

Key Clinical Message
We present a patient with a clinical diagnosis of Joubert syndrome with COACH phenotype who carries two TMEM67 variants of uncertain significance (VUS). One VUS can be reclassified as “likely pathogenic” by adding clinical data. As genetic testing becomes more accessible, more VUS will require clinical correlation for accurate classification.
Keywords: genetic testing, Joubert syndrome, molar tooth sign, TMEM67, variants of uncertain significance
1. INTRODUCTION
We present a patient with hypotonia, developmental delays, hepatomegaly, low platelet counts, and ocular colobomas, consistent with Joubert syndrome with COACH phenotype. She carries two variants in TMEM67. Reclassification in light of clinical information suggests pathogenicity of the c.517T>C (p.Cys173Arg) variant. Early MRI imaging missed a molar tooth sign.
Joubert syndrome (JS) is an autosomal recessive disorder characterized by a cerebellar and brainstem malformation called the “molar tooth sign,” hypotonia with eventual ataxia and developmental delays.1 The molar tooth sign involves hypoplasia of the cerebellar vermis, thick superior cerebellar peduncles, and a particularly deep interpeduncular fossa and is mandatory in the diagnosis of Joubert syndrome.2
Joubert syndrome is one of six subgroups of phenotypes that all present with the molar tooth sign. All six are classified in Joubert syndrome and related disorders (JSRD) group: (a) pure JS, (b) JS with ocular defect, (c) JS with renal defect, (d) JS with oculorenal defects, (e) JS with hepatic defect, and (f) JS with orofaciodigital defects.3 Although the true prevalence of JSRD is unknown, estimates range from 1:80 000 to 1:100 000.3, 4
The clinical heterogeneity of JSRD is reflected in significant genetic heterogeneity with some genotype‐phenotype correlations. Biallelic pathogenic variants have been identified in approximately 50% of patients with JSRD. Thirty‐four genes have been identified in the pathogenesis; alterations in the following genes are most commonly found: AHI1, CPLANE1, CC2D2A, CEP290, CSPP1, INPP5E, KIAA0586, MKS1, NPHP1, RPGRIP1L, TCTN2, TMEM67, and TMEM216.5 JSRD are ciliopathies as these genes encode ciliary structural and regulatory proteins.
Joubert syndrome with hepatic defect is also known as COACH syndrome: Cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma, and hepatic fibrosis.6 Mutations in TMEM67 (MKS3) are responsible for the majority of COACH syndrome, although mutations in CC2D2A and RPGRIP1L can also be causative.7 TMEM67/MKS3 is found on chromosome 8 (8q22.1) and encodes a 955‐amino acid seven‐transmembrane receptor protein that is thought to regulate cilia function and mutations in TMEM67/MKS3 can also cause Meckel‐Gruber syndrome (MKS), another autosomal recessive ciliopathy that is associated with renal cystic disease.8, 9, 10, 11 Although mutations in TMEM67/MKS3 are causative for both syndromes (accounting for 9% of JSRD cases and 16% of MKS cases), a correlation between missense mutations in exons 8‐15, particularly in combination with truncating mutations, and Meckel‐Gruber syndrome has been identified.7, 12
To evaluate pathogenicity of variants, laboratories rely on the current literature, functional validation and databases, and computational (in silico) predictive programs.13 However, as these variants may have been previously unseen or insufficient data have been collected on them, the absence of information results in variants being interpreted as neither benign nor pathogenic, but of unknown significance. Clinical correlation also contributes to variant classification, which can be difficult for the testing laboratory to obtain or to interpret. As genetic testing becomes more accessible, it is likely that more variants of unknown significance (VUS) will surface, requiring classification.
Here, we present a patient without a family history of Joubert syndrome who carries two heterozygous variants, classified by the testing laboratory as variants of uncertain significance, in TMEM67: c517T>C (p.Cys173Arg) and c.934T>C (p.Ser312Pro) and the following clinical findings: molar tooth sign, hypotonia, developmental delays, hepatomegaly, low platelet counts, and ocular colobomas, consistent with Joubert syndrome with the COACH phenotype.
