM SingleQuat Kit Suppl. & Growth Factors | Lonza | Cat#CC-4133 |
| Smooth Muscle Growth Supplement (SMGS) | Thermo Fisher | Cat#S00725 |
| Critical Commercial Assays | ||
| Senescence β-Galactosidase Staining Kit | Cell Signaling | 9860 |
| Fluorescein Di-β-D-Galactopyranoside | Invitrogen | F1179 |
| SPiDer β- galactosidase assay | Dojindo | SG02-10 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708890 |
| β-Hydroxybutyrate Colorimetric Assay Kit | BioVision | K632 |
| Annexin-V-FLUOS Staining Kit | Cell Signaling | 11988549001 |
| Mouse IL-6 ELISA MAX Deluxe | BioLegend | 431306 |
| Mouse IL-1α ELISA MAX Deluxe | BioLegend | 433406 |
| Experimental Models: Cell Lines | ||
| Human: Umbilical Vein Endo Cells (HUVECs) | Thermo Fisher | LSC0035C |
| Human: Aortic Smooth Muscle Cells (hASMCs) | Thermo Fisher | C0075C |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J (3 weeks) | The Jackson Lab | JAX000664; RRID: IMSR_JAX:000664 |
| Mouse: Aged C57BL/6J (64 weeks) | The Jackson Lab | JAX000664; RRID: IMSR_JAX:000664 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | Cat#48138 |
| CRISPR/Cas9-Oct4 | This paper | N/A |
| Oct4-myc | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad Software | http://graphpad.com |
| ImageJ Software | ImageJ | http://www.imagej.net |
| ChemBio Ultra 14.0 | PerkinElmer |
http://www.cambridgesoft.com om |
| Zen | ZEISS Microscope | https://www.zeiss.com |
| FlowJo | FlowJo LLC | |
| Deposited data | This paper | http://dx.doi.org/10.17632/t57mvdfz39.1 |
| Primer | Forward (5’ to 3’) | Reverse (5’ to 3’) |
|---|---|---|
| Oct4A | CGTGAAGCTGGAGAAGGAGAAGCTG | CAAGGGCCGCAGCTTACACATGTTC |
| Oct4B | ATGCATGAGTCAGTG AACAG | CCACATCGGCCTGTGTATAT |
| IL-1α | ATCAGTACCTCACGGCTGCT | TGGGTATCTCAGGCATCTCC |
| IL-6 | CCAGCTATGAACTCCTTCTC | GCTTGTTCCTCACATCTCTC |
| IL-8 | ATGACTTCCAAGCTGGCCGTGGCT | TCTCAGCCCTCTTCAAAAACTTCTC |
| SOX2 | GGGAAATGGGAGGGGTGCAAAAGAGG | TTGCGTGAGTGTGGATGGGATTGGTG |
| KLF4 | GTTTTGAGGAAGTGCTGAG | CAGTCACAGTGGTAAGGTTT |
| c-Myc | GTTGGTCAGGCTGGTCTTGAA | CATGCGCCTGTAATCCTAGCA |
| Nanog | ACCAGAACTGTGTTCTCTTCCACC | GGTTGCTCCAGGTTGAATTGTTCC |
| GAPDH | CAACTTTGGCATTGTGGAAGG | ACACATTGGGG GTAGGAACAC |
Highlights.
β-hydroxybutyrate prevents the vascular cell senescence.
β-hydroxybutyrate upregulates Oct4 expression via interacting with hnRNP A1.
Oct4-mediated quiescence is able to attenuate hallmarks of senescence.
Circulating β-hydroxybutyrate alleviates the senescence of mouse aorta.
ACKNOWLEDGEMENTS
This study was supported by the National Institutes of Health grants (HL07958 4, HL080499, HL089920, HL110488, HL128014, HL132500, HL137371, HL140954, HL142287, AG047776, and CA213022).
This work was, in part, supported by the Georgia Research Alliance. Dr. Zou is a Georgia Research Alliance Eminent Scholar in Molecular Medicine.