2. CASE PRESENTATION
The proband is a 22‐year‐old woman of Hispanic ancestry (Figure 1). The proband's family history includes cancer diagnoses in her grandparents’ generation, but no other history of Joubert syndrome and no consanguinity. According to the proband's mother, the proband had developmental delays starting at 2 months of age when she would not follow her toys with her eyes. Motor developmental delay, difficulty with balance, and learning delays were subsequently observed. Due to these developmental delays, the proband had multiple brain magnetic resonance imaging (MRI) and computerized tomography (CT) imaging studies performed during the first 3‐4 years of her life. However, the “molar tooth sign” characteristic of Joubert syndrome was not noted in any of them, and the proband was given the diagnosis of mild cerebral palsy.
Figure 1.
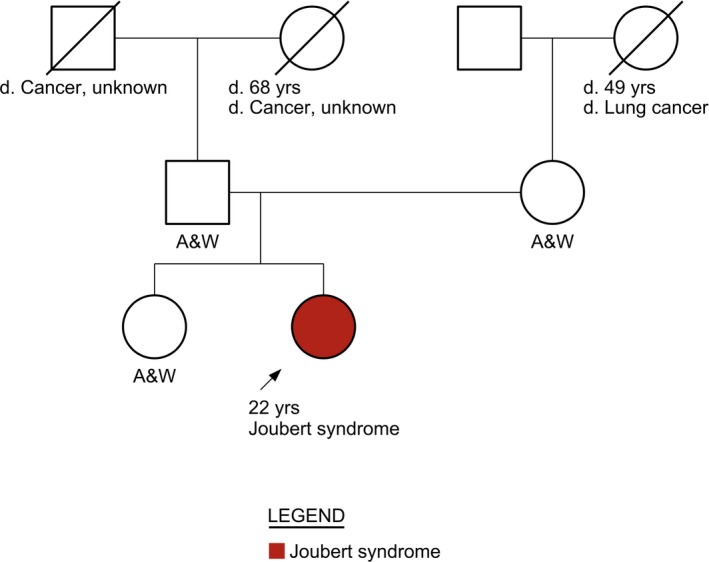
Family pedigree. Arrow depicts the proband. Women are represented by circles. Men are represented by squares. A diagonal slash indicates a deceased family member
When the proband turned 18 years old, her primary care provider referred her to the children's clinic. The brain MRI was repeated and the “molar tooth sign” was reported, with note made of hypoplasia of the cerebellar vermis, thick superior cerebellar peduncles, and a particularly deep interpeduncular fossa (Figure 2). The proband was then referred to genetics, which ordered genetic testing and subsequently diagnosed her with Joubert syndrome. Genetics noted her low platelet count and ordered an abdominal ultrasound that revealed hepatosplenomegaly. This was consistent with the COACH phenotype of Joubert syndrome, and genetic testing was ordered to evaluate genes known to be associated with this phenotype.
Figure 2.
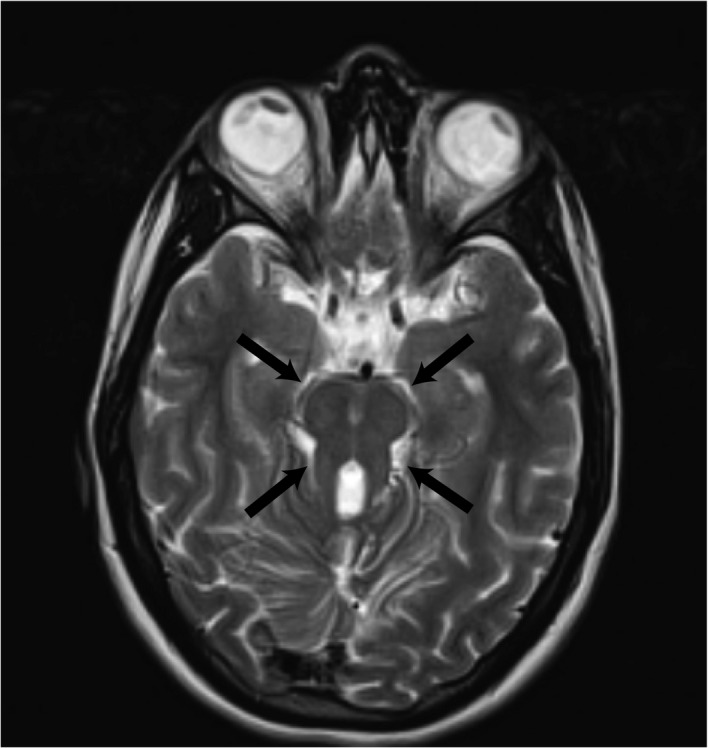
Axial T2‐weighted brain MRI taken when the patient was 19 years old. Black arrows point to “molar tooth” sign
Genetic testing was performed through a commercial genetic testing laboratory and revealed two missense variants in the TMEM67 gene, which they classified as variants of uncertain significance (VUS). The proband is heterozygous for both variants. Additional family samples could not be obtained for segregation analysis.
The first TMEM67 variant identified is a T to C substitution at nucleotide position 517 in exon 5 (dbSNP: 138783896; Table 1). This results in a change from the amino acid cysteine at position 173 to an arginine: c.517T>C; p.(Cys173Arg). This amino acid change falls in a cysteine‐rich domain in TMEM67 that spans amino acids 50‐18712 and mediates signaling through WNT5A interactions.8, 14 Mutations in similar regions in TMEM67 have been found in patients with Joubert syndrome and Joubert syndrome with the COACH (cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma, and hepatic fibrosis) phenotype. One patient with the COACH phenotype was found to carry a missense mutation at nucleotide position 515, with a G to A substitution (c.515G>A (p.Arg172Gln)).7 These are all consistent with this variant meeting ACMG PM1 criteria.13 This variant has been reported in population databases with a frequency in ExAC of 1 of 120 000, which is consistent with an autosomal recessive condition (ACMG PM2). In silico analyses carried out to predict the effect of this missense change on protein structure and function suggest pathogenicity (SIFT: “deleterious”; PolyPhen‐2: “Probably damaging”; CADD score for this variant is 27.2)15 and this site is entirely conserved evolutionarily (ACMG PP3). As we have shown above, the patient's family history and clinical phenotype are entirely consistent with the proposed disease (ACMG PP4). Thus, with two moderate (PM1 and PM2) and two supporting (PP3 and PP4) ACMG criteria, we argue that this VUS should be reclassified as “likely pathogenic.”13
Table 1.
Information on TMEM67 variants found in patient
| Gene | Variant | Zygosity | Variant Classification | In silico analysis | Population Database |
|---|---|---|---|---|---|
| TMEM67 | c.517T>C (p.Cys173Arg) | Heterozygous | VUS |
SIFT: deleterious PolyPhen‐2: probably damaging Align‐GVGD: Class C0 CADD: 27.2 |
Present, 0.0008% |
| TMEM67 | c.934T>C (p.Ser312Pro) | Heterozygous | VUS |
SIFT: likely tolerated PolyPhen‐2: likely tolerated Align‐GVGD: likely tolerated CADD: 2.2 |
Not present |
The second variant is a T to C substitution at nucleotide position 934 (dbSNP: 864622335; Table 1). This results in a change from the amino acid serine at position 312 to proline: c.934T>C; p.(Ser312Pro). Although this variant is not in a known functional domain, other patients with Joubert syndrome have been found to carry missense mutations at nearby nucleotide positions 903 and 986 (c.903C>G (p.Asp301Glu)) and (c.986A>C (p.Lys329Thr)).12, 16 This variant is a novel missense change that is not present in any population databases (ACMG PM2). In silico analyses to predict the effect of this missense change on protein structure and function all predict that this variant is likely to be tolerated; however, the predictions have not been confirmed by published functional studies. The CADD score for this variant is 2.2.15 This variant classifies as a VUS with ACMG PM2 and PP4 criteria being met.13
Thus, in addition to developmental delays, the proband's hypotonia, hepatomegaly, ocular colobomas, and low platelet counts point to Joubert syndrome with the COACH phenotype. Genetic testing identified two TMEM67 variants. Upon closer examination, the c.517T>C; p.(Cys173Arg) meets criteria for a likely pathogenic mutation, while the c.934T>C; p.(Ser312Pro) changes continue to be consistent with a VUS.