Footnotes
Lead Contact
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- An Z, Tassa A, Thomas C, Zhong R, Xiao G, Fotedar R, Tu BP, Klionsky DJ, and Levine B (2014). Autophagy is required for G(1)/G(0) quiescence in response to nitrogen starvation in Saccharomyces cerevisiae. Autophagy 10, 1702–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton SD, Karabetian C, Heekin K, and Leeuwenburgh C (2013). Caloric Restriction to Moderate Senescence: Mechanisms and Clinical Utility. Curr Transl Geriatr Exp Gerontol Rep 2, 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredesen DE, Amos EC, Canick J, Ackerley M, Raji C, Fiala M, and Ahdidan J (2016). Reversal of cognitive decline in Alzheimer's disease. Aging 8, 1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, and Jaenisch R (2013). Mechanisms and models of somatic cell reprogramming. Nat Rev Genet 14, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J, Weiss ML, and Rao MS (2004). In search of "sternness". Exp Hematol 32, 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, Alencar GF, Hess DL, Bevard MH, Greene ES, et al. (2016). Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med 22, 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TH, and Rando TA (2013). Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol 14, 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, and van Deursen JM (2016). Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Durik M, Baker DJ, and van Deursen JM (2015). Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21, 1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Lee HM, Jang YJ, Kim CH, and Ryu CJ (2013). Heterogeneous nuclear ribonucleoprotein A2/B1 regulates the self-renewal and pluripotency of human embryonic stem cells via the control of the G1/S transition. Stem Cells 31, 2647–2658. [DOI] [PubMed] [Google Scholar]
- Collado M, and Serrano M (2010). Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman RJ, Beasley TM, Kemnitz JW, Johnson SC, Weindruch R, and Anderson RM (2014). Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat Commun 5, 3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino S, Paneni F, and Cosentino F (2016). Ageing, metabolism and cardiovascular disease. J Physiol 594, 2061–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Serrano M, and Blasco MA (2007). The common biology of cancer and ageing. Nature 448, 767–774. [DOI] [PubMed] [Google Scholar]
- Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. [DOI] [PubMed] [Google Scholar]
- Guil S, Long JC, and Caceres JF (2006). hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol Cell Biol 26, 5744–5758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BJ, Burns CM, Nichols RC, and Rigby WF (1997). Modulation of AUUUA response element binding by heterogeneous nuclear ribonucleoprotein A1 in human T lymphocytes. The roles of cytoplasmic location, transcription, and phosphorylation. J Biol Chem 272, 28732–28741. [DOI] [PubMed] [Google Scholar]
- Henics T, Sanfridson A, Hamilton BJ, Nagy E, and Rigby WF (1994). Enhanced stability of interleukin-2 mRNA in MLA 144 cells. Possible role of cytoplasmic AU-rich sequence-binding proteins. J Biol Chem 269, 5377–5383. [PubMed] [Google Scholar]
- Ito K, Sportoletti P, Clohessy JG, Silvia G, and Pandolfi PP (2010). The Role of Nucleophosmin In Hematopoietic Stem Cells and the Pathogenesis of Myelodysplastic Syndrome. Blood 116, 48–48. [Google Scholar]
- Jean-Philippe J, Paz S, and Caputi M (2013). hnRNP A1: the Swiss army knife of gene expression. International journal of molecular sciences 14, 18999–19024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellner S, and Kikyo N (2010). Transcriptional regulation of the Oct4 gene, a master gene for pluripotency. Histol Histopathol 25, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Chen CC, Monzon RI, and Lau LF (2013). Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol 33, 2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacic JC, Moreno P, Hachinski V, Nabel EG, and Fuster V (2011). Cellular senescence, vascular disease, and aging: Part 1 of a 2-part review. Circulation 123, 1650–1660. [DOI] [PubMed] [Google Scholar]
- Lengner CJ, Welstead GG, and Jaenisch R (2008). The pluripotency regulator Oct4: a role in somatic stem cells? Cell Cycle 7, 725–728. [DOI] [PubMed] [Google Scholar]
- Liu R, Liu H, Ha Y, Tilton RG, and Zhang W (2014). Oxidative stress induces endothelial cell senescence via downregulation of Sirt6. Biomed Res Int 2014, 902842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Mattson MP, and Maudsley S (2006). Caloric restriction and intermittent fasting: two potential diets for successful brain aging. Ageing research reviews 5, 332–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, Chen S, Maya-Mendoza A, Lovric J, Sims PF, and Jackson DA (2009). Lamin B1 maintains the functional plasticity of nucleoli. Journal of cell science 122, 1551–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, and Komuro I (2007). Vascular cell senescence: contribution to atherosclerosis. Circ Res 100, 15–26. [DOI] [PubMed] [Google Scholar]