3. DISCUSSION
We present a patient with a clinical diagnosis of Joubert syndrome with the COACH phenotype who carries two TMEM67 missense variants that were classified by the commercial testing laboratory as variants of uncertain significance. With additional clinical evaluation, one of these meets ACMG criteria for classification as likely pathogenic. Our study demonstrates (a) the value of repeated MRI evaluation and/or communication with the radiology department to ensure that subtle findings are reported since the molar tooth sign was missed for >10 years on repeated imaging and (b) the need for clinical correlation in the evaluation of rare variants. Making this diagnosis identified the cause of hepatomegaly and low platelet counts, consistent with Joubert syndrome with the COACH (cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma, and hepatic fibrosis) phenotype, avoiding the need of extensive additional evaluation and ensuring appropriate referral to hepatology for monitoring.
This case emphasizes the importance of assessing genetic information in the setting of clinical data and of communication between specialists. Here, a clear clinical phenotype consistent with Joubert syndrome clarifies the meaning of an unclear genotype, and recognition of a molar tooth sign could have hastened diagnosis by a decade.
4. METHODS
4.1. Mutation identification
Next‐generation sequencing was performed by a commercial laboratory to identify mutations in a panel that included the following genes: AHI1, ARL13B, B9D1, CC2D2A, CEP290, INPP5E, MKS1, NPHP1, OFD1, RPGRIP1L, TCTN1, TCTN2, TMEM216, TMEM67, and TTC21B.
ACKNOWLEDGMENTS
We wish to thank Laurel Johnstone for assistance in variant reclassification.
AUTHORSHIP
JMH: analyzed and interpreted the data, drafted the manuscript, and critically revised the manuscript. MG: involved in patient management and collected the data. CML: involved in patient management, analyzed and interpreted the data, drafted the manuscript, and critically revised the manuscript. JMH, MG, and CML: approved the final manuscript.
CONFLICT OF INTEREST
None declared.
Huynh JM, Galindo M, Laukaitis CM. Missense variants in TMEM67 in a patient with Joubert syndrome. Clin Case Rep. 2018;6:2189–2192. 10.1002/ccr3.1748
REFERENCES
- 1. Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43(4):726‐731. [DOI] [PubMed] [Google Scholar]
- 2. Maria BL, Hoang KB, Tusa RJ, et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12(7):423‐430. [DOI] [PubMed] [Google Scholar]
- 3. Brancati F, Dallapiccola B, Valente EM. Joubert syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet. 2007;15(5):511‐521. [DOI] [PubMed] [Google Scholar]
- 5. Parisi M, Glass I. Joubert Syndrome and Related Disorders In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, eds. GeneReviews(R). Seattle, WA: University of Washington; 1993:1993‐2003. [Google Scholar]
- 6. Verloes A, Lambotte C. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. Am J Med Genet. 1989;32(2):227‐232. [DOI] [PubMed] [Google Scholar]
- 7. Doherty D, Parisi MA, Finn LS, et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet. 2010;47(1):8‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Smith UM, Consugar M, Tee LJ, et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel‐Gruber syndrome and the wpk rat. Nat Genet. 2006;38(2):191‐196. [DOI] [PubMed] [Google Scholar]
- 9. Dawe HR, Smith UM, Cullinane AR, et al. The Meckel‐Gruber Syndrome proteins MKS1 and meckelin interact and are required for primary cilium formation. Hum Mol Genet. 2007;16(2):173‐186. [DOI] [PubMed] [Google Scholar]
- 10. Leightner AC, Hommerding CJ, Peng Y, et al. The Meckel syndrome protein meckelin (TMEM67) is a key regulator of cilia function but is not required for tissue planar polarity. Hum Mol Genet. 2013;22(10):2024‐2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baala L, Romano S, Khaddour R, et al. The Meckel‐Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80(1):186‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iannicelli M, Brancati F, Mougou‐Zerelli S, et al. Novel TMEM67 mutations and genotype‐phenotype correlates in meckelin‐related ciliopathies. Hum Mutat. 2010;31(5):E1319‐E1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abdelhamed ZA, Natarajan S, Wheway G, et al. The Meckel‐Gruber syndrome protein TMEM67 controls basal body positioning and epithelial branching morphogenesis in mice via the non‐canonical Wnt pathway. Dis Model Mech. 2015;8(6):527‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Otto EA, Ramaswami G, Janssen S. Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet. 2011;48(2):105‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]