- Minor RK, Allard JS, Younts CM, Ward TM, and de Cabo R (2010). Dietary interventions to extend life span and health span based on calorie restriction. J Gerontol A Biol Sci Med Sci 65, 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning YC, Cai GY, Zhuo L, Gao JJ, Dong D, Cui S, Feng Z, Shi SZ, Bai XY, Sun XF, et al. (2013). Short-term calorie restriction protects against renal senescence of aged rats by increasing autophagic activity and reducing oxidative damage. Mech Ageing Dev 134, 570–579. [DOI] [PubMed] [Google Scholar]
- North BJ, and Sinclair DA (2012). The intersection between aging and cardiovascular disease. Circ Res 110, 1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoli A, Bianco A, Damiani E, and Bosco G (2014). Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed Res Int 2014, 474296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M, Dillon C, Narita M, Young AR, Cairney CJ, Godwin LS, Torrance CJ, Bennett DC, Keith WN, and Narita M (2015). Cell-based screen for altered nuclear phenotypes reveals senescence progression in polyploid cells after Aurora kinase B inhibition. Mol Biol Cell 26, 2971–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivia M, Ceballos-Perez G, Bart JM, and Navarro M (2016). The AMPKalpha1 Pathway Positively Regulates the Developmental Transition from Proliferation to Quiescence in Trypanosoma brucei. Cell reports 17, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmenpera P, Karhemo PR, Rasanen K, Laakkonen P, and Vaheri A (2016). Fibroblast spheroids as a model to study sustained fibroblast quiescence and their crosstalk with tumor cells. Exp Cell Res 345, 17–24. [DOI] [PubMed] [Google Scholar]
- Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, and Goldman RD (2011). The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 25, 2579–2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring DR (2005). Chemical genetics to chemical genomics: small molecules offer big insights. Chem Soc Rev 34, 472–482. [DOI] [PubMed] [Google Scholar]
- Stone GW, Lansky AJ, Pocock SJ, Gersh BJ, Dangas G, Wong SC, Witzenbichler B, Guagliumi G, Peruga JZ, Brodie BR, et al. (2009). Paclitaxel-eluting stents versus bare-metal stents in acute myocardial infarction. N Engl J Med 360, 1946–1959. [DOI] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Tchkonia T, Zhu Y, van Deursen J, Campisi J, and Kirkland JL (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123, 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian XL, and Li Y (2014). Endothelial cell senescence and age-related vascular diseases. J Genet Genomics 41, 485–495. [DOI] [PubMed] [Google Scholar]
- Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy Rv et al. (2003). D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest 112, 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uryga AK, and Bennett MR (2016). Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J Physiol 594, 2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM (2014). The role of senescent cells in ageing. Nature 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, and Bennett M (2012). Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res 111, 245–259. [DOI] [PubMed] [Google Scholar]
- Will B, Vogler TO, Bartholdy B, Garrett-Bakelman F, Mayer J, Barreyro L, Pandolfi A, Todorova TI, Okoye-Okafor UC, Stanley RF, et al. (2013). Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nature immunology 14, 437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AB, and Schumacher B (2016). p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Lu LY, and Yu X (2010). The role of BRCA1 in DNA damage response. Protein Cell 1, 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhou Y, Coughlan KA, Ding Y, Wang S, Wu Y, Song P, and Zou MH (2015). AMPKalpha1 deficiency promotes cellular proliferation and DNA damage via p21 reduction in mouse embryonic fibroblasts. Biochim Biophys Acta 1853, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, and Brody LC (2002). BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet 30, 285–289. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Armstrong JL, Tchkonia T, and Kirkland JL (2014). Cellular senescence and the senescent secretory phenotype in age-related chronic diseases. Curr Opin Clin Nutr Metab Care 17, 324–328. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.